

Abstract
Rosiglitazone is an insulin-sensitizing agent that has recently been shown to exert beneficial effects on atherosclerosis. In addition to peroxisome proliferator–activated receptor (PPAR)-γ, rosiglitazone can affect other targets, such as directly inhibiting recombinant long-chain acyl-CoA synthetase (ACSL)-4 activity. Because it is unknown if ACSL4 is expressed in vascular cells involved in atherosclerosis, we investigated the ability of rosiglitazone to inhibit ACSL activity and fatty acid partitioning in human and murine arterial smooth muscle cells (SMCs) and macrophages. Human and murine SMCs and human macrophages expressed Acsl4, and rosiglitazone inhibited Acsl activity in these cells. Furthermore, rosiglitazone acutely inhibited partitioning of fatty acids into phospholipids in human SMCs and inhibited fatty acid partitioning into diacylglycerol and triacylglycerol in human SMCs and macrophages through a PPAR-γ–independent mechanism. Conversely, murine macrophages did not express ACSL4, and rosiglitazone did not inhibit ACSL activity in these cells, nor did it affect acute fatty acid partitioning into cellular lipids. Thus, rosiglitazone inhibits ACSL activity and fatty acid partitioning in human and murine SMCs and in human macrophages through a PPAR-γ–independent mechanism likely to be mediated by ACSL4 inhibition. Therefore, rosiglitazone might alter the biological effects of fatty acids in these cells and in atherosclerosis.
Long-chain acyl-CoA synthetases (ACSLs) (E.C.6.2.1.3) catalyze esterification of long-chain fatty acids, mediating the partitioning of fatty acids in mammalian cells. ACSL isoforms (ACSL1, ACSL3, ACSL4, ACSL5, and ACSL6) generate bioactive fatty acyl-CoAs from CoA, ATP, and long-chain (C12–C20) fatty acids (1). ACSLs exhibit different tissue distribution, subcellular localization, fatty acid preference, responsiveness to pharmacological inhibitors, and transcriptional regulation. Previous studies have characterized these enzymes in different tissues (2); however, expression levels and function in vascular cells are unknown. Until recently, inconsistencies in the ACSL nomenclature led to confusion in the literature. The standard nomenclature now uses ACSL1 and ACSL3–6 for human and Acsl1 and Acsl3–6 for murine members of this family (3).
Thiazolidinediones (TZDs) are a class of oral insulin-sensitizing agents, extensively used in the treatment of type 2 diabetes. Whereas members of this class of agents, such as rosiglitazone and pioglitazone, have been demonstrated to provide effective glycemic control in patients with diabetes (4), their effect on lipid metabolism is less clear (5). Perhaps more interesting are the recent findings that rosiglitazone treatment results in fewer inflammatory cells and increased collagen content in advanced atherosclerotic plaques (6,7; rev. in 8), suggesting that rosiglitazone might exert a stabilizing effect on atherosclerotic plaques.
The mechanism of TZD action is mediated largely through the activation of PPAR-γ, a member of the super family of ligand-activated nuclear transcription factors. In vascular tissue, peroxisome proliferator–activated receptor (PPAR)-γ is expressed in smooth muscle cells, macrophages, and endothelial cells. TZDs may also exert nongenomic effects. For example, rosiglitazone has been shown to activate 5′-AMP protein kinase (AMPK) through a PPAR-γ–independent mechanism (9). With regard to Acsl isoforms, in vitro studies of rat recombinant proteins have demonstrated that TZDs can directly inhibit the activity of one of the gene products, Acsl4 (10).
Because of the strong association among increased fatty acid load, atherosclerosis, and cardiovascular disease, particularly in the setting of type 2 diabetes, we investigated the ability of rosiglitazone to inhibit ACSL activity and acute fatty acid partitioning in smooth muscle cells (SMCs) and macrophages. We show that human and murine SMCs and human macrophages express a number of ACSL isoforms and that ACSL4, the rosiglitazone-sensitive isoform, is expressed in these cells. We also show that rosiglitazone inhibits ACSL activity and fatty acid partitioning in human and murine SMCs and in human macrophages through a PPAR-γ–independent mechanism likely to be mediated by direct inhibition of ACSL4. Furthermore, rosiglitazone might exert effects on fatty acid partitioning in human macrophages that are not mimicked by mouse models, because murine macrophages are devoid of Acsl4.
RESEARCH DESIGN AND METHODS
Oleic acid (OA) (sodium salt) was purchased from Nucheck Prep (Elysian, MN). RPMI-1640, fetal bovine serum, and penicillin/streptomycin were obtained from Gibco/Invitrogen (Grand Island, NY). CoA, ATP, essentially fatty acid–free BSA, sodium thioglycollate, dithiothreitol, EDTA, EGTA, leupeptin, aprotinin, pepstatin, phenylmethylsulfonyl fluoride, and benzamidine were obtained from Sigma-Aldrich (St. Louis, MO). Triascin C, T0070907, 5-aminoimidazole-4-carboxaide 1-β-d-ribo-furanoside (AICAR), 15-deoxy-Δ12,14 prostaglandin J2 (15-dPGJ2), and Wy14643 were from Biomol Research Laboratories (Plymouth Meeting, PA). Rosiglitazone was obtained from Alexis Biochemical (San Diego, CA). Anti-rabbit IgG horseradish peroxidase–linked antibodies, [9,10(n)-3H]-OA (259 GBq/mmol), [1-14C]-OA (2.07 GBq/mmol), and [U-14C]-glycerol (5.9 GBq/mmol) were obtained from Amersham Biosciences (Piscataway, NJ). Arachidonic acid (AA) [5,6,8,9,11,12,14,15,3H(N)] (6.66–8.88 TBq/mmol, 9.25 MBq) was obtained from Perkin Elmer Life and Analytical Sciences (Wellesley, MA). Recombinant human platelet-derived growth factor-BB was from Upstate Biotechnology (Lake Placid, NY).
Isolation and culture of human SMCs
Normal human aortic smooth muscle cells (hSMCs) were isolated by an explant method and grown as previously described (11). Incubation with fatty acids was performed in serum-free medium in the presence of 0.5% fatty acid–free BSA. All investigation on human cells was conducted according to the principles of the Declaration of Helsinki.
Isolation of human monocyte–derived macrophages
Monocytes were purified from buffy coats obtained from human donors by the Red Cross (Portland, OR), as described (12). Monocytes were purified by using density-gradient centrifugation with Ficoll-Paque Plus (Amersham Pharmacia Biotech), followed by positive selection with magnetic CD-14 microbeads (Miltenyi Biotec, Auburn, CA). Monocytes were differentiated in RPMI medium 1640 supplemented with penicillin/streptomycin and 10% fetal bovine serum and buffered with 10 mmol/l HEPES. The monocytes were differentiated to macrophages at a concentration of 5 × 105 cells per milliliter with 100 ng/ml granulocyte macrophage colony stimulating factor (Leukine; Drug Services, University of Washington).
Isolation and culture of murine SMCs
Normal murine SMCs were isolated from the thoracic aorta of male C57BL/6 mice via enzymatic digestion (13).
