

Abstract
A total synthesis of the Aspidosperma alkaloid quebrachamine in racemic form is first described. A key catalytic ring-closing metathesis of an achiral triene is used to establish the all-carbon quaternary stereogenic center and the tetracyclic structure of the natural product; the catalytic transformation proceeds with reasonable efficiency through the use of existing achiral Ru or Mo catalysts. Ru- or Mo-based chiral olefin metathesis catalysts have proven to be inefficient and entirely nonselective in cases where the desired product is observed. In the present study, the synthesis route thus serves as a platform for the discovery of new olefin metathesis catalysts that allow for efficient completion of an enantioselective synthesis of quebrachamine. Accordingly, on the basis of mechanistic principles, stereogenic-at-Mo complexes bearing only monodentate ligands have been designed. The new catalysts provide significantly higher levels of activity than observed with the previously reported Ru- or Mo-based complexes. Enantiomerically enriched chiral alkylidenes are generated through diastereoselective reactions involving achiral Mo-based bispyrrolides and enantiomerically pure silyl-protected binaphthols. Such chiral catalysts initiate the key enantioselective ring-closing metathesis step in the total synthesis of quebrachamine efficiently (1 mol % loading, 22 °C, 1 h, >98% conversion, 84% yield) and with high selectivity (98:2 er, 96% ee).
Introduction
Herein, we first outline a scheme for a total synthesis of the Aspidosperma alkaloid quebrachamine, conceived specifically to challenge the state-of-the-art in catalytic olefin metathesis,1 with particular emphasis on enantioselective transformations.2 We detail a concise synthesis of racemic quebrachamine3,4 by a route that underscores the need for substantially more efficient and selective olefin metathesis catalysts. In the second section, we illustrate how, guided by mechanistic principles and inspired by recent theoretical investigations,5 we have been able to design, synthesize, and develop an exceptionally effective new class of stereogenic-at-Mo olefin metathesis catalysts that readily promote a highly enantioselective synthesis of the target alkaloid.6
Several aspects of the investigations described in this account are noteworthy: (1) Synthesis of the chiral catalysts, which bear a stereogenic metal center, is accomplished through an unprecedented diastereoselective desymmetrization of Mo-based bispyrrolides, carried out by selective protonation of one of the two Mo–N bonds. (2) The stereogenic-at-Mo complexes developed are, to the best of our knowledge, the first effective chiral catalysts that bear a stereogenic metal center and contain only monodentate ligands. (3) The new catalysts represent a rare case of the successful use of a monodentate O-based chiral ligand in enantioselective catalysis [i.e., >95:5 enantiomer ratio (er) obtained].
Enantioselective preparation of a complex molecule provides an invaluable framework for the design of new methods and catalysts for efficient and selective chemical synthesis. It is often in the course of a total synthesis that shortcomings in the existing repertoire of protocols and/or catalysts become evident. Target-molecule synthesis offers the means to illustrate the advantages and limitations of a catalytic protocol, but more importantly, it can furnish a springboard for avenues of research that would otherwise remain unexplored.7 As part of our program directed toward discovery and development of catalysts that promote a range of enantioselective C–C bond formation reactions, we have adopted the general strategy of designing synthesis routes that challenge the effectiveness of the existing catalysts and methods; synthesis plans that require catalyst innovation to achieve a successful enantioselective synthesis are thus favored.
Our interest in the development of more effective chiral catalysts for enantioselective olefin metathesis spans more than a decade of investigations, during which time we have introduced a number of chiral Mo- and Ru-based complexes that promote highly enantioselective ring-closing8,9 as well as ring-opening metathesis reactions.10 Such transformations offer unique opportunities for enantioselective synthesis of small organic molecules that would not be accessible by alternative approaches, such as those involving the use of achiral olefin metathesis catalysts and enantiomerically pure or highly enriched substrates secured through alternative protocols. Nonetheless, application of catalytic enantioselective olefin metathesis to natural product synthesis remains relatively uncommon, particularly in comparison with the myriad complex-molecule syntheses achieved through utilization of the corresponding achiral catalysts.11 One rationale for the aforementioned paucity is that target molecules traditionally are selected subsequent to method development; as a result, a protocol may not provide an ideal solution for application to any particular total synthesis problem. In our judgment, it would be more expedient to first secure the most direct synthesis route that benefits from catalytic enantioselective processes. The effectiveness of the existing chiral complexes would then be evaluated, and if necessary, any attendant shortcomings would be addressed by new catalyst design.
Our decision to focus on the adrenergic blocking agent quebrachamine (Scheme 1),12,3,4 a tetracycle from the Aspidosperma group of alkaloids,13 two other members of which are also illustrated in Scheme 1, was based on several factors. First, a catalytic olefin metathesis reaction would have to be designed that allows for an effective transformation, likely involving sterically congested alkenes.14 Second, the optimal chiral catalyst must allow for control of the absolute stereochemistry of an all-carbon quaternary stereogenic center, which represents a difficult and important challenge in modern chemical synthesis.15 Third, an effective catalyst would have to remain operative in the presence of the Lewis basic amine, a functional unit that has been shown to cause deactivation of Mo alkylidenes8c as well as Ru-based carbenes.16
Scheme 1.
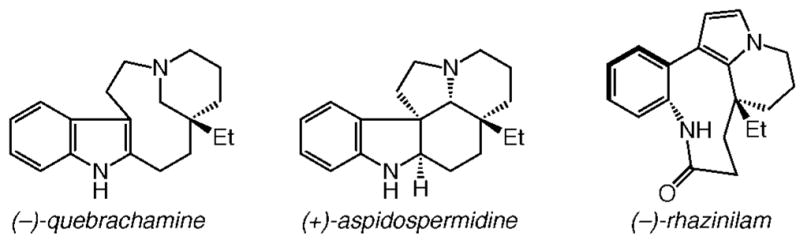
Representative Members of the Aspidosperma Group of Alkaloid Natural Products
Results and Discussion
I. Total Synthesis of rac-Quebrachamine
1. Retrosynthesis Plan
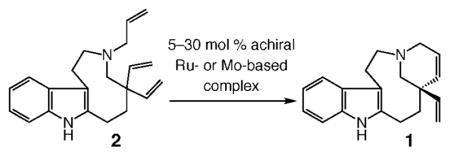
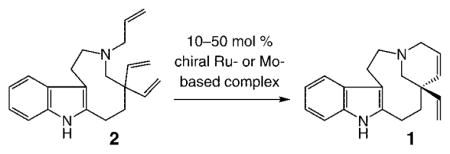
The retrosynthesis plan adopted in this investigation is outlined in Scheme 2. We envisioned that formation of the tetracyclic moiety of the target alkaloid could be effected through enantioselective ring-closing metathesis (RCM) of triene 2, concomitant with control of the all-carbon quaternary stereogenic center. Catalytic hydrogenation of the resulting diene 1 would deliver enantiomerically enriched quebrachamine.
Scheme 2.

Retrosynthesis Analysis for Enantioselective Preparation of Quebrachamine
To prepare the relatively strained nine-membered ring of the target, we took inspiration from a report by Calverley17 and planned to access the requisite medium ring by reductive cleavage of the C–N bond (shown in red) of indole-based tertiary amine 3.18 The tetracycle required for the Calverley-type ring expansion would be synthesized through condensation of divinyl aldehyde 4 with commercially available and inexpensive tryptamine (5). We surmised that catalytic ring opening/cross metathesis (ROCM) of spirocyclic lactone 6 in the presence of ethylene would present a concise method for the preparation of 4, an early intermediate that seemed simple but was nontrivial because of the lack of protocols for efficient synthesis of gem-divinyl functionality at an all-carbon quaternary center.19 Successful application of catalytic ROCM would allow for direct synthesis of the requisite carbon framework through a relatively unusual cyclopropenation.
