

Abstract
Chicken ovalbumin upstream promoter transcription factor (COUP-TF)-interacting proteins 1 and 2 (CTIP1 and CTIP2) enhance transcriptional repression mediated by COUP-TF II and have been implicated in hematopoietic cell development and malignancies. CTIP1 and CTIP2 are also sequence-specific DNA-binding proteins that repress transcription through direct, COUP-TF-independent binding to a GC-rich response element. CTIP1- and CTIP2-mediated transcriptional repression is insensitive to trichostatin A, an inhibitor of known class I and II histone deacetylases. However, chromatin immunoprecipitation assays revealed that expression of CTIP2 in mammalian cells resulted in deacetylation of histones H3 and/or H4 that were associated with the promoter region of a reporter gene. CTIP2-mediated transcriptional repression, as well as deacetylation of promoter-associated histones H3/H4 in CTIP2-transfected cells, was reversed by nicotinamide, an inhibitor of class III histone deacetylases such as the mammalian homologs of yeast Silent Information Regulator 2 (Sir2). The human homolog of yeast Sir2, SIRT1, was found to interact directly with CTIP2 and was recruited to the promoter template in a CTIP2-dependent manner. Moreover, SIRT1 enhanced the deacetylation of template-associated histones H3/H4 in CTIP2-transfected cells, and stimulated CTIP2-dependent transcriptional repression. Finally, endogenous SIRT1 and CTIP2 co-purified from Jurkat cell nuclear extracts in the context of a large (1–2 mDa) complex. These findings implicate SIRT1 as a histone H3/H4 deacetylase in mammalian cells and in transcriptional repression mediated by CTIP2.
CTIP11 (EVI9 or BCL11A) and CTIP2 (BCL11B) are two related C2H2 zinc finger proteins that were originally isolated and identified as COUP-TF-interacting proteins (1). CTIP12 (1) and CTIP2 both enhance COUP-TFII-mediated transcriptional repression in transfected cells independently of trichostatin A (TSA)-sensitive histone deacetylation. Both CTIPs are expressed in hematopoietic cells of lymphoid origin,3 which is of interest because lymphoid-derived cells are devoid of transcripts encoding COUP-TF family members.3 Thus, it is likely that CTIPs either function with other nuclear receptors in cells of lymphoid origin or act as COUP-TF-independent transcription factors in these cells. The latter appears to be true as CTIP1 and CTIP2 have been demonstrated to repress expression of a reporter gene through direct binding to a recently identified binding site, 5′-GGCCGGAGG-3′ (upper strand) (2). This repression was observed in the absence of cotransfected COUP-TF proteins and was insensitive to reversal by TSA (2). These findings indicate that CTIP proteins may regulate transcription independently of COUP-TF proteins and TSA-sensitive HDACs in some cell types and/or promoter contexts.
CTIP1 and CTIP2 have been implicated in the etiology of both myeloid and lymphoid malignancies. Overexpression of the CTIP1 gene following proviral integration in murine hematopoietic cells results in the generation of myeloid leukemia (3). Similarly, dysregulation of the human CTIP1 locus (2p13) either by amplification or translocation appears to result in B cell chronic lymphocytic leukemia and immunocytoma (4). The human CTIP2 locus (14q32) has been associated with a translocation event, t(5;14)(q35;q32), which results in acute T lymphoblastic leukemia (5). More recently, CTIP2 loss of function mutations were found to contribute to mouse lymphoma genesis, leading the authors to speculate that CTIP2 may function as a tumor suppressor protein (6). The analysis of both CTIP1-(7) and CTIP2- (8) null animals has demonstrated a role for each in hematopoiesis and postnatal development. Although human CTIP1 and CTIP2 are expressed in early, multipotent hemopoietic progenitors, neither the physiological function(s) nor the contribution of these proteins to neoplastic processes in these cells is known (5, 9). Accordingly, it is important to elucidate the mechanism(s) underlying transcriptional repression mediated by CTIP proteins to understand the biological properties of these proteins, and ultimately, their cellular function(s).