Isolation of murine peritoneal macrophages
Peritoneal macrophages were collected from male and female C57BL/6 (25–30 g) mice 5 days after intraperitoneal injection of 2 ml thioglycollate (40 mg/ml), as described previously (14). The harvested cells were incubated in an erythrocyte lysis buffer (155 mmol/l NH4Cl, 10 mmol/l KHCO3, 0.1 mmol/l EDTA) for removal of residual erythrocytes before plating. All animal work was approved and performed by following the guidelines for the use and care of laboratory animals at the University of Washington.
Reverse transcriptase and PCR
Total RNA was isolated from quiesced hSMCs (passage 4–11), murine SMCs (passage 2–5), human monocyte–derived macrophages, or murine peritoneal macrophages, using an RNeasy Mini Kit (Qiagen, Valencia, CA). A total amount of 250 ng RNA was reverse-transcribed using random hexamers and Omniscript reverse transcriptase (0.2 units/μl; Invitrogen, Grand Island, NY). The mixture was incubated for 60 min at 37°C and heat-inactivated for 5 min at 94°C. The resultant cDNA template was amplified by PCR with specific oligonucleotide primers targeted for human and murine ACSL1, ACSL3, ACSL4, ACSL5, and ACSL6 (Table 1). Primers were designed to recognize all known splice variants, including the newly described splice variants for human ACSL1 and ACSL6 (15). PCR was performed using a GeneAmp PCR system (Applied Biosystems, Foster City, CA) with a primer concentration of 400 nmol/l. PCR products were then separated via gel electrophoresis in 2% agarose gels and visualized with ethidium bromide. Murine brain and liver were used as positive controls for the Acsl3, Acsl5, and Acsl6 primers. A human neuroblastoma cell line (SH-SY5Y) was used as a positive control for human ACSL6.
TABLE 1.
ACSL primer sequences
| Target mRNA | Primer sequence | PCR product (bp) | Annealing temperature |
|---|---|---|---|
| Human ACSL isoforms | |||
| ACSL1_v1–2 | |||
| 5′primer | 5′-TGCAGCACTCACCACCTTC-3′ | 582 | 60°C |
| 3′primer | 5′-TAGGCATCCATGACAACTA-3′ | ||
| ACSL1_v1–3 | |||
| 5′primer | 5′-GAAGAGCCAACAGACGGAAG-3′ | 245′ACSL1_v3 | 54°C |
| 3′primer | 5′- GACAGTGGGTTGAAGCACCT-3′ | 326 ACSL1_v1-2 | 54°C |
| ACSL3 | |||
| 5′primer | 5′-CARCGCCATCTTCTGTGAG-3′ | 243 | 62°C |
| 3′primer | 5′-CGTGGCTTTCCATCAACAG-3′ | ||
| ACSL4 | |||
| 5′primer | 5′-CCGACCTAAGGGAGTGATGA-3′ | 169 | 62°C |
| 3′primer | 5′-CCTGCAGCCATAGGTAAAGC-3′ | ||
| ACSL5 | |||
| 5′primer | 5′-TTCCAAAGAGGACTCGCTG-3′ | 215 | 60°C |
| 3′primer | 5′-CAAGCCAATTCGGAGATGA-3′ | ||
| ACSL6 | |||
| 5′primer | 5′-CAGCCTGATGACCTCTCCAT-3′ | 101 | 55°C |
| 3′primer | 5′-CCTGAGAAATCAGCCACCAC-3′ | ||
| Murine Acsl isoforms | |||
| Acsl1 | |||
| 5′primer | 5′-GAACAGAGTGAAGCCCAAGC-3′ | 463 | 55°C |
| 3′primer | 5′-CAGGCTGTTTCTGACGA-3′ | ||
| Acsl3 | |||
| 5′primer | 5′-ATGATCGCTGCACAGGCGT-3′ | 369 | 51°C |
| 3′primer | 5′-C/GCTTTGGAATTCCTGTGGACC-3′ | ||
| Acsl4 | |||
| 5′primer | 5′-CGTTCCTCCAAGTAGACCAAC-3′ | 273 | 45°C |
| 3′primer | 5′-CCTTACACTGTCTGACCAGTC-3′ | ||
| Acsl5 | |||
| 5′primer | 5′-CAGAATGCACAGGTGGATG-3′ | 372 | 55°C |
| 3′primer | 5′-CAGGAGCAGACCAGTATTGC-3′ | ||
| Acsl6 | |||
| 5′primer | 5′-CCCGATGACCTCTCCATCGTG-3′ | 619 | 46°C |
| 3′primer | 5′-GGTGTACCTTCACAACGCCAG-3′ | ||
ACSL1_v, ACSL1 variant.
Western blot analysis of ACSL1 and ACSL4
Lysates were prepared as previously described (12,14,16), separated using 7.5% Tris-glycine SDS-PAGE, and transferred to polyvinylidene difluoride membranes. The membranes were then incubated overnight at 4°C with rabbit polyclonal anti-rat Acsl1 or Acsl4 antibodies (1:10,000) generated against the amino acid sequences MEVHELFRYFRMPELIDIR and EIHSMQSVEELGSKPENSSI, respectively (17). Horseradish peroxidase–linked anti-rabbit secondary antibodies were used at a final dilution of 1:5,000 for 1 h at room temperature. Signal detection was performed by enhanced chemiluminescence. Recombinant rat Acsl4 (3 μg) was used as a positive control. Rat liver high-speed supernatant was used as a positive control for ACSL1 expression. Briefly, rat liver was homogenized (1:3; weight:volume) using a Potter-Elvehjem homogenizer in a 320 mmol/l sucrose buffer containing Tris-HCl, pH 7.4 (20 mmol/l), EDTA (1 mmol/l), EGTA (1 mmol/l), leupeptin (20 μg/ml), benzamidine (1 mmol/l), aprotinin (10 μg/ml), pepstatin (5 μg/ml), dithiothreitol (1 mmol/l), and phenylmethylsulfonyl fluoride (0.1 mmol/l). The homogenate was centrifuged at 7,700g for 2 min. The supernatant was then centrifuged at 12,000g for 12 min and then at 177,000g for 1 h. The resulting high-speed supernatant was used for analyses.
Analysis of ACSL activity
Total acyl-CoA synthetase activity was measured via a modified protocol by Kim et al. (10), involving the formation of OA-CoA from OA. Briefly, cells were scraped into ice-cold cell sonication buffer containing 50 mmol/l potassium phosphate (pH 7.4), 10% glycerol, 1 mmol/l EDTA, 20 μg/ml leupeptin, 5 μg/ml pepstatin, 10 μg/ml aprotinin, and 5 mmol/l benzamidine and sonicated with two bursts of 10-s duration at 50 W. Protein concentration was determined via BCA protein assays (Pierce, Rockford, IL). The lysate (200 μg/sample) was then incubated in the presence of increasing concentrations of rosiglitazone, triacsin C, 15-dPGJ2, AICAR, or Wy16463 for 20 min at 37°C in a reaction mixture containing 175 mmol/l Tris (pH 7.4), 8 mmol/l MgCl2, 5 mmol/l dithiothreitol, 10 mmol/l ATP, and 250 μmol/l CoA with 50 μmol/l OA trace-labeled with [3H]-OA (1 μCi) in a final volume of 200 μl. The reaction was initiated by the addition of total cell lysate and terminated by the addition of 1 ml Doles reagent (2-propanol:hexane:H2SO4; 80:20:2), followed by 2 ml heptane, 0.5 ml water, and vigorous vortexing. The upper layer was removed and the lower (aqueous) phase was washed with 2 ml heptane. The radioactivity of the lower phase was evaluated by a liquid scintillation counter. The radioactivity measured was used to calculate the total enzymatic activity and was expressed as picomoles of OA-CoA ester formed per unit time. The results were corrected for blanks (samples without cell lysates added and samples analyzed in the absence of CoA or ATP) and for protein content. All reactions were confirmed to occur in the linear range.