The brevity of the initial synthesis plan notwithstanding, we considered the route shown in Scheme 2 attractive for an additional and important reason: the tetracyclic diene 1, accessed through catalytic enantioselective RCM, does more than serve as a precursor to the naturally occurring quebrachamine; because of its two differentiable alkenes, 1 might be utilized to access derivatives of this class of biologically active alkaloids.13
2. Synthesis of Triene 2 (Substrate for Catalytic Ring-Closing Metathesis)
a. Preparation of Divinylamide Fragment 4
Synthesis of divinylamide fragment 4 began with treatment of commercially available α-acetylbutyrolactone 7 with NaN3, Tf2O, and 1.0 mol % tetrabutylammonium bromide (TBAB) (0 °C, 30 min) to afford lactone 8 in 66% yield after deacylative Regitz diazo transfer (Scheme 3).20 The in situ generation of the diazo transfer agent under phase-transfer conditions obviated the need for preparation and handling of trifluoromethanesulfonyl azide; gram-scale synthesis of 8 could therefore be safely performed. It should be noted that cyclopropenations of diazo compounds bearing α-hydrogens are scarce since such processes typically suffer from adventitious elimination and dimerization pathways.21 Efficient synthesis of Si-substituted cyclopropene 9, however, was readily achieved by slow addition of 8 to a solution of Rh2(OAc)4 in trimethylsilylacetylene (recovered and used as the solvent for multiple transformations),22 allowing us to secure the desired product in 64% yield after purification. Subjection of vinylsilane 9 to 3.7 mol % KOH in EtOH for 5 min at 0 °C furnished spirocyclic cyclopropene 6. Catalytic ROCM involving 6 and ethylene in the presence of 5.0 mol % of Ru carbene 1023 led to the formation of divinyl lactone 11 in 70% yield. The four-step procedure beginning with 7 constitutes a straightforward route for the preparation of lactone 11, which contains the critical quaternary carbon center, in 27% overall yield.
Scheme 3.
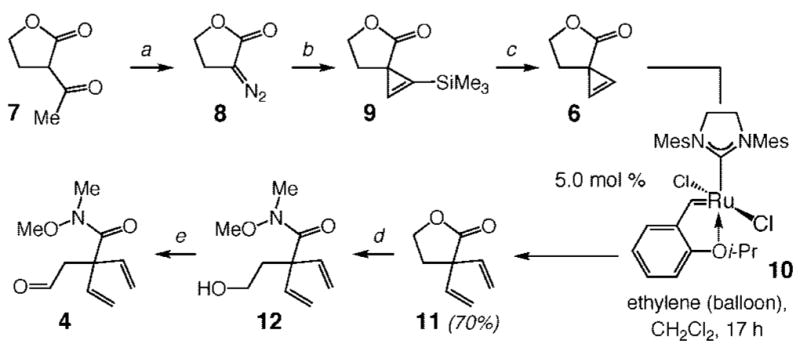
Synthesis of Divinylamide Fragment 4 through Ru-Catalyzed ROCMa
a (a) 4.0 equiv of NaN3, 2.0 equiv of Tf2O, 1.0 mol % TBAB, 2.0 M NaOH, petroleum ether/MeCN, 0 °C, 30 min; 66% yield. (b) 1.0 mol % Rh2(OAc)4, HCCSiMe3, 55 °C, 20 h; 64% yield. (c) 3.7 mol % KOH, EtOH, 0 °C, 5 min; 93% yield. (d) 1.3 equiv of HCl · HNMe(OMe), 2.6 equiv of i-PrMgCl, THF, 0 °C →22 °C, 30 min; pH 7 buffer. (e) 2.3 equiv of DMSO, 2.0 equiv of (COCl)2, CH2Cl2, 7.4 equiv of Et3N, −78 °C → 22 °C; 86% overall yield for two steps.
Attempts to carry out a saponification/oxidation sequence were complicated by a relatively facile reconversion of derivatives of 12 to cyclic lactone 11. We established, however, that treatment of 11 with the magnesium amide of N-methoxy-N-methylamine,24 followed by quenching with a pH 7 buffer solution, furnished the desired alcohol 12. Immediate oxidation of 12 provided aldehyde 4 in 86% overall yield.
b. Preparation of Tricyclic Triene 2 through Reductive C–N Bond Cleavage and Pd-Catalyzed Decarboxylation of an Allylcarbamate
As illustrated in Scheme 4, synthesis of triene 2 began with Pictet–Spengler condensation of 4 with commercially available tryptamine 5 in the presence of HOAc at 80 °C, affording tetracyclic amide 13 in 79% yield after silica gel chromatography. Amine 3 was subsequently obtained in 97% yield by subjection of 13 to lithium aluminum hydride. Reductive cleavage of the C–N bond in 3 required initial activation through formation of the corresponding quaternary carbamate. The tertiary carbamate obtained as a result of the reductive C–N bond rupture would then be converted to the derived secondary amine, which could be alkylated to afford the desired allylamine 2. Although the Calverley protocol17 calls for use of ethyl chloroformate, we surmised that activation of the amine unit in 3 in the form of an allylcarbamate, followed by reductive C–N cleavage to afford 14, would be more efficient. We considered the possibility that a Pd-catalyzed protocol based on recent studies by Tunge and co-workers25 might afford allyl amine 2 directly, without the need to resort to carbamate deprotection. The above scenario, as depicted in Scheme 4, is viable, affording a direct route to 2. Thus, treatment of 3 with allyl chloroformate (−78 °C, 1 h) followed by addition of purified NaCNBH3 resulted in the generation of nine-membered ring 14 in 94% yield after purification. Allyl carbamate 14 is susceptible to decomposition upon storage for reasons that are unclear at the present time; accordingly, direct subjection of the allyl carbamate to 2.0 mol % Pd(PPh3)4 at 22 °C for 20 min was used to secure 2 in 88% yield.
Scheme 4.

Synthesis of the RCM Precursor, Tricyclic Triene 2a
a (a) HOAc, toluene, 80 °C, 2 h; 79% yield (1.5 equiv of 5 used). (b) 5.0 equiv of LAH, THF, 0 °C → 65 °C, 1 h; 97% yield. (c) 10 equiv of allyl chloroformate, THF, −78 °C, 1 h; 7.0 equiv of NaCNBH3, THF, −78 °C, 2 h; warm to 0 °C; 94% yield. (d) 2.0 mol % Pd(PPh3)4, CH2Cl2, 22 °C, 20 min; 88% yield.
II. Evaluation of Existing Achiral and Chiral Catalysts in Effecting a Total Synthesis of Quebrachamine
1. Examination of Existing Catalysts to Promote Ring-Closing Metathesis of Triene 2
Having established an efficient route for gram-scale synthesis of tricyclic triene 2, we began to evaluate the ability of achiral and chiral Ru- and Mo-based complexes to promote its transformation to tetracyclic diene 1.
a. Catalytic RCM of Triene 2 with Achiral Catalysts
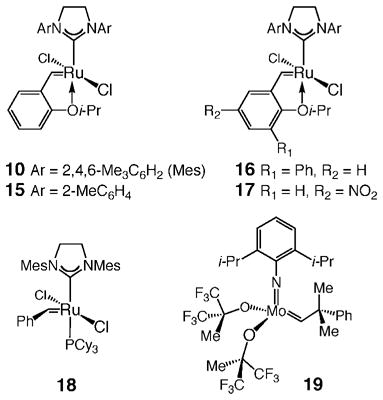
The results of studies regarding catalytic RCM reactions of tricyclic triene 2 with various achiral complexes are summarized in Table 1. In the case of Ru complex 10 (entry 1, Table 1), with 5 mol % catalyst loading (22 °C), there was 75% conversion after 6 h, and the desired product was obtained in 61% yield after purification; extended reaction time (9 h) did not lead to further conversion, an observation suggesting that in the presence of tertiary amine 2, the typically robust 10 undergoes decomposition after several hours. Therefore, to achieve complete substrate consumption, 7.5 mol % of 10 was required (entry 2, Table 1). With the modified Ru carbene 1526 bearing less-substituted N-heterocyclic carbene aryl substituents (entry 3, Table 1), significantly less reaction was achieved in the same amount of time (5 mol %, 6 h, 46% conv, 36% yield); the lower level of conversion to 1 exhibited by 15 is likely a result of a faster rate of decomposition of this more sterically exposed Ru carbene. Thus, as summarized in entries 4 and 5 of Table 1, with sterically modified derivative 1627 and electronically altered carbene 17,28 both of which are complexes that initiate faster than the parent complex 10, higher conversion was obtained (91% and >98% conv, respectively); in the case of complex 17 (entry 5, Table 1), reduced reaction times (i.e., <6 h) translated to substantial amounts of recovered substrate. The phosphine-containing Ru complex 1829 (entry 6, Table 1) showed nearly the same activity and level of efficiency as 10 (entry 1, Table 1).
Table 1.