A number of mammalian HDACs have been identified and divided into three classes based on their similarity to yeast enzymes. Mammalian class I HDACs, such as HDAC 1–3, 8, and possibly 11, are similar to yeast Rpd3, whereas class II HDACs (HDAC 4–7, 9, and 10) are most similar to the yeast enzyme Hda1 (10–13). All known members of class I and II HDAC families are sensitive to inhibition by TSA to varying degrees (10–15). Histone deacetylation is believed to promote the repressed transcriptional state by facilitating formation of a compacted form of chromatin, thereby making genes less accessible to transcriptional activators and/or the general transcription machinery (16). Moreover, TSA-sensitive histone deacetylation is required for the initial steps of heterochromatin formation leading to gene silencing (17). HDAC complexes are recruited to the nucleosomal template via specific interaction with DNA-binding repressor proteins such as Mad (18, 19), YY1 (20), Ume6 (21), REST/NRSF (22), and BTEB3 (23), or with their associated corepressors such as N-CoR/SMRT (24–26) and Sin3A (27).
A third class of HDACs whose members are structurally and catalytically distinct from class I and class II enzymes is represented by the Sir2-like proteins (reviewed in Refs. 10 and 11). Yeast Sir2 and its mammalian homologs are nicotinamide adenine dinucleotide (NAD+)-dependent deacetylases (28), which distinguishes them from class I and II HDACs. To date, seven human homologs of class III HDACs, known as Sirtuins (SIRT1–7), have been isolated (29, 30). Based on protein sequence homology, SIRT1 and its mouse ortholog Sir2α are the closest homologs to yeast Sir2 (11, 29). Although the substrate specificity of mammalian sirtuins has not been rigorously investigated, mouse Sir2α has been demonstrated to deacetylate lysine 9 and 14 of histone H3 and lysine 16 of H4 in the context of synthetic, acetylated peptides (28). SIRT1 has also been demonstrated to deacetylate p53 and attenuate p53-mediated functions (31, 32), and to antagonize PML/p53-induced cellular senescence (33). In addition, SIRT1 has been shown to repress p53-mediated transcriptional activation in mammalian cells (31–33), providing evidence that SIRT1, at least in part, may play a role in transcriptional repression of specific genes. Although various in vitro inhibitors of SIRT isozymes have been described, the catalytic activity of class III HDACs is not inhibited by TSA but may be inhibited by nicotinamide, a product of the SIRT-mediated deacetylation reaction (28, 34, 35).
Although CTIPs repress transcription in a TSA-insensitive manner (1, 2), we found that transient transfection of CTIP2 resulted in deacetylation of histone H3 and/or H4 that were associated with the promoter region of a target gene. Subsequently, we found that nicotinamide reversed both CTIP2-mediated transcriptional repression and deacetylation of promoter-associated histone H3/H4 in CTIP2-transfected cells. These findings led us to investigate the possibility that a class III HDAC may be involved in CTIP-mediated transcriptional repression. CTIP2 and the class III HDAC SIRT1 co-immuno-precipitated from extracts of transfected, co-transfected and untransfected cells, and the two proteins were shown to interact directly in vitro. Moreover, SIRT1 potentiated both transcriptional repression and histone H3/H4 deacetylation in cells transfected with CTIP2. These findings suggest a role for the NAD+-dependent histone deacetylase SIRT1 in the transcriptional repression activity of CTIP2 in mammalian cells.
Materials and Methods
Constructs
The (17-mer)4-tk-CAT reporter construct was a kind gift from Dr. Ming-jer Tsai (Baylor College of Medicine). FLAG-CTIP2 construct was prepared by PCR amplification of the CTIP2 open reading frame (1) with appropriate primers and insertion into pcDNA3(+) (In-vitrogen). The Gal4 DBD-CTIP2 construct was prepared by PCR amplification with appropriate primers followed by insertion into a commercially available vector (pM; Clontech). Myc-SIRT1, Myc-SIRT1 H363Y, and GST-SIRT1 constructs (33) were kind gifts from Dr. T. Kouzarides (University of Cambridge, Cambridge, UK). All vectors encoding GST fusion proteins were prepared by PCR amplification of appropriate templates followed by insertion into pGEX-2T (Amersham Biosciences). The constructs used for generating [35S]methionine-labeled proteins were prepared by PCR amplification with primers containing appropriate restriction sites for insertion into pcDNA3(+) or pcDNA3.1/His (Invitrogen). All constructs were verified by complete DNA sequencing.