Fatty acid partitioning into phospholipids and neutral lipids
Analysis of fatty acid partitioning into cellular lipids was measured as [14C]-OA, [14C]-glycerol, and [3H]-AA incorporation into phospholipids, diacylglycerol (DAG), and triacylglycerol (TAG) after separation by thin-layer chromatography, as previously described (16). For analysis of AA incorporation, neutral and phospholipid spots were visualized by exposure to iodine vapor, scraped, and quantified via liquid scintillation spectroscopy.
Adenoviral infection of human SMCs
The dominant-negative form of PPAR-γ harbors mutations of Leu468 and Glu471 of the full-length PPAR-γ1 sequence into Ala and has been shown to block the effects of endogenous PPAR-γ in different cells (18,19). This PPAR-γ mutant retains ligand and DNA binding, but exhibits markedly reduced transactivation due to impaired co-activator recruitment (18). The dominant-negative PPAR-γ was subcloned into a recombinant type 5 adenovirus (Adx-D/N-PPAR-γ). An adenovirus expressing the LacZ gene was used as a control vector. Human SMCs were infected with 40 plaque-forming units/cell via a modified protocol by Bruemmer et al. (20).
Statistical analysis
Values are shown as means ± SE. Curve fitting and data analysis were performed using GraphPad Prism (GraphPad, San Diego, CA). Statistical significance of difference was assessed by either one-way or two-way ANOVA and, in some experiments, the Student’s t test. Simultaneous multiple comparisons were based on a post hoc comparison test using a Student-Newman-Keuls test.
RESULTS
ACSL expression profiles in SMCs and macrophages
To evaluate the role of ACSLs in fatty acid partitioning in SMCs and macrophages, we characterized gene expression of ACSL isoforms present in human and murine SMCs, human monocyte–derived macrophages, and murine peritoneal macrophages. Using primers specific for each isoform (Table 1), we amplified mRNA for the different ACSL isoforms. Human SMCs and monocyte-derived macrophages expressed ACSL1, ACSL3, ACSL4, and ACSL5 mRNA (Fig. 1A). In hSMCs, the ACSL1 splice variant 1 and 2 primers detected a band of the correct size, whereas ACSL1 variant 3 was not detected (data not shown). Detectable levels of ACSL5 mRNA were found in hSMCs at cycle numbers above 30 (data not shown). Murine SMCs expressed Acsl1 and Acsl4 mRNA, and murine peritoneal macrophages expressed Acsl1, Acsl3, and Acsl4 mRNA. None of the Acsl6 splice variants were detected in any of the cell types studied (Fig. 1A).
FIG. 1.

Expression of ACSL isoforms in human and murine SMCs and macrophages. Semiquantitative RT-PCR analysis of ACSL isoforms (A) was performed on total RNA. A total of 28 cycles of PCRs were performed, using the primers and annealing temperatures shown in Table 1. The products were resolved on 2% agarose gels and visualized via ethidium bromide staining. ACSL1 (B) and ACSL4 (C) protein expression was evaluated by Western blot analysis. Cell lysates (60 μg/lane) were separated using 7.5% SDS-PAGE and probed with anti-rat Acsl1 or Acsl4 antibodies (1:10,000). The blots were re-probed with an anti–β-actin antibody (Sigma) at a 1:10,000 dilution (C, lower panel). Recombinant rat Acsl4 (rAcsl4) was used as a positive control in C. Molecular weight markers are indicated by arrows. Analyses were repeated at least three times with similar results. HSS, high-speed supernatant; Mφ, macrophages.
Antibodies that recognize human and murine ACSLs are not yet available for all isoforms. However, an anti-rat Acsl1 antibody recognized a band of the predicted 68 kDa in high-speed supernatant from rat liver (Fig. 1B) and in human and murine SMCs, monocyte-derived macrophages, and murine peritoneal macrophages (Fig. 1B). The Acsl4 antibody recognized a band of 74 kDa in hSMCs that co-migrated with recombinant rat Acsl4 (Fig. 1C; upper panel). ACSL4 was expressed in hSMCs and monocyte-derived macrophages and in murine SMCs, while no band was seen in murine peritoneal macrophages (Fig. 1C; lower panel).
Analysis of total ACSL activity in SMCs and macrophages
Previous studies have demonstrated that TZDs directly inhibit recombinant Acsl4 activity (10,21). This effect is selective to the Acsl4 isoform, because recombinant Acsl1, Acsl3, Acsl5, and Acsl6 are not inhibited by rosiglitazone until concentrations of >10 μmol/l are reached (10,21). Furthermore, rosiglitazone has not been reported to affect other enzymes with acyl-CoA synthetase activity, such as the fatty acid transport proteins. Triacsin C, an alkenyl-N-hydrozytrazene fungal metabolite, inhibits Acsl1, Acsl3, and Acsl4, but not Acsl5 and Acsl6 activity, at concentrations <10 μmol/l (10,21).
Initially, the ability of rosiglitazone to inhibit ACSL activity in cell lysates was evaluated. Rosiglitazone dose-dependently inhibited ACSL activity in both human and murine SMCs (Fig. 2A and E). The non-TZD PPAR-γ ligand 15dPGJ2, the PPAR-α agonist Wy14643, and the AMPK activator AICAR had no effect on ACSL activity at the concentrations tested (Table 2). Rosiglitazone did not inhibit ACSL activity in murine peritoneal macrophages at concentrations up to 100 μmol/l, consistent with the nondetectable expression of Acsl4 protein in these cells (Fig. 2G). However, contrary to the effects seen in murine macrophages, rosiglitazone inhibited ACSL activity in human monocyte–derived macrophages (Fig. 2C).
FIG. 2.