Screening of Achiral Ru- and Mo-based Catalysts for Conversion of 2 to 1 through RCMa
 | |||||
|---|---|---|---|---|---|
| entry | complex | mol % | time (h) | conv (%)b | yield (%)c |
| 1 | 10 | 5 | 6 | 75 | 61d |
| 2 | 10 | 7.5 | 6 | 94 | 83 |
| 3 | 15 | 5 | 6 | 46 | 36d |
| 4 | 16 | 5 | 6 | 91 | 49d |
| 5 | 17 | 5 | 6 | >98 | 65 |
| 6 | 18 | 5 | 6 | 67 | 57d |
| 7 | 19 | 30 | 2 | >98 | 59 |
 | |||||
Reactions were performed under N2 atmosphere; see the Supporting Information for experimental details.
All conversions >98% (based on the amounts of unreacted substrate and product formed), as determined through analysis of 400 MHz 1H NMR spectra of the unpurified mixtures.
Yields of purified products.
Yields were estimated from purified mixtures of 1 and 2, which are inseparable by silica gel chromatography.
Larger amounts (30 mol %) of the Mo-based alkylidene 192 were required to achieve >98% conversion of the substrate (entry 7, Table 1); the high catalyst loading is likely linked to the faster (vs Ru carbenes) rate of decomposition of 19. It should be noted that in many of the cases shown in Table 1, and particularly with Ru complexes 16 and 17 and Mo complex 19 (entries 4, 5, and 7, Table 1), there is a significant difference between the percent conversion and the percent yield of isolated product; this discrepancy is due to formation of byproducts that we have not as yet been able to identify.
b. Enantioselective RCM
The chiral Ru-based (20–23) and Mo-based (24–30) complexes examined for catalytic enantioselective RCM of 2 to afford enantiomerically enriched 1 are illustrated in Scheme 5; the corresponding data are summarized in Table 2.
Scheme 5.

Ru- and Mo-Based Chiral Catalysts Examined for RCM of Triene 2 to Afford Diene 1
Table 2.
Screening of Chiral Ru- and Mo-Based Catalysts for Conversion of 2 to 1 through Enantioselective RCMa
 | ||||||
|---|---|---|---|---|---|---|
| entry | catalyst | mol % | temp (°C) | time (h) | conv (%)b | ee (%)c |
| 1 | 20 | 15 | 80 | 12 | >98d | <5 |
| 2 | 21a | 15 | 80 | 12 | >98e | <5 |
| 3 | 21b | 16 | 80 | 18 | 50 | <10 |
| 4 | 22a | 10 | 22 | 12 | 50 | <5 |
| 5 | 22b | 10 | 22 | 24 | <10 | nd |
| 6 | 23a | 15 | 22 | 12 | 50 | 10 |
| 7 | 23b | 15 | 35 | 16 | 17 | nd |
| 8 | 24a | 50 | 80 | 12 | <5 | – |
| 9 | 24b | 50 | 80 | 12 | <5 | – |
| 10 | 24c | 50 | 80 | 12 | <5 | – |
| 11 | 25 | 10 | 22 | 12 | <5 | – |
| 12 | 26 | 50 | 80 | 12 | <5 | – |
| 13 | 27 | 50 | 80 | 18 | <5 | – |
| 14 | 28 | 10 | 80 | 18 | <5 | – |
| 15 | 29a | 10 | 80 | 18 | <5 | – |
| 16 | 29b | 10 | 80 | 18 | <5 | – |
| 17 | 30 | 10 | 80 | 18 | <5 | – |
Reactions were performed under N2 atmosphere; see the Supporting Information for experimental details. nd = not determined.
Conversions (based on the amounts of unreacted substrate and product formed) were determined through analysis of 400 MHz 1H NMR spectra of the unpurified mixtures.
Enantioselectivities (ee) were determined by HPLC analysis; see the Supporting Information for details.
Product isolated in 54% yield.
Product isolated in 51% yield.
When chiral Ru catalysts bearing bidentate9a,30 or monodentate chiral N-heterocyclic carbenes9b (NHCs) were used (entries 1–7, Table 2), regardless of whether these were the typically more active chlorides or the less effective but more discriminating iodides, low enantioselectivity was observed (<10% ee). The Ru chlorides bearing bidentate carbenes (i.e., 20 and 21a) were more active than their corresponding iodide salts (e.g., 21b); nonetheless, chloride complexes 20 and 21a (entries 1 and 2, Table 2) required elevated temperatures (80 °C) to promote >98% conversion to 1 after 12 h. Reactions with Ru complexes bearing a dissymmetric (22a and 22b;31 entries 4 and 5, Table 2) or C2-symmetric (23a and 23b;9b entries 6 and 7, Table 2) monodentate chiral NHC also furnished nearly racemic 1 (≤10% ee).
Mo-based diolates 24–302 (Scheme 5), as evident from the data summarized in Table 2, were entirely ineffective in promoting the conversion of 2 to 1. Regardless of whether we resorted to high catalyst loadings (e.g., entries 8–10 and 12–13 in Table 2) or elevated reaction temperatures (e.g., entries 8–10 and 12–17, Table 2), none of the desired ring-closed tetracyclic product (1) could be observed.
The above investigations, involving the entire range of available chiral olefin metathesis catalysts, clearly indicated that a new and significantly more effective catalyst system would be required if an efficient and enantioselective synthesis of 1 through catalytic enantioselective RCM of 2 were to be realized. The studies outlined below describe the realization of this goal.
III. Design of New Chiral Stereogenic-at-Mo Catalysts for Olefin Metathesis
1. Mechanistic Considerations and Design Criteria
As the first step toward designing and/or identifying a chiral catalyst that would convert tricyclic triene 2 to tetracyclic diene 1 efficiently as well as enantioselectively, we considered the structural aspects of the existing catalysts that required alteration. Since Mo-based complexes have thus far generally proven to be more effective than Ru-based carbenes in catalyzing enantioselective RCM reactions8,9 (vs enantioselective ring-opening metathesis processes),10 we decided to focus our attention on this class of complexes.
a. Reducing the Strain of Metallacyclobutanes
We began by considering the reason for the higher activity of achiral Mo alkylidene 19 (Table 1), which bears two monodentate hexafluoro-t-butoxide ligands, versus complexes 24–30 (Scheme 5), which contain various bidentate ligands (i.e., biphenoxides in 24–26, binaphtholates in 27, 29, and 30, or a tetrahydrobinaphtholate in 28). We noted that the difference in reactivity is not likely due to the higher Lewis acidity of Mo in diolates [for aryl alcohol and hexafluoro-t-butanol, pKa ≈ 9–10 (measured in H2O)].32 We reasoned that the significantly higher (albeit less than desired) activity of the achiral Mo-based complex 19 compared with the chiral variants might originate from the structural rigidity of diolates 24–30. That is, we surmised that the strain existing within the initiating alkylidenes that carry a bidentate bisaryloxide (vs the relatively flexible alkoxides in 19) might be exacerbated by the formation of the spirometallacyclobutane intermediates (i.e., I and II vs III and IV in Figure 1), causing a rise in the activation barrier for a key step of the catalytic cycle and, as a result, a diminution in the rate of the RCM. The variation of the O–Mo–O angle in alkylidene 24a (O–Mo–O = 127°)33 from that in a closely related tungstacyclobutane analogous to IV (O–W–O = 98°)34 indicates the potentially significant structural change involved in such transformations.
Figure 1.

The relatively rigid diolate ligands in Mo-based complexes translate to high-energy metallacyclobutane intermediates. Abbreviations: G = C(Ph)Me2; TBP = trigonal bipyramidal; SP = square pyramidal.
The proposal that a structurally more flexible Mo diolate might generate a higher level of catalytic activity led us to prepare complex 31, which bears a chiral taddolate.35 We predicted that the seven-membered-ring bisalkoxide in 31, which is built from sp3-hybridized atoms, could more readily transform into the required metallacyclobutane than could complexes 24–30 (Scheme 5), each of which contains a seven-membered ring diolate with four sp2-hybridized carbon atoms. Thus, as illustrated in eq 1, we found that in the presence of 20 mol % chiral complex 31, there was 23% conversion of triene 2 to tetracyclic 1, albeit with minimal enantioselectivity; under identical conditions, the chiral Mo diolates shown in Scheme 5 delivered <2% of the desired product. It should be noted that the increase in catalytic activity manifested by 31 occurs in spite of the lower Lewis acidity of its Mo center, which is due to a less electron-withdrawing diolate ligand (vs the bisaryloxides in 24–30).