Antibodies
The rabbit anti-SIRT1 antibody was described previously (33). Purified rabbit anti-Sir2α (which cross-reacts with human SIRT1; data not shown) and anti-acetylated-histone H3 and -histone H4 were obtained from Upstate. Mouse anti-FLAG and -Myc monoclonal antibodies were purchased from Sigma and Oncogene, respectively. The rat anti-CTIP2 monoclonal antibody (25B6) was raised against a recombinant GST fusion protein by the Monoclonal Antibody Facility, Institute for Neuroscience, University of Oregon, Eugene, OR. Epitope mapping has revealed that the 25B6 anti-CTIP2 monoclonal antibody recognizes an antigen within the 150 amino acids located at the extreme amino terminus of the protein, CTIP2-(1–150).4
Chromatin Immunoprecipitation (ChIP) Assays
ChIP assays were performed on transfected cells essentially as described by the Dean laboratory (36) with slight modifications. HEK293 cells were co-transfected at 60% confluency (10-cm plates) with 5 μg of the (17-mer)4-tk-CAT reporter, 1.5–10 μg of Gal4-CTIP2, 0.5 μg of Myc-SIRT1, and/or the parent control vectors using the calcium phosphate method. After 48 h, cells were washed twice with phosphate-buffered saline and cross-linked with 1% formaldehyde in phosphate-buffered saline at room temperature. Cells were washed twice with ice-cold phosphate-buffered saline buffer and collected in harvesting buffer (100 mm Tris-HCl, pH 9.4, containing 10 mm dithiothreitol). The cells were lysed in lysis buffer (50 mm Tris-HCl, pH 8.1, containing 1% SDS, 10 mm EDTA, and a protease inhibitor mixture). The sonicated lysates were then cleared by centrifugation and diluted 2.5-fold with ChIP dilution buffer (16.7 mm Tris-HCl, pH 8.1, containing 0.01% SDS, 1.1% Triton X-100, 1.2 mm EDTA, 167 mm NaCl, and a protease inhibitor mixture). One-tenth of the diluted lysate was reserved as an input sample to determine the total amount of reporter plasmid in transfected cells for subsequent normalization procedures. Two equal aliquots of the remaining lysate were used for immunoprecipitation with and without the addition of antibodies against K9-, K16-di-acetylated histone H3, and K5-, K8-, K12-, K16-tetra-acetylated histone H4 (Upstate; 5 μg of each antibody per immunoprecipitation reaction). Immune complexes were recovered with Protein A-Sepharose (Amersham Biosciences) and washed under stringent conditions. Chromatin complexes were eluted with the freshly prepared elution buffer (0.1 m NaHCO3 containing 1% SDS). The eluates and the above input samples were subjected to an overnight reversal of cross-links at 65 °C, followed by treatment with Proteinase K at 45 °C for 1 h. DNA was recovered by using a Nucleospin PCR extraction kit (Clontech) and amplified using a forward primer (5′-GGCATCAGAGCAGATTGTACT-3′) upstream of the multimerized 17-mer and a reverse primer (5′-CCTTAGCTCCTGAAAATCTCG-3′) downstream of the tk promoter but upstream of the transcriptional start site. The resulting PCR product (327 bp) was analyzed by agarose gel electrophoresis and ethidium bromide staining. Experiments were performed three to five times.
Transfection and Reporter Assays
HEK293 cells were transfected and harvested as described above. Where indicated, TSA (100 ng/ml) and nicotinamide (10 mm) treatments were initiated 24 h after transfection, and cells were harvested 24 h later. A β-galactosidase expression vector (pCMV-Sport-βGal, Invitrogen) was cotransfected as an internal control, and β-galactosidase activity was used to normalized CAT activity as described (37).
Coimmunoprecipitation Assays
HEK293 cells were transfected as described above with 10 μg each of expression vectors encoding FLAG-CTIP2 and/or Myc-SIRT1. Forty-eight hours after transfection the cells were lysed in NET-N buffer (20 mm Tris-HCl, pH 8, containing 150 mm NaCl, 0.5% Nonidet P-40, 10% glycerol, 1 mm EDTA, and a protease inhibitor mixture) by agitation at 4 °C for 30 min. After a brief sonication, lysates were cleared by centrifugation and immunoprecipitated as described previously (38) using the antibodies described above. Nuclear extracts from Jurkat cells were prepared using standard techniques (39), and immunoprecipitated (500 μg of protein per reaction) with purified anti-CTIP2 monoclonal (2 μg) or anti-Sir2α (0.5–2.5 μg) antibodies. All immunoprecipitates were analyzed by immunoblotting with appropriate antibodies.