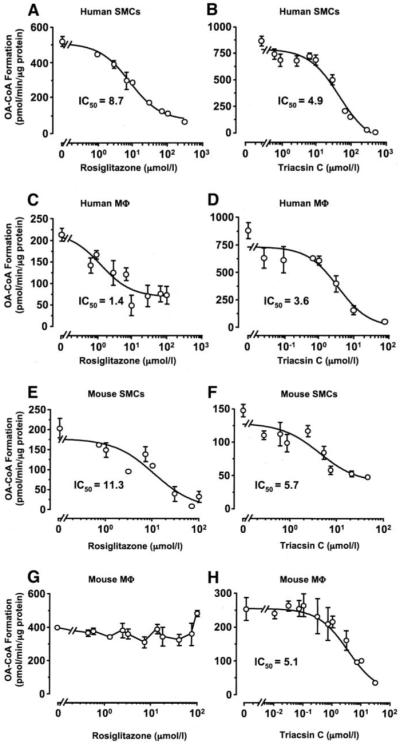
Rosiglitazone inhibits ACSL activity in SMCs and human macrophages, but not in murine macrophages. ACSL activity was analyzed in rosiglitazone or triacsin C–treated cell lysates. Lysates (200 μg total protein) from human SMCs (A and B), human macrophages (C and D), murine SMCs (E and F), and murine macrophages (G and H) were incubated in the presence of [3H]-OA, ATP, CoA, and rosiglitazone or triacsin C for 20 min at 37°C. Generated [3H]-OA-CoA was separated, and the radioactivity was determined via a scintillation counter. The results are the representative means of picomoles oleoyl-CoA formed/minute ± SE, performed in triplicates. The experiments were repeated three times with similar results. Mφ, macrophages.
TABLE 2.
Concentrations of agents resulting in IC50 of total ACSL activity in human SMC lysates
| IC50 value | pD2 value | |
|---|---|---|
| Triascin C | 2.74 μmol/l | 5.81 ± 0.23 |
| Rosiglitazone | 15.53 μmol/l | 4.86 ± 0.10 |
| Wy14643 | >70 μmol/l | ND |
| 15d-PGJ2 | >100 μmol/l | ND |
| AICAR | >10 mmol/l | ND |
Data are means (n = 6). Negative logarithms of the IC50 data (pD2) are expressed as means ± SE. ND, not determined.
Triacsin C dose-dependently inhibited total ACSL activity in both human and murine SMCs and macrophages, with similar half-maximal inhibition (IC50) values (Fig. 2B, D, F, and H).
Fatty acid partitioning in SMCs and macrophages
Because ACSL4 is likely to play an important role in fatty acid partitioning, we next investigated the effect of rosiglitazone on fatty acid partitioning in SMCs and macrophages. We had previously demonstrated, using thin-layer and high-performance liquid chromatography, that in SMCs, OA is incorporated into phospholipids (phosphati-dylcholine, phosphatidylserine, and phosphatidylethanolamine), monoglycerides, DAG, and TAG (16,22). In this study, we analyzed the effects of rosiglitazone and triacsin C on the kinetics of OA incorporation into these lipid pools. As seen in our previous studies, [14C]-OA was incorporated into cellular phospholipids, DAG, TAG, and, to a small degree, cholesteryl esters in hSMCs (Fig. 3A and B). However, the kinetics of OA incorporation into these pools were distinctly different. OA incorporation into phospholipids occurred rapidly (<2 min), while incorporation into DAG was first observed at the 5-min time point and reached a maximum after 4 h (Fig. 3A and B). Incorporation into TAG was first observed at the 1-h time point (Fig. 3A and B) and reached a maximum at 6 h (data not shown).
FIG. 3.
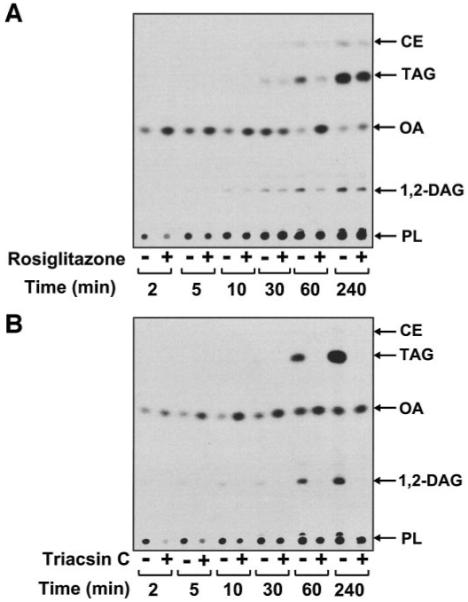
Analysis of OA partitioning in human SMCs. SMCs were pretreated for 30 min with 10 μmol/l rosiglitazone (A)or1 μmol/l triacsin C (B) before being labeled with [14C]-OA (1 μCi; 1 μmol/l) for the indicated times. The lipids were then extracted and separated by thin-layer chromatography. The plates were exposed to a PhosphorImager. CE, cholesteryl esters; PL, phospholipids. Representative experiments are shown, n = 3–5.
In hSMCs, preincubation with rosiglitazone caused an initial decrease of ~30% in OA incorporation into phospholipids (2–5 min after addition of OA; Figs. 3A and 4). This effect is most likely due to inhibition of phospholipid re-acylation because de novo phospholipid synthesis was undetectable at these time points (data not shown). Furthermore, in hSMCs (Figs. 3A and 5A) and macrophages (Fig. 5C), rosiglitazone inhibited [14C]-OA incorporation into DAG and TAG. Similarly, [14C]-glycerol incorporation into TAG was also inhibited by rosiglitazone in SMCs (65.2 ± 3.3% of untreated control, P < 0.01), indicating the attenuation observed is due to a decrease in de novo TAG formation. Similar results were seen with [3H]-AA–treated hSMCs (Fig. 5B). The non-TZD PPAR-γ ligand 15-dPGJ2 (Fig. 5A, □), the PPAR-α ligand Wy14643, and the AMPK activator AICAR (data not shown) were without effect. Thus, rosiglitazone results in a transient inhibition of phospholipid reacylation and a more sustained inhibition of DAG and TAG synthesis in hSMCs. As expected, rosiglitazone had no effect on [14C]-OA incorporation into these lipid pools in murine peritoneal macrophages (Fig. 5D).
FIG. 4.
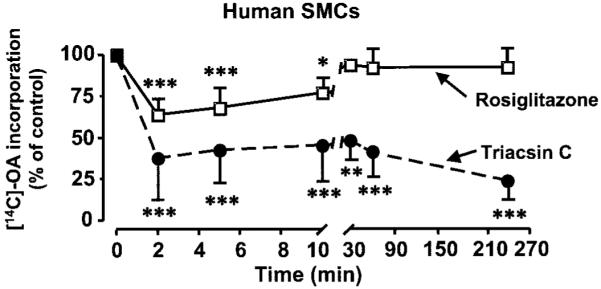
Rosiglitazone transiently inhibits OA partitioning into phospholipids, whereas triacsin C exerts a sustained inhibition in human SMCs. Human SMCs were pretreated for 30 min with 10 μmol/l rosiglitazone (□)or1 μmol/l triacsin C (●) before being labeled with [14C]-OA (1 μCi; 1 μmol/l) for the indicated times. The lipids were then extracted and separated by thin-layer chromatography, and the radioactivity in the phospholipid spot was quantified. Values are means ± SE; n = 3–5; ***P < 0.001, **P < 0.01, *P < 0.05 compared with untreated cells (two-way ANOVA).
FIG. 5.