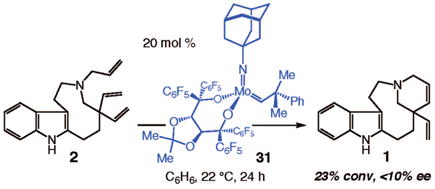 |
(1) |
The above analysis led us to consider that Mo-based catalysts bearing monodentate ligands, either both chiral (same enantiomer, nonstereogenic-at-Mo) or one achiral and one chiral (stereogenic-at-Mo), would be more effective as chiral catalysts for olefin metathesis. Additional considerations, as detailed below, pointed to the significantly less explored alternative: a stereogenic-at-Mo complex.36,37
b. Incorporation of Stereoelectronic Effects at the Metal Center: Nonstereogenic-at-Mo or Stereogenic-at-Mo?
Recent theoretical studies by Eisenstein and co-workers5 present the case that high-oxidation-state complexes that bear a stereogenic metal center would be exceptionally effective olefin metathesis catalysts (Figure 2). First, their theoretical investigations suggest that the presence of one acceptor ligand (A in V, Figure 2) is required in order to ensure that the metal center possesses sufficient Lewis acidity to provide effective binding to olefins. Furthermore, and more importantly, the presence of a donor ligand (D in V, Figure 2) is favorable as it causes the geometrically distorted VI to be energetically more accessible. In contrast to tetrahedral V, which cannot directly coordinate with an alkene (structural distortion is required), transition complex VI easily provides a ligation site for an olefin. A critical aspect of the proposed scenario is that there is maximum stabilization of distorted VI (Figure 2) when the donor ligand D is associated with the energetically most favorable metal orbital, namely, the one that generates minimal electronic repulsion (trans effect). Accordingly, ligand D should occupy an apical site in VI (Figure 2) with the alkylidene, acceptor, and imido ligands constituting the equatorial plane. Consequently, the weakly coordinated olefin preferably approaches the metal center trans to D. The suggested coordination of the substrate olefin trans to the donor ligand suggests that in the design of an enantiomerically enriched stereogenic-at-Mo complex, it might be preferable that the acceptor ligand A be chiral rather than the donor ligand D, since the substrate most likely approaches the catalyst syn to the former, leading to higher enantioselectivity.
Figure 2.

Electronic dissymmetry at the metal facilitates olefin coordination and metallacyclobutane collapse.
Another consequence of the above stereoelectronic principles might be that the resulting trigonal bipyramidal complex VIII (Figure 2) can undergo facile degradation to afford IX, since the carbons constituting the releasing olefin are situated trans to ligand D. Thus, the electronic effects caused by the presence of a donor and an acceptor ligand (a stereogenic-at-Mo complex) further facilitate catalytic turnover by elevating the energy (trans effect) of the metallacyclobutane complex VIII (thereby lowering the activation barrier for metallacyclobutane decomposition to afford IX).
The abovementioned hypotheses regarding the higher catalytic activity of a Mo-based catalyst that bears a donor and an acceptor ligand (vs two donor or two acceptor ligands) finds support in the remarkably facile reaction shown in eq 2: 1 mol % rac-3238 promotes formation of racemic 1 within only 1 h. This level of activity is in stark contrast to that of achiral Mo complex 19 (see Table 1), a complex with two hexafluoro-tert-butoxide ligands. It merits mention that although RCM of 2 with complex 32 is more efficient than with the achiral Ru and Mo complexes shown in Table 1, there is still ~20% yield of unidentified byproducts generated in the transformation illustrated in eq 2.
2. Stereoselective Synthesis of Stereogenic-at-Mo Complexes
The investigations and mechanistic considerations discussed above point to structurally fluxional stereogenic-at-Mo complexes bearing appropriate donor and acceptor ligands as the
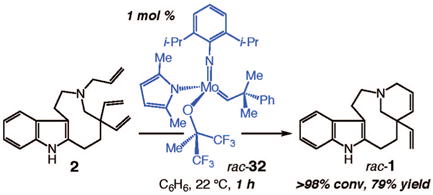 |
(2) |
preferred class of chiral olefin metathesis catalysts. That is, to achieve an enantioselective synthesis of quebrachamine, we were required to identify an effective, enantiomerically enriched version of complex 32 (see eq 2). To achieve this goal, one possible strategy would be to use the same class of Mo alkylidene precursors that deliver monopyrrolide 32 (see below). The challenging task of identifying an appropriate set of chiral monodentate alcohol ligands that would allow for efficient and diastereoselective synthesis of stereogenic-at-Mo alkylidenes, however, remained to be accomplished.
a. A Suitable Mo Alkylidene Precursor
Recently, in connection with a program directed toward the development of practical methods for synthesis of chiral Mo diolate catalysts, we prepared complexes 33a and 33b.39 These complexes, which are easily accessed from the reaction of a Mo bistriflate with an appropriate lithium pyrrolide, are converted to the desired diolates (e.g., complexes in Scheme 5) upon exposure to chiral biphenols or binaphthols; the resulting diolates can be used without isolation or purification to promote enantioselective olefin metathesis reactions with efficiencies and selectivities similar to those observed with purified catalysts.40 The above investigations indicated that pyrrole molecules generated via protonation by the chiral diols do not cause diminution of catalyst activity or reaction selectivity. We thus surmised that one approach toward preparation of stereogenic-at-Mo catalysts would involve diastereoselective reaction of 33a and 33b with 1 equiv of a chiral enantiomerically pure alcohol. An attractive feature of such an approach would be that chiral catalysts might be generated and used in situ to promote olefin metathesis reactions.
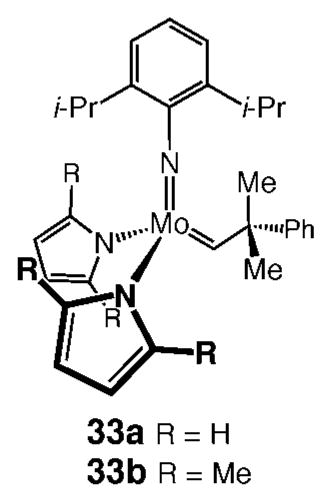
b. Identification of a Suitable Class of Enantiomerically Pure Chiral Alcohols to Serve As Chiral Monodentate O-Based Ligands
Although a variety of diols have been utilized as ligands in enantioselective catalysis,41 and whereas mono-dentate phosphines42 or N-heterocyclic carbenes43 can be found in the structures of chiral catalysts, monodentate O-based ligands have scarcely been used in this context.44 As the first step toward identification of an effective class of chiral monodentate alcohols, we adopted the simple strategy that such chiral ligands could simply be derived from protection of one of the hydroxyl units of a chiral diol. As such, we would be able to select from a readily available class of ligands (i.e., chiral diols). The above considerations led us to identify binaphthol-derived alcohols as the most desirable. As illustrated in Scheme 6, monoprotected binaphthols benefit from a number of attributes that are critical to chiral catalyst development: (a) ease and low cost of synthesis through protection of commercially available and inexpensive binaphthol (e.g., 34 → A in Scheme 6) and (b) the facility of ligand modification for fine-tuning the steric and electronic attributes of the stereogenic-at-Mo catalysts, as represented by functionalization at C3 and C3′ sites and partial hydrogenation of the aromatic rings45 (as illustrated by A → C in Scheme 6.) Since bisaryloxides have proven to be effective ligands for Mo-based olefin metathesis catalysis, we judged monoaryloxides should prove to be effective catalysts as well.
Scheme 6.

Monoprotected Binaphthols: A Versatile Class of Chiral Monodentate Ligands
c. Preparation of Stereogenic-at-Mo Complexes through Diastereoselective Protonation of Mo Bispyrrolides
Having identified Mo-based precursors and a readily available class of chiral monodentate alcohols, we turned our attention to stereo-selective synthesis of stereogenic-at-Mo complexes. We initiated our studies by examining the reactions of bispyrrolides 33a and 33b with several monoprotected diols derived from commercially available enantiomerically pure binaphthol 34. These investigations led us to establish that, as illustrated in Scheme 7, treatment of 33a with 1 equiv of enantiomerically pure bisbromide monoprotected binaphthol 35 leads to the formation of 36a with 19:1 diastereoselectivity. When complex 33b bearing 2,5-dimethylpyrrolide ligands is used, 36b is generated with lower selectivity as a 7:1 mixture (Scheme 7).