GST Pull-down Experiments
GST pull-down experiments were conducted as described previously (40). Briefly, equivalent amounts of GST or GST-SIRT1 fusion proteins were bound to glutathione-Sepharose (Amersham Biosciences) and incubated with [35S]methionine-labeled proteins (CTIP2 or CTIP2 truncation mutants) prepared using the TnT transcription-translation system (Promega). The reactions were washed five times with binding buffer (10 mm Na-HEPES containing 10% glycerol, 1 mm EDTA, 1 mm dithiothreitol, 150 mm NaCl, and 0.05% Nonidet P-40) and bound proteins were eluted and resolved on denaturing SDS-PAGE gels for analysis by autoradiography.
Column Chromatography
Nuclear extracts were prepared from 30 liters of Jurkat cells (∼1.5 × 1010 cells; provided by the National Cell Culture Center, Minneapolis, MN) using the Dignam method (39) with the following minor modifications: Buffer C contained 720 mm NaCl instead of 420 mm, and Buffer D was replaced with Buffer 100 (20 mm HEPES, pH 7.9,100 mm NaCl, 0.2 mm EDTA, 0.5 mm dithiothreitol, and 10% glycerol). CTIP2 immunoreactivity was found only in the nuclear fraction using this protocol (data not shown). Jurkat nuclear extract (100 mg protein) was applied to a 1.5 × 8-cm phosphocellulose P11 column (Whatman) that had been equilibrated in Buffer 100 and running at 80 ml/h. The column was washed with Buffer 100 until the A280 returned to baseline and then eluted in a stepwise fashion with Buffer 100 containing 300, 600, and 1000 mm NaCl. The A280 of the eluate was allowed to return to baseline at each step. CTIP2 and SIRT1 immuno-reactivities were analyzed in the step fractions by immunoblotting with appropriate antibodies and chemiluminescence detection. The 600 mm step off the phosphocellulose column (16 mg of protein) was dialyzed against Buffer 100 and loaded onto a 2.5 ml of DEAE Bio-Gel A column (Bio-Rad) that had been equilibrated with Buffer 100. After collecting the flow-through fraction, the column was eluted with Buffer 100 containing 200, 400, and 1000 mm NaCl in a stepwise fashion, and the steps were analyzed by immunoblotting as described above. The 200 mm step off the DEAE column, which contained both CTIP2 and SIRT1 immunoreactivity, was concentrated to 1 ml (3.1 mg of protein) using a Milipore Ultrafree centrifugal filter device (30-kDa nominal molecular mass limit), and loaded onto a 1.6 × 90-cm Superose 6 size exclusion column (Amersham Biosciences) that was running at a flow rate of 0.4 ml/min. The Superose 6 column had been previously equilibrated with Buffer 100 and calibrated with protein standards (thyroglobulin, 669 kDa; catalase, 232 kDa; bovine serum albumin, 67 kDa; blue dextran, 2000 kDa; all from Amersham Biosciences). Fractions (5.0 min, 2.0 ml) were analyzed for the presence of CTIP2 and SIRT1 by immunoblotting.
Results
CTIP2-mediated Transcriptional Repression Is Partially Reversed by Nicotinamide, an Inhibitor of NAD+-dependent Deacetylases
Previous studies indicated that CTIP1-mediated transcriptional repression was independent of TSA-sensitive histone deacetylation (1, 2). In the present study, transcriptional repression mediated by a Gal4-CTIP2 fusion protein was found to be similarly insensitive to reversal with TSA in HEK293 cells (Fig. 1A, compare lanes 4 and 5 with 7 and 8). Considered together, these findings suggest that neither CTIP1- nor CTIP2-mediated transcriptional repression involves recruitment of class I or class II TSA-sensitive HDACs to the template. However, nicotinamide, an inhibitor of NAD+-dependent, class III HDACs, such as the SIRT family of proteins (29, 30), was found to inhibit CTIP2-mediated transcriptional repression, at least partially (∼70% reversal of repression; Fig. 1A, compare lanes 7 and 9 with lane 4).
Fig. 1. CTIP2-mediated transcriptional repression is partially reversed by nicotinamide (NAM), an inhibitor of NAD+-dependent deacetylases.