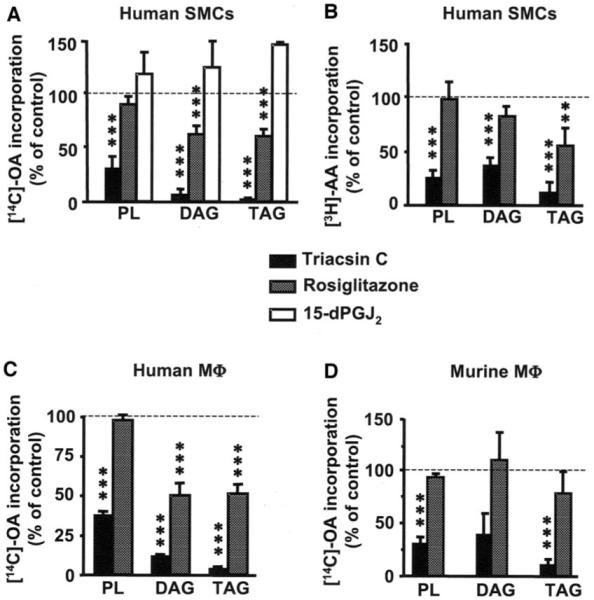
Rosiglitazone inhibits fatty acid partitioning into DAG and TAG in human SMCs and macrophages, but not in murine macrophages. SMCs and macrophages were pretreated for 30 min with 10 μmol/l rosiglitazone, 1 μmol/l triacsin C (A and B), 5 μmol/l triacsin C (C), or 25 μmol/l 15-dPGJ2 (A) before being labeled with [14C]-OA or [3H]-AA (1 μCi; 1 μmol/l) for 4 h. The lipids were then extracted and separated via thin-layer chromatography. Values are means ± SE, n = 3–7; ***P < 0.001, **P < 0.01, *P < 0.05 compared with nontreated controls (one-way ANOVA). The 15-dPGJ2 experiments were repeated twice and at a 24-h time point (n = 4) with similar results.
Triacsin C was used as a positive control to investigate the role of triacsin C–sensitive ACSL isoforms in SMCs and macrophages. Preincubation with triacsin C dramatically attenuated OA and AA incorporation into the DAG, TAG, and phospholipids (Figs. 3B, 4, and 5A and B) in hSMCs. As with rosiglitazone, the inhibitory effects of triacsin C on [14C]-OA incorporation into phospholipids were seen early (2–5 min) after addition of the fatty acid, at a time when [14C]-glycerol incorporation was not detectable, indicating that this effect was due to inhibition of phospholipid reacylation (Figs. 3B and 4). At later times, preincubation with triacsin C also attenuated [14C]-glycerol incorporation into phospholipids (8.6 ± 2.8% of control; P < 0.05), indicating that both reacylation and de novo pathways of phospholipid synthesis were inhibited.
As in hSMCs, triacsin C significantly attenuated [14C]-OA incorporation into the phospholipid, DAG, and TAG pools in human monocyte–derived macrophages (Fig. 5C). Similarly, in murine peritoneal macrophages, triacsin C inhibited [14C]-OA incorporation into phospholipids and TAG (Fig. 5D). These results suggest that triacsin C mimics some of the effects of rosiglitazone, but in addition has more dramatic effects on fatty acid incorporation into lipid pools, consistent with its ability to inhibit several ACSL isoforms.
PPAR-γ–independent effects of rosiglitazone on lipid partitioning
To confirm that the effects of rosiglitazone on OA incorporation into DAG and TAG in hSMCs were not mediated by PPAR-γ activation, we used two approaches. First, hSMCs were treated with T0070907, a noncompetitive antagonist of PPAR-γ (23). Rosiglitazone caused a 50% decrease in OA incorporation into DAG, an effect that was not influenced by pretreatment with T0070907 (Fig. 6A). Interestingly, while treatment with the noncompetitive inhibitor alone attenuated OA incorporation into TAG, it had no effect on the rosiglitazone-induced effects. Second, we transfected hSMCs with an adenovirus containing a dominant-negative PPAR-γ mutant. Similar to T0070907, neither treatment with an adenovirus-containing LacZ, nor the dominant-negative form of PPAR-γ, reversed the inhibitory effects of rosiglitazone on OA incorporation into DAG and TAG (Fig. 6B). Conversely, the dominant-negative PPAR-γ mutant resulted in a significant stimulation of platelet-derived growth factor-BB–mediated [3H]-thymidine incorporation into DNA in these cells (27,580 ± 765 cpm/mg protein in hSMCs expressing LacZ versus 64,343 ± 5,087 cpm/mg protein in SMCs expressing dominant-negative PPAR-γ; n = 3; P < 0.05), consistent with the growth inhibitory actions of PPAR-γ (20).
FIG. 6.
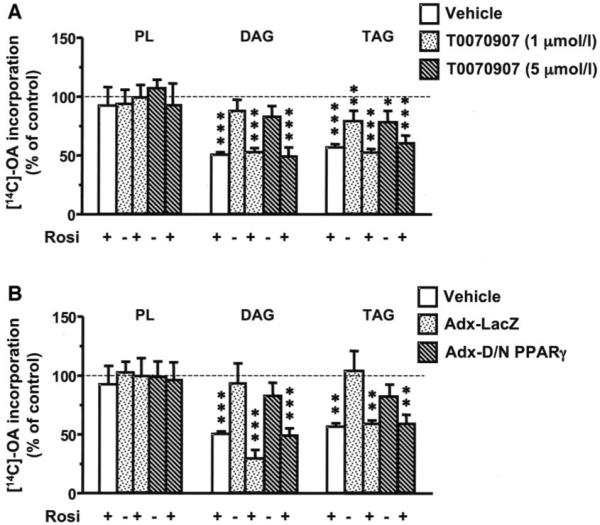
Inhibition of PPAR-γ does not block the inhibitory effect of rosiglitazone on fatty acid partitioning into DAG and TAG. Human SMCs were pretreated for 30 min with the indicated concentrations of the PPAR-γ inhibitor T0070907 (A) or were infected 48 h before analysis with 40 plaque-forming units/cell of a dominant-negative PPAR-γ mutant (Adx-D/N-PPARγ) or control (Adx-LacZ) (B). The SMCs were then incubated in the absence or presence of 10 μmol/l rosiglitazone and then in the presence of [14C]-OA (1 μCi; 1 μmol/l) for 4 h. The lipids were extracted and separated via thin-layer chromatography. Values are means ± SE; n = 3; ***P < 0.001, **P < 0.01, *P < 0.05 compared with nontreated controls (one-way ANOVA). PL, phospholipids.