Scheme 7.

Diastereoselective Synthesis of Stereogenic-at-Mo Complexes
Our previous studies involving Mo diolates indicated that complexes bearing a tetrahydrobinaphtholate ligand (e.g., 28 in Scheme 5) might exhibit different and at times superior reactivity and selectivity levels in comparison with binaphtholates (e.g., 27, 29, and 30 in Scheme 5) or biphenolates (e.g., 24–26 in Scheme 5).46 Structurally, tetrahydrobinaphtholates contain features of both Mo biphenolates (tetraalkyl-substituted biphenyl) and the corresponding binaphtholates. The observed differences in the activities and enantioselectivities of catalysts bearing the partially reduced ligands47 may be attributed to differences in the dihedral angle of the two aromatic planes.
Chiral Mo aryloxides 37a and 37b were prepared by the same procedures as described above, with nearly identical results (Scheme 7). We established the identity of the major diastereomer of 37b through X-ray crystallography.6 The diastereoselective desymmetrizations depicted in Scheme 7 are highly efficient: the reactions proceed to >95% conversion at 22 °C within 1 h.
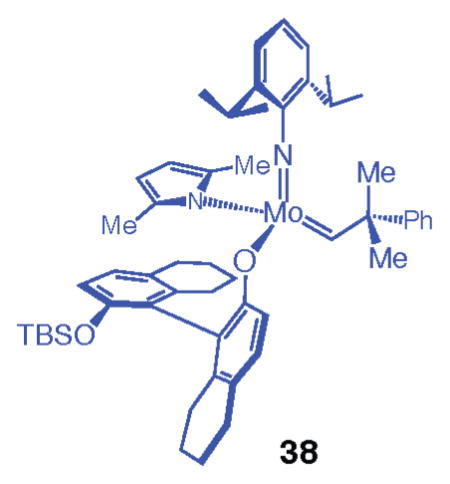
The presence of the bromide substituents at the C3 and C3′ positions of the chiral ligand are necessary for clean generation of the desired monoaryloxides. Attempts to prepare stereogenic-at-Mo complexes derived from reactions of unsubstituted monosilyl-protected binaphthol under conditions used to access 36a, 36b, 37a, and 37b (with 1 equiv of alcohol) gave rise to a mixture containing a substantial amount of the bisaryloxide. When 1 equiv of monosilyl-protected tetrahydrobinaphthol is used instead of the corresponding binaphthol, however, the desired monoaryloxide (38) can be obtained selectively.
IV. Total Synthesis of Quebrachamine through Efficient Enantioselective RCM of Triene 2
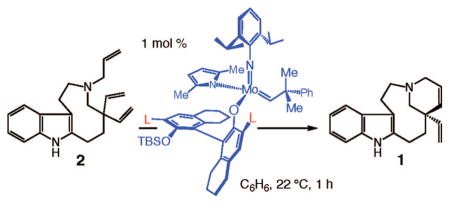
With several stereogenic-at-Mo complexes available (Scheme 7), we began to examine the ability of these systems to promote enantioselective RCM of 2. For ease of operation, the in situ-prepared complexes were used in the initial studies. That is, in all cases, reactions were performed in the presence of diastereomeric mixtures of stereogenic-at-Mo complexes. Catalytic enantioselective RCM in the presence of 1 mol % 36a, as shown in entry 1 of Table 3, gave rise to <10% conversion of 2 to the desired tetracycle 1 in 1 h; longer reaction times (e.g., 6 h) did not lead to further transformation. Higher reaction efficiency (24% conversion to 1) was observed with the 2,5-dimethylpyrrolide complex 36b under identical conditions (entry 2, Table 3). When 5 mol % 36b was used (entry 3, Table 3), RCM proceeded to complete conversion, affording 2 as an 83.5:16.5 mixture of enantiomers (67% ee). As in the case of complex 36a, the presence of 1 mol % unsubstituted monopyrrolide 37a did not lead to formation of the desired product after 1 h (entry 4, Table 3). As shown in entry 5 of Table 3, however, with 1 mol % 2,5-dimethylpyrrolide complex 37b, RCM of triene 2 was complete (>98% conv) within 1 h, affording diene 1 in 95% ee (97.5: 2.5 er) in 83% yield after purification. As illustrated in entry 6 of Table 3, with monoaryloxide 38, a complex that lacks the bromide substituents at the C3 and C3′ positions, <20% conversion to 1 was observed. It is also important to note that the bisaryloxides derived from reactions of pyrrolide complex 33a with (mono-t-BuMe2Si)binaphthol or the derived tetrahydrobinaphthol were not effective in catalyzing the conversion of 2 to 1 (~5% conv at 5 mol % loading, 1 h, 22 °C).
Table 3.
Catalytic Enantioselective RCM of Triene 2 Promoted by Stereogenic-at-Mo Complexesa
 | ||||
|---|---|---|---|---|
| entry | chiral complex; mol % | conv (%);b yield (%)c | erd | ee (%)d |
| 1 | 36a; 1 | <10; nd | nd | nd |
| 2 | 36b; 1 | 24; nd | nd | nd |
| 3 | 36b; 5 | >98; 60 | 83.5:16.5 | 67 |
| 4 | 37a; 1 | <5; nd | – | – |
| 5 | 37b; 1 | >98; 83 | 97.5:2.5 | 95 |
| 6 | 38; 1 | 19; nd | nd | nd |
Reactions were performed under N2 atmosphere; see the Supporting Information for experimental details. nd = not determined.
Conversions (based on the amounts of unreacted substrate and product formed) were determined through analysis of 400 MHz 1H NMR spectra of the unpurified mixtures.
Yield of 1 after purification.
Enantiomer ratios (er) and enantioselectivities (ee) were determined by HPLC analysis; see the Supporting Information for details.
A mechanistically critical aspect of the highly enantioselective process promoted by chiral complex 37b prepared and used in situ as a 7:1 mixture of diastereomers relates to the relative activity of each catalyst isomer. Initial studies (analysis by 400 MHz 1H NMR spectroscopy) indicate that <5% of the minor complex diastereomer is initiated, allowing only the major isomer to perform the enantioselective RCM reaction. Furthermore, when a sample of diastereomerically pure 37b prepared after a single recrystallization was utilized to promote conversion of triene 2 to tetracycle 1, nearly identical levels of reactivity and enantioselectivity were observed as when the 7:1 mixture was used directly. Additional details will be provided in separate accounts along with the results of mechanistic investigations.
Next, we investigated the effect of the halide substituents of the chiral monodentate ligand on the enantioselective RCM of 2; the results of these studies are summarized in Table 4. Reaction in the presence of bisfluoride complex 39 was equally efficient as that with 37b (entry 3) but delivered 1 with lower selectivity (92:8 er, 84% ee vs 97.5:2.5 er, 95% ee). Dichloride 40 (entry 2, Table 4) and diiodide 41 (entry 4, Table 4) promoted efficient reactions (>98% conv, 1 mol %, 1 h, 22 °C), and furnished 1 with high enantioselectivities (96 and 93% ee, respectively), with the former complex delivering a slightly higher level of enantiodifferentiation (compare entries 2 and 4, Table 4). It is also noteworthy that the yields of isolated 1 in Table 4 are in some instances significantly higher than those obtained with achiral catalysts or rac-32 (see Table 1 and eq 2, respectively); smaller amounts of the aforementioned unidentified products are generated in the transformations promoted by enantiomerically pure chiral Mo monoaryloxides.
Table 4.
Effect of Halogen Substituents of the Monodentate Chiral Ligand on Catalytic Enantioselective RCM of Triene 2a
 | ||||
|---|---|---|---|---|
| entry | chiral complex; L | conv (%);b yield (%)c | erd | ee (%)d |
| 1 | 39; F | >98; 80 | 92:8 | 84 |
| 2 | 40; Cl | >98; 84 | 98:2 | 96 |
| 3 | 37b; Br | >98; 83 | 97.5:2.5 | 95 |
| 4 | 41; I | >98; 93 | 96.5:3.5 | 93 |
Reactions were performed under N2 atmosphere; see the Supporting Information for experimental details.
Conversions (based on the amounts of unreacted substrate and product formed) were determined through analysis of 400 MHz 1H NMR spectra of the unpurified mixtures.
Yield of 1 after purification.
Enantiomer ratios (er) and enantioselectivities (ee) were determined by HPLC analysis; see the Supporting Information for details.