DISCUSSION
Rosiglitazone directly inhibits ACSL activity and fatty acid partitioning into DAG and TAG through a PPAR-γ–independent mechanism
The main finding of this report is that rosiglitazone inhibits ACSL activity and fatty acid partitioning into DAG and TAG in human SMCs and macrophages through a PPAR-γ–independent mechanism. Several findings point to a direct inhibition of ACSL4 enzymatic activity as a mechanism of action of rosiglitazone on fatty acid partitioning. First, recombinant rat Acsl4 activity is inhibited by TZDs through a mixed-type inhibition with respect to all of its substrates: fatty acid, CoA, and ATP (10). Of all the ASCL isoforms, only ACSL4 is inhibited by rosiglitazone (10,21). Second, human SMCs and macrophages and murine SMCs express ACSL4, and rosiglitazone inhibited ACSL activity in cell lysates, whereas murine macrophages did not express Acsl4 and were insensitive to the effects of rosiglitazone on acute fatty acid partitioning. Third, the effect of rosiglitazone on ACSL activity and fatty acid partitioning into DAG and TAG was not mimicked by the PPAR-γ agonist 15-dPGJ2, the PPAR-α agonist Wy14643, or the AMPK activator AICAR. Finally, the inhibitory effects of rosiglitazone on OA partitioning into DAG and TAG were not blocked by pharmacological inhibition of PPAR-γ nor by inhibition of PPAR-γ action by expression of a dominant-negative PPAR-γ mutant. Thus, in addition to the well-described ability of rosiglitazone and other TZDs to activate AMPK (9,24) and p38 mitogen-activated protein kinase (25), ACSL4 appears to be an important contributor to the PPAR-γ–independent effects of rosiglitazone, at least in human and murine SMCs and in human macrophages. In fact, it is possible that the ability of rosiglitazone to activate AMPK might be due to its inhibitory action on ACSL4. ACSL4 is highly expressed in peroxisomes and mitochondrial-associated membranes and may be important in activating fatty acids for peroxisomal oxidation (26). Activity of AMPK, which is regulated by changes in the AMP:ATP ratio, might be upregulated by a reduced ACSL4 activity, leading to decreased fatty acid oxidation.
Rosiglitazone-induced inhibition of ACSL4 may have important biological effects in human macrophages and SMCs. Interestingly, AA is one of the preferred substrates for recombinant ACSL4 (27), and uterine tissues isolated from Acsl4+/− mice show increased levels of prostaglandin E2 (PGE2) and 6-keto-PGF1α (prostaglandin F1α) (28). It is therefore possible that inhibition of AA re-acylation by rosiglitazone-mediated ACSL4 inhibition may lead to increased levels of bioactive eicosanoids in the vascular wall. Indeed, rosiglitazone increases lipopolysaccaride-induced prostanoid production in the human monocytic cell line U937 (29). On the other hand, the PPAR-γ–mediated effects of rosiglitazone are likely to increase cellular fatty acid uptake. For example, in adipocytes, rosiglitazone induces the expression of oxidized LDL receptor-1, which not only increases LDL uptake but also increases fatty acid uptake (30). CD36 is another PPAR-γ target gene induced by rosiglitazone in macrophages (31). CD36 can act as a fatty acid transporter (32), and increased expression therefore might increase fatty acid uptake. Overall, these studies show that several different pathways are likely to mediate the effects of rosiglitazone on fatty acid metabolism in macrophages and SMCs.
The concentrations of rosiglitazone used in the present study are similar to maximal plasma concentrations (1.1 μmol/l) achieved in humans by oral dosing of 8 mg/day (33) and are also similar to concentrations found to activate AMPK. Thus, rosiglitazone inhibits ACSL activity with an average IC50 of 15.5 μmol/l in hSMCs (this study) and activates AMPK in the skeletal muscle cell line H-2Kb above 5 μmol/l (9). Therefore, the clinical effects of rosiglitazone, as well as other TZDs, may well be mediated not only by activation of PPAR-γ, but also by activation of AMPK and inhibition of ACSL4.
Do the different ACSL isoforms have distinct functions in SMCs and macrophages?
Based on differences in subcellular localization, regulation, tissue distribution, and fatty acid substrate preferences, it has been proposed that the different ACSL gene products channel fatty acids to different fates in the cell (34). Although this hypothesis will have to be carefully addressed, our data appear to support this notion, but also indicate that there is no absolute specificity in ACSL isoform function. Inhibition of ACSL4 by rosiglitazone resulted in an inhibition of fatty acid partitioning into DAG and TAG in both human SMCs and macrophages. Inhibition of ACSL4 has only a minor and transient effect on fatty acid partitioning into phospholipids. This appears to be primarily an effect on the re-acylation pathway, as our experiments demonstrated no [14C]-glycerol incorporation at these early time points (data not shown). In contrast, triacsin C, which inhibits ACSL1 and ACSL3 in addition to ACSL4, had dramatic effects on fatty acid partitioning into all lipid pools, including phospholipids. Although triacsin C might affect other fatty acid–binding proteins in the cell, these results suggest that ACSL4 is involved in de novo synthesis of DAG and fatty acid storage in TAG and that ACSL1 and/or ACSL3 have additional effects on fatty acid partitioning into phospholipids.
However, our results on murine macrophages show that in some cell types, one ACSL isoform appears sufficient to mediate fatty acid partitioning into all lipid pools, in addition to other fates, such as peroxisomal and mitochondrial β-oxidation. In these cells, although Acsl1 is the principal isoform expressed at detectable levels, murine macrophages efficiently incorporate fatty acids into all lipid pools.
There are important species differences in ACSL expression profiles in macrophages
Our results also demonstrate that human monocyte–derived macrophages express ACSL4 and exhibit sensitivity to rosiglitazone-inhibited fatty acid partitioning, whereas murine thioglycollate–elicited macrophages do not. While this study compared human monocyte–derived macrophages and murine thioglycollate–elicited peritoneal macrophages, preliminary studies in our laboratory show that bone marrow–derived macrophages from mice exhibit an Acsl expression profile very similar to that of murine peritoneal macrophages, indicating that the source of macrophages does not significantly alter the Acsl expression profile (J.E. Kanter and K.E. Bornfeldt, unpublished data). Important species differences have previously been demonstrated for other enzymes, such as the cyclic nucleotide phosphodiesterases (35).
TZDs have been shown to reduce atherosclerosis in diabetic and insulin-resistant mice (36,37). However, while rosiglitazone reduced the incidence of type 2 diabetes, it did not reduce the rate of myocardial infarction or stroke in the DREAM trial (38). On the contrary, patients receiving rosiglitazone developed more frequent congestive heart failure (38). One could speculate that the fact that rosiglitazone has several targets in addition to PPAR-γ, including ACSL4, might potentially explain the lack of beneficial cardiovascular effects in humans despite promising effects on atherosclerosis in mice (36,37). It will therefore be important to verify results obtained in murine models on isolated human macrophages and in clinical trials.
ACKNOWLEDGMENTS
This study was supported by National Institutes of Health Grants HL62887 and HL076719 to K.E.B., by DK21723 and HL 44948 to J.A.B., and by DK59935 to R.A.C.
We would like to thank Dr. Harald Frankowski for providing us with RNA from SH-SY5Y cells and Dr. Xia Shen for designing the ACSL1_v1-2 and ACSL1_v3 primers.
Glossary
- 15-dPGJ2
15-deoxy-Δ12,14 prostaglandin J2
- AA
arachidonic acid
- ACSL
long-chain acyl-CoA synthetase
- AICAR
5-aminoimidazole-4-carboxaide 1-β-d-ribo-furanoside
- DAG
diacylglycerol
- hSMC
human aortic smooth muscle cell
- IC50
half-maximal inhibition
- OA
oleic acid
- SMC
smooth muscle cell
- TAG
triacylglycerol
- TZD
thiazolidinedione
REFERENCES
- 1.Kornberg A, Pricer WE., Jr Enzymatic synthesis of the coenzyme A derivatives of long chain fatty acids. J Biol Chem. 1953;204:329–343. [PubMed] [Google Scholar]
- 2.Coleman RA, Van Horn CG, Gonzalez-Baró MR, Lewin TM. Long-chain acyl CoA synthetase isoforms and their functions. In: Haldar D, Das SK, editors. Lipids: Glycerolipid Metabolizing Enzymes. Research Signpost; Trivandrum, India: 2002. pp. 1–15. [Google Scholar]
- 3.Mashek DG, Bornfeldt KE, Coleman RA, Berger J, Bernlohr DA, Black P, DiRusso CC, Farber SA, Guo W, Hashimoto N, Khodiyar V, Kuypers FA, Maltais LJ, Nebert DW, Renieri A, Schaffer JE, Stahl A, Watkins PA, Vasiliou V, Yamamoto TT. Revised nomenclature for the mammalian long-chain acyl-CoA synthetase gene family. J Lipid Res. 2004;45:1958–1961. doi: 10.1194/jlr.E400002-JLR200. [DOI] [PubMed] [Google Scholar]
- 4.Grinberg A, Park KW. Nuclear peroxisome proliferator-activated receptors and thiazolidinediones. Int Anesthesiol Clin. 2005;43:1–21. doi: 10.1097/01.aia.0000157485.91241.0b. [DOI] [PubMed] [Google Scholar]
- 5.Chiquette E, Ramirez G, DeFronzo R. A meta-analysis comparing the effects of thiazolidinediones on cardiovascular risk factors. Arch Intern Med. 2004;164:2097–2104. doi: 10.1001/archinte.164.19.2097. [DOI] [PubMed] [Google Scholar]
- 6.Meisner F, Walcher D, Gizard F, Kapfer X, Huber R, Noak A, Sunder-Plassmann L, Bach H, Haug C, Bachem M, Stojakovic T, Marz W, Hombach V, Koenig W, Staels B, Marx N. Effect of rosiglitazone treatment on plaque inflammation and collagen content in nondiabetic patients: data from a randomized placebo-controlled trial. Arterioscler Thromb Vasc Biol. 2006;26:845–850. doi: 10.1161/01.ATV.0000203511.66681.7f. [DOI] [PubMed] [Google Scholar]
- 7.Marfella R, D’Amico M, Di Filippo C, Baldi A, Siniscalchi M, Sasso FC, Portoghese M, Carbonara O, Crescenzi B, Sangiuolo P, Nicoletti GF, Rossiello R, Ferraraccio F, Cacciapuoti F, Verza M, Coppola L, Rossi F, Paolisso G. Increased activity of the ubiquitin-proteasome system in patients with symptomatic carotid disease is associated with enhanced inflammation and may destabilize the atherosclerotic plaque: effects of rosiglitazone treatment. J Am Coll Cardiol. 2006;47:2444–2455. doi: 10.1016/j.jacc.2006.01.073. [DOI] [PubMed] [Google Scholar]
- 8.Blaschke F, Spanheimer R, Khan M, Law RE. Vascular effects of TZDs: new implications. Vascul Pharmacol. 2006;45:3–18. doi: 10.1016/j.vph.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 9.Fryer LG, Parbu-Patel A, Carling D. The anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J Biol Chem. 2002;277:25226–25232. doi: 10.1074/jbc.M202489200. [DOI] [PubMed] [Google Scholar]
- 10.Kim JH, Lewin TM, Coleman RA. Expression and characterization of recombinant rat acyl-CoA synthetases 1, 4, and 5: selective inhibition by triacsin C and thiazolidinediones. J Biol Chem. 2001;276:24667–24673. doi: 10.1074/jbc.M010793200. [DOI] [PubMed] [Google Scholar]
- 11.Suzuki LA, Poot M, Gerrity RG, Bornfeldt KE. Diabetes accelerates smooth muscle accumulation in lesions of atherosclerosis: lack of direct growth-promoting effects of high glucose levels. Diabetes. 2001;50:851–860. doi: 10.2337/diabetes.50.4.851. [DOI] [PubMed] [Google Scholar]
- 12.Bender AT, Ostenson CL, Giordano D, Beavo JA. Differentiation of human monocytes in vitro with granulocyte-macrophage colony-stimulating factor and macrophage colony-stimulating factor produces distinct changes in cGMP phosphodiesterase expression. Cell Signal. 2004;16:365–374. doi: 10.1016/j.cellsig.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 13.Wong ST, Baker LP, Trinh K, Hetman M, Suzuki LA, Storm DR, Bornfeldt KE. Adenylyl cyclase 3 mediates prostaglandin E2-induced growth inhibition in arterial smooth muscle cells. J Biol Chem. 2001;276:34206–34212. doi: 10.1074/jbc.M103923200. [DOI] [PubMed] [Google Scholar]
- 14.Lamharzi N, Renard CB, Kramer F, Pennathur S, Heinecke JW, Chait A, Bornfeldt KE. Hyperlipidemia in concert with hyperglycemia stimulates the proliferation of macrophages in atherosclerotic lesions. Diabetes. 2004;53:3217–3225. doi: 10.2337/diabetes.53.12.3217. [DOI] [PubMed] [Google Scholar]
- 15.Soupene E, Kuypers FA. Multiple erythroid isoforms of human long-chain acyl-CoA synthetases are produced by switch of the fatty acid gate domains. BMC Mol Biol. 2006;7:21. doi: 10.1186/1471-2199-7-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Askari B, Carroll MA, Capparelli M, Kramer F, Gerrity RG, Bornfeld KE. Oleate and linoleate enhance the growth-promoting effects of insulin-like growth factor-I through a phospholipase D-dependent pathway in arterial smooth muscle cells. J Biol Chem. 2002;277:36338–36344. doi: 10.1074/jbc.M205112200. [DOI] [PubMed] [Google Scholar]
- 17.Lewin TM, Kim JH, Granger DA, Vance JE, Coleman RA. Acyl-CoA synthetase isoforms 1, 4, and 5 are present in different subcellular membranes in rat liver and can be inhibited independently. J Biol Chem. 2001;276:24674–24679. doi: 10.1074/jbc.M102036200. [DOI] [PubMed] [Google Scholar]
- 18.Gurnell M, Wentworth JM, Agostini M, Adams M, Collingwood TN, Provenzano C, Browne PO, Rajanayagam O, Burris TP, Schwabe JW, Lazar MA, Chatterjee VKK. A dominant-negative peroxisome proliferator-activated receptor γ (PPAR γ) mutant is a constitutive repressor and inhibits PPAR γ-mediated adipogenesis. J Biol Chem. 2000;275:5754–5759. doi: 10.1074/jbc.275.8.5754. [DOI] [PubMed] [Google Scholar]
- 19.Eibl G, Takata Y, Boros LG, Liu J, Okada Y, Reber HA, Hines OJ. Growth stimulation of COX-2-negative pancreatic cancer by a selective COX-2 inhibitor. Cancer Res. 2005;65:982–990. [PubMed] [Google Scholar]