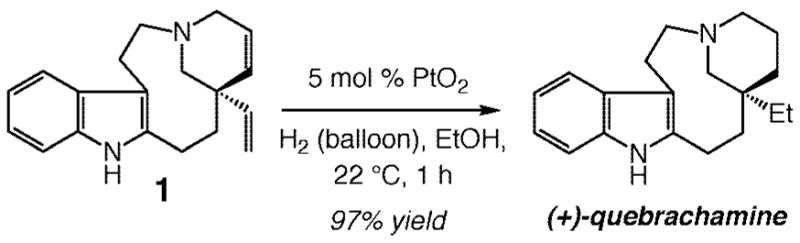
With an efficient catalytic enantioselective RCM of 2 in hand, we turned our attention to the completion of the total synthesis of (+)-quebrachamine. As illustrated in Scheme 8, catalytic hydrogenation in the presence of 5 mol % PtO2 under H2 atmosphere (balloon) afforded the desired target molecule in 97% yield after purification. This final step thus completes a total synthesis of the natural alkaloid in 12% overall yield through a scheme containing two separate six-step linear sequences.
Scheme 8.
Completion of the Enantioselective Total Synthesis of Quebrachamine
Conclusions and Future Research
The stereogenic-at-metal complexes designed, synthesized, and examined in the course of this study represent a new approach to the development of catalysts for efficient and stereoselective olefin metathesis.48 Specific electronic factors incorporated within the structure of the chiral catalysts as a result of previous theoretical studies are largely responsible for the exceptional levels of reactivity and selectivity observed. Stereoelectronic and conformational mobility considerations, together with the tetrahedral structure of high-oxidation-state alkylidenes, led us to consider designing chiral complexes that bear a stereogenic metal center and only monodentate ligands.
Development of stereogenic-at-metal catalysts presents complications that must be addressed if such complexes are to be employed effectively in promoting catalytic enantioselective transformations. First, stereoselective synthesis of the catalyst and control of metal stereogenicity might be required. We have addressed this problem by a diastereoselective process through the use of a readily available enantiomerically pure monoprotected binaphthol-based ligand. The mechanistic details of stereoselective reactions of Mo-based bispyrrolides with various chiral aryloxides and the rationale for the lower rate of initiation of the minor Mo alkylidene diastereomer are subjects of ongoing investigations.
Second, the stereogenic-at-Mo complex must avoid uncontrolled stereomutation in the course of a catalytic process. Otherwise, low enantioselectivity is likely to result if both diastereomers are reasonably reactive or do not easily isomerize; in the case that the minor chiral complex diastereomer is unreactive, there would be reduced activity unless higher catalyst loadings are used. Such considerations bring forth a notable advantage of this class of high-oxidation-state complexes for the design of stereogenic-at-metal catalysts: the absence of coordinatively labile ligands (e.g., phosphines) minimizes the possibility of complex planarization and loss of stereochemical integrity.49 An important attribute of an olefin metathesis reaction, one that becomes crucial in cases where stereogenic-at-metal complexes are involved, is that the catalyst’s metal center undergoes inversion with each olefin metathesis event (see Figure 2 and related discussions).5,37g A ring-closing process involves two olefin metathesis events, however, so the original isomer of a chiral complex should emerge after completion of each catalytic cycle (double inversion).50 Stereomutation through degenerate olefin metathesis can therefore be a possible pitfall in the use of conformationally flexible stereogenic-at-metal complexes. On the other hand, if one diastereomer is lower in energy and catalytic activity, degenerate processes can furnish a “correcting mechanism” and thus prove to be beneficial. Studies that clarify the above mechanistic subtleties are in progress.
The stereogenic-at-Mo complexes presented herein offer fresh opportunities for addressing a significant number of unresolved problems in catalytic olefin metathesis; such complexes are expected to serve not only as uniquely effective catalysts for enantioselective synthesis but also as highly efficient complexes that provide unprecedented activity (see eq 2). Investigations along these lines and applications of the new chiral catalysts to additional enantioselective RCM processes as well as other types of olefin metathesis reactions will be the focus of our future studies. As was the case in the studies presented herein, we will continue to be guided by the demands presented to us in the course of our efforts in complex molecule total synthesis.
Supplementary Material
Acknowledgments
Financial support was generously provided by the NIH (Grant GM-59426 to A.H.H. and R.R.S.) and the NSF (CHE-0715138 to A.H.H.). We are grateful to Mr. A. Zhugralin (Boston College) for numerous invaluable discussions regarding theoretical and mechanistic issues as well as synthesis and characterization of the chiral Ru-based complexes 22a and 22b and to Dr. A. Hock, Dr. T. Pilyugina, and Dr. R. Singh (MIT) for helpful experimental suggestions. We are grateful to Dr. B. Bailey and Mr. K. Wampler (MIT) for assistance in obtaining the X-ray structure of the major diastereomer of Mo complex 37b. Mass spectrometry facilities at Boston College are supported by the NSF (DBI-0619576).
Footnotes
Supporting Information Available: Experimental procedures and spectral data for substrates and products. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Grubbs RH, editor. Handbook of Metathesis. Wiley-VCH; Weinheim, Germany: 2003. [Google Scholar]; (b) Hoveyda AH, Zhugralin AR. Nature. 2007;450:243–251. doi: 10.1038/nature06351. [DOI] [PubMed] [Google Scholar]
- 2.Schrock RR, Hoveyda AH. Angew Chem, Int Ed. Vol. 42. 2003. pp. 4592–4633. [DOI] [PubMed] [Google Scholar]
- 3.For previous racemic total syntheses of quebrachamine, see: Stork G, Dolfini JE. J Am Chem Soc. 1963;85:2872–2873.Wenker E, Garratt S, Dave KG. Can J Chem. 1964;42:489–490.Kutney JP, Abdurahman N, Le Quesne P, Piers E, Vlattas I. J Am Chem Soc. 1966;88:3656–3657. doi: 10.1021/ja00709a052.Ziegler FE, Kloek JA, Zoretic PA. J Am Chem Soc. 1969;91:2342–2346.Takano S, Hatakeyama S, Ogasawara K. J Am Chem Soc. 1979;101:6414–6420. doi: 10.1021/ja00426a062.Wenkert E, Halls TDJ, Kwart LD, Magnusson G, Showalter HDH. Tetrahedron. 1981;37:4017–4025.Ban Y, Yoshida K, Goto J, Oishi T. J Am Chem Soc. 1981;103:6990–6992.Coldham I, Burrell AJM, White LE, Adams H, Oram N. Angew Chem, Int Ed. 2007;46:6159–6162. doi: 10.1002/anie.200701943.
- 4.For previous enantioselective total syntheses of quebrachamine, see: Takano S, Yonaga M, Ogasawara K. J Chem Soc, Chem Commun. 1981:1153–1155.Node M, Nagasawa H, Fuji K. J Am Chem Soc. 1987;109:7901–7903.Temme O, Taj SA, Andersson PG. J Org Chem. 1998;63:6007–6015. doi: 10.1021/jo9807417.Kozmin SA, Iwama T, Huang Y, Rawal VH. J Am Chem Soc. 2002;124:4628–4641. doi: 10.1021/ja017863s.Amat M, Lozano O, Escolano C, Molins E, Bosch J. J Org Chem. 2007;72:4431–4439. doi: 10.1021/jo070397q.
- 5.(a) Solans-Monfort X, Clot E, Copéret C, Eisenstein O. J Am Chem Soc. 2005;127:14015–14025. doi: 10.1021/ja053528i. [DOI] [PubMed] [Google Scholar]; (b) Poater A, Solans-Monfort X, Clot E, Copéret C, Eisenstein O. J Am Chem Soc. 2007;129:8207–8216. doi: 10.1021/ja070625y. [DOI] [PubMed] [Google Scholar]
- 6.For the preliminary account of this study, see: Malcolmson SJ, Meek SJ, Sattely ES, Schrock RR, Hoveyda AH. Nature. 2008;456:933–937. doi: 10.1038/nature07594.
- 7.For recent examples in which a natural product total synthesis effort resulted in the development of a new catalytic enantioselective protocol (in addition to the application of an already existing method), see: Gillingham DG, Hoveyda AH. Angew Chem, Int Ed. 2007;46:3860–3864. doi: 10.1002/anie.200700501.Brown MK, Hoveyda AH. J Am Chem Soc. 2008;130:12904–12906. doi: 10.1021/ja8058414.