- 20.Bruemmer D, Berger JP, Liu J, Kintscher U, Wakino S, Fleck E, Moller DE, Law RE. A non-thiazolidinedione partial peroxisome proliferator-activated receptor γ ligand inhibits vascular smooth muscle cell growth. Eur J Pharmacol. 2003;466:225–234. doi: 10.1016/s0014-2999(03)01556-5. [DOI] [PubMed] [Google Scholar]
- 21.Van Horn CG, Caviglia JM, Li LO, Wang S, Granger DA, Coleman RA. Characterization of recombinant long-chain rat acyl-CoA synthetase isoforms 3 and 6: identification of a novel variant of isoform 6. Biochemistry. 2005;44:1635–1642. doi: 10.1021/bi047721l. [DOI] [PubMed] [Google Scholar]
- 22.Renard CB, Askari B, Suzuki LA, Kramer F, Bornfeldt KE. Oleate, not ligands of the receptor for advanced glycation end-products, promotes proliferation of human arterial smooth muscle cells. Diabetologia. 2003;46:1676–1687. doi: 10.1007/s00125-003-1247-9. [DOI] [PubMed] [Google Scholar]
- 23.Lee G, Elwood F, McNally J, Weiszmann J, Lindstrom M, Amaral K, Nakamura M, Miao S, Cao P, Learned RM, Chen JL, Li Y. T0070907, a selective ligand for peroxisome proliferator-activated receptor γ, functions as an antagonist of biochemical and cellular activities. J Biol Chem. 2002;277:19649–19657. doi: 10.1074/jbc.M200743200. [DOI] [PubMed] [Google Scholar]
- 24.Lebrasseur NK, Kelly M, Tsao TS, Farmer SR, Saha AK, Ruderman NB, Tomas E. Thiazolidinediones can rapidly activate AMP-activated protein kinase (AMPK) in mammalian tissues. Am J Physiol Endocrinol Metab. 2006;291:E175–E181. doi: 10.1152/ajpendo.00453.2005. [DOI] [PubMed] [Google Scholar]
- 25.Gardner OS, Shiau CW, Chen CS, Graves LM. Peroxisome proliferator-activated receptor gamma-independent activation of p38 MAPK by thiazo-lidinediones involves calcium/calmodulin-dependent protein kinase II and protein kinase R: correlation with endoplasmic reticulum stress. J Biol Chem. 2005;280:10109–10118. doi: 10.1074/jbc.M410445200. [DOI] [PubMed] [Google Scholar]
- 26.Lewin TM, Van Horn CG, Krisans SK, Coleman RA. Rat liver acyl-CoA synthetase 4 is a peripheral-membrane protein located in two distinct subcellular organelles, peroxisomes, and mitochondrial-associated membrane. Arch Biochem Biophys. 2002;404:263–270. doi: 10.1016/s0003-9861(02)00247-3. [DOI] [PubMed] [Google Scholar]
- 27.Kang MJ, Fujino T, Sasano H, Minekura H, Yabuki N, Nagura H, Iijima H, Yamamoto TT. A novel arachidonate-preferring acyl-CoA synthetase is present in steroidogenic cells of the rat adrenal, ovary, and testis. Proc Natl Acad Sci U S A. 1997;94:2880–2884. doi: 10.1073/pnas.94.7.2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cho YY, Kang MJ, Sone H, Suzuki T, Abe M, Igarashi M, Tokunaga T, Ogawa S, Takei YA, Miyazawa T, Sasano H, Fujino T, Yamamoto TT. Abnormal uterus with polycysts, accumulation of uterine prostaglandins, and reduced fertility in mice heterozygous for acyl-CoA synthetase 4 deficiency. Biochem Biophys Res Commun. 2001;284:993–997. doi: 10.1006/bbrc.2001.5065. [DOI] [PubMed] [Google Scholar]
- 29.Tsukamoto H, Hishinuma T, Suzuki N, Tayama R, Hiratsuka M, Yoshihisa T, Mizugaki M, Goto J. Thiazolidinediones increase arachidonic acid release and subsequent prostanoid production in a peroxisome proliferator-activated receptor γ-independent manner. Prostaglandins Other Lipid Mediat. 2004;73:191–213. doi: 10.1016/j.prostaglandins.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 30.Chui PC, Guan HP, Lehrke M, Lazar MA. PPARγ regulates adipocyte cholesterol metabolism via oxidized LDL receptor 1. J Clin Invest. 2005;115:2244–2256. doi: 10.1172/JCI24130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liang CP, Han S, Okamoto H, Carnemolla R, Tabas I, Accili D, Tall AR. Increased CD36 protein as a response to defective insulin signaling in macrophages. J Clin Invest. 2004;113:764–773. doi: 10.1172/JCI19528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Febbraio M, Guy E, Coburn C, Knapp FF, Jr, Beets AL, Abumrad NA, Silverstein RL. The impact of overexpression and deficiency of fatty acid translocase (FAT)/CD36. Mol Cell Biochem. 2002;239:193–197. [PubMed] [Google Scholar]
- 33.Miller AK, Inglis AM, Culkin KT, Jorkasky DK, Freed MI. The effect of acarbose on the pharmacokinetics of rosiglitazone. Eur J Clin Pharmacol. 2001;57:105–109. doi: 10.1007/s002280100275. [DOI] [PubMed] [Google Scholar]
- 34.Coleman RA, Lewin TM, Van Horn CG, Gonzalez-Baró MR. Do long-chain acyl-CoA synthetases regulate fatty acid entry into synthetic versus degradative pathways? J Nutr. 2002;132:2123–2126. doi: 10.1093/jn/132.8.2123. [DOI] [PubMed] [Google Scholar]
- 35.Rybalkin SD, Bornfeldt KE, Sonnenburg WK, Rybalkina IG, Kwak KS, Hanson K, Krebs EG, Beavo JA. Calmodulin-stimulated cyclic nucleotide phosphodiesterase (PDE1C) is induced in human arterial smooth muscle cells of the synthetic, proliferative phenotype. J Clin Invest. 1997;100:2611–2621. doi: 10.1172/JCI119805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li AC, Brown KK, Silvestre MJ, Willson TM, Palinski W, Glass CK. Peroxisome proliferator-activated receptor γ ligands inhibit development of atherosclerosis in LDL receptor-deficient mice. J Clin Invest. 2000;106:523–531. doi: 10.1172/JCI10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Calkin AC, Forbes JM, Smith CM, Lassila M, Cooper ME, Jandeleit-Dahm KA, Allen TJ. Rosiglitazone attenuates atherosclerosis in a model of insulin insufficiency independent of its metabolic effects. Arterioscler Thromb Vasc Biol. 2005;25:1903–1909. doi: 10.1161/01.ATV.0000177813.99577.6b. [DOI] [PubMed] [Google Scholar]
- 38.DREAM (Diabetes REduction Assessment with ramipril and rosiglitazone Medication) Trial Investigators. Gerstein HC, Yusuf S, Bosch J, Pogue J, Sheridan P, Dinccag N, Hanefeld M, Hoogwerf B, Laakso M, Mohan V, Shaw J, Zinman B, Holman RR. Effect of rosiglitazone on the frequency of diabetes in patients with impaired glucose tolerance or impaired fasting glucose: a randomised controlled trial. Lancet. 2006;368:1096–1105. doi: 10.1016/S0140-6736(06)69420-8. [DOI] [PubMed] [Google Scholar]