- 8.For Mo-catalyzed enantioselective RCM processes that deliver N-containing heterocycles, see: Dolman SJ, Sattely ES, Hoveyda AH, Schrock RR. J Am Chem Soc. 2002;124:6991–6997. doi: 10.1021/ja012534l.Dolman SJ, Schrock RR, Hoveyda AH. Org Lett. 2003;5:4899–4902. doi: 10.1021/ol036026n.Sattely ES, Cortez GA, Moebius DC, Schrock RR, Hoveyda AH. J Am Chem Soc. 2005;127:8526–8533. doi: 10.1021/ja051330s.. For Mo-catalyzed enantioselective RCM reactions that afford other types of ring structures, see: Alexander JB, La DS, Cefalo DR, Hoveyda AH, Schrock RR. J Am Chem Soc. 1998;120:4041–4042.La DS, Alexander JB, Cefalo DR, Graf DD, Hoveyda AH, Schrock RR. J Am Chem Soc. 1998;120:9720–9721.Zhu SS, Cefalo DR, La DS, Jamieson JY, Davis WM, Hoveyda AH, Schrock RR. J Am Chem Soc. 1999;121:8251–8259.Cefalo DR, Kiely AF, Wuchrer M, Jamieson JY, Schrock RR, Hoveyda AH. J Am Chem Soc. 2001;123:3139–3140.Kiely AF, Jernelius JA, Schrock RR, Hoveyda AH. J Am Chem Soc. 2002;124:2868–2869. doi: 10.1021/ja012679s.Jernelius JA, Schrock RR, Hoveyda AH. Tetrahedron. 2004;60:7345–7351.Lee AL, Malcolmson SJ, Puglisi A, Schrock RR, Hoveyda AH. J Am Chem Soc. 2006;128:5153–5157. doi: 10.1021/ja058428r.
- 9.For enantioselective RCM reactions promoted by Ru-based catalysts, see: Van Veldhuizen JJ, Gillingham DG, Garber SB, Kataoka O, Hoveyda AH. J Am Chem Soc. 2003;125:12502–12508. doi: 10.1021/ja0302228.. For related Ru-catalyzed processes disclosed by other laboratories, see: Seiders TJ, Ward DW, Grubbs RH. Org Lett. 2001;3:3225–3228. doi: 10.1021/ol0165692.Funk TW, Berlin JM, Grubbs RH. J Am Chem Soc. 2006;128:1840–1846. doi: 10.1021/ja055994d.Fournier PA, Collins SK. Organometallics. 2007;26:2945–2949.
- 10.For Mo-catalyzed enantioselective ring-opening/cross-metathesis and/or ring-opening/ring-closing reactions, see: Weatherhead GS, Ford JG, Alexanian EJ, Schrock RR, Hoveyda AH. J Am Chem Soc. 2000;122:1828–1829.La DS, Sattely ES, Ford JG, Schrock RR, Hoveyda AH. J Am Chem Soc. 2001;123:7767–7778. doi: 10.1021/ja010684q.Cortez GA, Schrock RR, Hoveyda AH. Angew Chem, Int Ed. 2007;46:4534–4538. doi: 10.1002/anie.200605130.. For Ru-catalyzed enantioselective ring-opening/cross-metathesis reactions, see: Gillingham DG, Kataoka O, Garber SB, Hoveyda AH. J Am Chem Soc. 2004;126:12288–12290. doi: 10.1021/ja0458672.. For a comparison of Mo- and Ru-based enantioselective ring-opening/cross-metathesis processes, see: Cortez GA, Baxter CA, Schrock RR, Hoveyda AH. Org Lett. 2007;9:2871–2874. doi: 10.1021/ol071008h.. For applications of catalytic enantioselective ring-opening/cross-metathesis reactions to natural product total synthesis, see: Weatherhead GS, Cortez GA, Schrock RR, Hoveyda AH. Proc Natl Acad Sci USA. 2004;101:5805–5809. doi: 10.1073/pnas.0307589101.. (g) See ref 7a. For Ru-catalyzed enantioselective ring-opening/cross-metathesis reactions developed in other laboratories, see: Berlin JM, Goldberg SD, Grubbs RH. Angew Chem, Int Ed. 2006;45:7591–7595. doi: 10.1002/anie.200602469.
- 11.For recent reviews regarding the use of catalytic olefin metathesis in natural product syntheses, see: Deiters A, Martin SF. Chem Rev. 2004;104:2199–2238. doi: 10.1021/cr0200872.Nicolaou KC, Bulger PG, Sarlah D. Angew Chem, Int Ed. 2005;44:4490–4527. doi: 10.1002/anie.200500369.
- 12.For biological activity of quebrachamine, see: Deutsch HF, Evenson MA, Drescher P, Sparwasser C, Madsen P. J Pharm Biomed Anal. 1994;12:1283–1287. doi: 10.1016/0731-7085(94)00066-2.
- 13.Saxton JE. In: The Alkaloids Alkaloids of the Aspidospermine Group. Cordell GA, editor. Vol. 51. Academic Press; New York: 1998. Chapter 1. [Google Scholar]
- 14.For a total synthesis of (–)-aspidospermine that involves a (non-enantioselective) catalytic RCM related to the strategy employed in this study, see: Fukuda Y-i, Shindo M, Shishido K. Org Lett. 2003;5:749–751. doi: 10.1021/ol034020s.
- 15.Christophers J, Baro A. Quaternary Stereocenters: Challenges and Solutions for Organic Synthesis. Wiley-VCH; Weinheim, Germany: 2006. [Google Scholar]
- 16.For representative examples, see: Lee KL, Goh JB, Martin SF. Tetrahedron Lett. 2001;42:1635–1638.Wipf P, Rector SR, Takahashi H. J Am Chem Soc. 2002;124:14848–14849. doi: 10.1021/ja028603t.Wipf P, Spencer SR. J Am Chem Soc. 2005;127:225–235. doi: 10.1021/ja044280k.
- 17.(a) Calverley MJ. J Chem Soc, Chem Commun. 1981:1209–1210. [Google Scholar]; (b) Calverley MJ. J Chem Res, Miniprint. 1983:1848–1890. [Google Scholar]
- 18.For additional examples of related reductive C–N bond cleavage, see: Bonjoch J, Fernandez JC, Valls N. J Org Chem. 1998;63:7338–7347. doi: 10.1021/jo980909o. and references cited therein.
- 19.For an example of metal-mediated preparation of a gem-divinyl unit incorporated at an ester-substituted quaternary carbon, see: Arisawa M, Miyagawa C, Yoshimura S, Kido Y, Yamaguchi M. Chem Lett. 2001:1080–1081.
- 20.The procedure used to prepare diazo lactone 8 is based on previously reported protocols. See: Brown RCD, Bataille CJR, Brunton G, Hinks JD, Swain NA. J Org Chem. 2001;66:6719–6728. doi: 10.1021/jo015829q.
- 21.Panne P, Fox JM. J Am Chem Soc. 2007;129:22–23. doi: 10.1021/ja0660195. [DOI] [PubMed] [Google Scholar]
- 22.See the Supporting Information for complete experimental details.
- 23.Garber SB, Kingsbury JS, Gray BL, Hoveyda AH. J Am Chem Soc. 2000;122:8168–8179.. For an overview of Ru-based carbenes bearing bidentate styrene ether ligands, see: Hoveyda AH, Gillingham DG, Van Veldhuizen JJ, Kataoka O, Garber SB, Kingsbury JS, Harrity JPA. Org Biomol Chem. 2004;2:8–23. doi: 10.1039/b311496c.
- 24.Williams JM, Jobson RB, Yasuda N, Marchesini G, Dolling UH, Grabowski EJJ. Tetrahedron Lett. 1995;36:5461–5464. [Google Scholar]
- 25.Mellegaard-Waetzig SR, Rayabarapu DK, Tunge JA. Synlett. 2005:2759–2762. [Google Scholar]
- 26.Stewart IC, Douglas CJ, Grubbs RH. Org Lett. 2008;10:441–444. doi: 10.1021/ol702624n. [DOI] [PubMed] [Google Scholar]
- 27.Wakamatsu H, Blechert S. Angew Chem, Int Ed. 2002;41:2403–2405. doi: 10.1002/1521-3773(20020703)41:13<2403::AID-ANIE2403>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 28.Bieniek M, Michrowska A, Usanov DL, Grela K. Chem – Eur J. 2008;14:806–818. doi: 10.1002/chem.200701340. [DOI] [PubMed] [Google Scholar]
- 29.Scholl M, Ding S, Lee CW, Grubbs RH. Org Lett. 1999;1:953–956. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
- 30.Van Veldhuizen JJ, Campbell JE, Giudici RE, Hoveyda AH. J Am Chem Soc. 2005;127:6877–6882. doi: 10.1021/ja050179j. [DOI] [PubMed] [Google Scholar]
- 31.Zhugralin AR, Hoveyda AH. unpublished results. [Google Scholar]
- 32.Filler R, Schure RM. J Org Chem. 1967;32:1217–1219. [Google Scholar]
- 33.Alexander JB, Schrock RR, Davis WM, Hultzsch KC, Hoveyda AH, Houser JH. Organometallics. 2000;19:3700–3715. [Google Scholar]
- 34.Tsang WCP, Hultzsch KC, Alexander JB, Bonitatebus PJ, Jr, Schrock RR, Hoveyda AH. J Am Chem Soc. 2003;125:2652–2666. doi: 10.1021/ja0210603. [DOI] [PubMed] [Google Scholar]
- 35.For a previous example of a chiral Mo-based taddolate complex, see: McConville DH, Wolf JR, Schrock RR. J Am Chem Soc. 1993;115:4413–4414.
- 36.For overviews regarding stereogenic-at-metal complexes, see: Brunner H. Angew Chem, Int Ed. 1999;38:1194–1208. doi: 10.1002/(SICI)1521-3773(19990503)38:9<1194::AID-ANIE1194>3.0.CO;2-X.Fontecave M, Hamelin O, Ménage S. Top Organomet Chem. 2005;15:271–288.
- 37.For enantiomerically pure stereogenic-at-Ru catalysts used in enantioselective olefin metathesis that bear a bidentate chiral ligand, see: Van Veldhuizen JJ, Garber SB, Kingsbury JS, Hoveyda AH. J Am Chem Soc. 2002;124:4954–4955. doi: 10.1021/ja020259c.. (b) See ref 9a. (c) See ref 10d. (d) See ref 7a. Giudici RE, Hoveyda AH. J Am Chem Soc. 2007;129:3824–3825. doi: 10.1021/ja070187v.. For stereogenic-at-Ru olefin metathesis catalysts that have been used in olefin metathesis but only in the racemic form, see: Fürstner A, Liebl M, Lehmann CW, Picquet M, Kunz R, Bruneau C, Touchard D, Dixneuf PH. Chem – Eur J. 2000;6:1847–1857. doi: 10.1002/(sici)1521-3765(20000515)6:10<1847::aid-chem1847>3.0.co;2-1.Bornand M, Chen P. Angew Chem, Int Ed. 2005;44:7909–7911. doi: 10.1002/anie.200502606.Conrad JC, Parnas HH, Snelgrove JL, Fogg DE. J Am Chem Soc. 2005;127:11882–11883. doi: 10.1021/ja042736s.Bornand M, Torker S, Chen P. Organometallics. 2007;26:3585–3596.
- 38.Singh R, Schrock RR, Müller P, Hoveyda AH. J Am Chem Soc. 2007;129:12654–12655. doi: 10.1021/ja075569f. [DOI] [PubMed] [Google Scholar]
- 39.(a) Hock AS, Schrock RR, Hoveyda AH. J Am Chem Soc. 2006;128:16373–16375. doi: 10.1021/ja0665904. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Singh R, Czekelius C, Schrock RR, Müller P, Hoveyda AH. Organometallics. 2007;26:2528–2539. doi: 10.1021/om061134+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pilyugina T, Schrock RR, Hoveyda AH. unpublished results. [Google Scholar]
- 41.For reviews regarding the use of binaphthol and its derivatives as chiral ligands, see: Chen Y, Yekta S, Yudin AK. Chem Rev. 2003;103:3155–3212. doi: 10.1021/cr020025b.Brunel JM. Chem Rev. 2005;105:857–898. doi: 10.1021/cr040079g.. For an overview of taddol-type chiral ligands in stereoselective synthesis, see: Seebach D, Beck AK, Heckel A. Angew Chem, Int Ed. 2001;40:92–138.Rauniyar V, Zhai H, Hall DG. J Am Chem Soc. 2008;130:8481–8490. doi: 10.1021/ja8016076.. For a review that includes diols as chiral Brønsted acids, see: Akiyama T. Chem Rev. 2007;107:5744–5758. doi: 10.1021/cr068374j.
- 42.For use of monodentate phosphoramidites as ligands in enantioselective catalysis, see: Feringa BL. Acc Chem Res. 2000;33:346–353. doi: 10.1021/ar990084k.Leitner A, Shekhar S, Pouy MJ, Hartwig JF. J Am Chem Soc. 2005;127:15506–15514. doi: 10.1021/ja054331t.Minnaard AJ, Feringa BL, Lefort L, de Vries JG. Acc Chem Res. 2007;40:1267–1277. doi: 10.1021/ar7001107.
- 43.For monodentate chiral NHCs used as ligands in enantioselective catalysis, see: César V, Bellemin-Laponnaz S, Gade LH. Chem Soc Rev. 2004;33:619–636. doi: 10.1039/b406802p.Gade LH, Bellemin-Laponnaz S. Top Organomet Chem. 2007;21:117–157.Douthwaite RE. Coord Chem Rev. 2007;251:702–717.
- 44.We are aware of only two examples where a chiral O-based monodentate ligand was systematically used in enantioselective catalysis. In both cases, low enantioselectivity (up to 72% ee, based on optical rotation values) was observed. See: Hashimoto S, Komeshima N, Koga K. J Chem Soc, Chem Commun. 1979:437–438.Tayama E, Saito A, Ooi T, Maruoka K. Tetrahedron. 2002;58:8307–8312.
- 45.Cram DJ, Helgeson RC, Peacock SC, Kaplan LJ, Domeier L, Moreau P, Koga K, Mayer JM, Chao Y, Siegel MG, Hoffman DH, Sogah GDY. J Org Chem. 1978;43:1930–1946. [Google Scholar]
- 46.For examples of tetrahydrobinaphthol-based chiral Mo diolate complexes, see: Aeilts SL, Cefalo DR, Bonitatebus PJ, Jr, Houser JH, Hoveyda AH, Schrock RR. Angew Chem, Int Ed. 2001;40:1452–1456.Schrock RR, Jamieson JY, Dolman SJ, Miller SA, Bonitatebus PJ, Jr, Hoveyda AH. Organometallics. 2002;21:409–417.. For a review regarding the differences between binaphthol and its hydrogenated derivatives, see: Au-Yeung TTL, Chan SS, Chan ASC. Adv Synth Catal. 2003;345:537–555.
- 47.For examples where tetrahydrobinaphthol-based Mo-diolate complexes outperform other olefin metathesis catalysts, see refs 8a–c.
- 48.Stereogenic-at-metal Ru-based catalysts for enantioselective olefin metathesis have been developed in these laboratories; such complexes, however, bear a bidentate chiral ligand. See refs 37a–e.
- 49.For a stereogenic-at-Fe complex that undergoes racemization faster than it promotes ketone hydrosilylation, see: Brunner H, Fisch K. Angew Chem, Int Ed. 1990;29:1131–1132.
-
50.As illustrated in the hypothetical case below, each catalytic RCM process involves two distinct olefin metathesis reactions, one involving formation of the substrate-bound Mo alkylidene (or Ru carbene) and the other the ring closure:
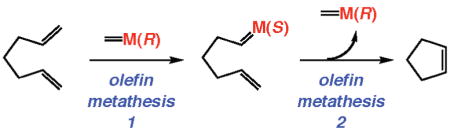 Thus, in the case of a stereogenic-at-metal complex in the absence of alternative stereomutation mechanisms (e.g., by a degenerate olefin metathesis reaction), each catalytic cycle regenerates the original catalyst isomer. Further discussions will be provided in the full account of the mechanistic aspects of the new class of stereogenic-at-Mo complexes.
Thus, in the case of a stereogenic-at-metal complex in the absence of alternative stereomutation mechanisms (e.g., by a degenerate olefin metathesis reaction), each catalytic cycle regenerates the original catalyst isomer. Further discussions will be provided in the full account of the mechanistic aspects of the new class of stereogenic-at-Mo complexes.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.