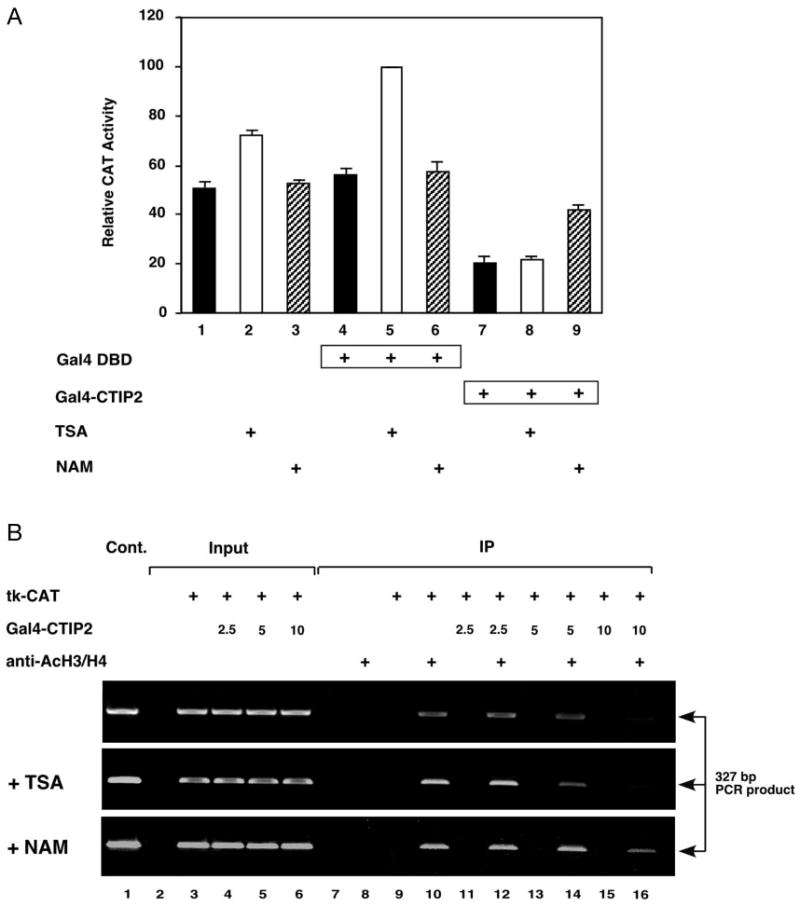
A, HEK293 cells were transiently transfected with 5 μg of the (17-mer)4-tk-CAT reporter along with 1.5 μg of expression vectors encoding either Gal4-CTIP2 or Gal4 DBD as indicated. Twenty-four hours after transfection, cells were treated or not (solid bars) with histone deacetylase inhibitors, TSA (100 ng/ml; open bars) and nicotinamide (NAM, 10 mm; hatched bars), for 24 h before collection. Transfection efficiency was normalized by β-galactosidase activity produced by a cotransfected β-galactosidase expression vector. CAT activity determined in the presence of Gal4 DBD and TSA (lane 5) was taken to be maximal and that against which all other determined CAT activities were compared. The results presented represent the mean (±S.E.) of three independent experimental determinations. B, HEK293 cells were transfected with 5 μg of (17-mer)4-tk-CAT reporter and increasing amounts (2.5, 5.0, and 10 μg) of an expression vector encoding Gal4-CTIP2 as indicated. The treatments with TSA and nicotinamide were carried out as described above. Transfection efficiency was normalized by the total amount of the transfected (17-mer)4-tk-CAT reporter as determined by PCR amplification (input lanes 3–6; 2.5% of total). Lanes 7–16 represent template amplification reactions from samples immunoprecipitated (IP) with or without antibodies specific for acetylated histone H3/H4 as indicated. Amplification reactions were separated on a 1% agarose gel that was stained with ethidium bromide to visualize DNA products. The indicated band is the expected, 327-bp amplification product from the reporter gene template. Lane 1 corresponds to a positive control in which the reporter plasmid was used as the template. Results are representative of three independent experiments.
These data indicated a possible role of a nicotinamide-sensitive HDAC(s) in the transcriptional repression mechanism of CTIP2 in mammalian cells. If true, transfection of Gal4-CTIP2 would be expected to result in recruitment of a nicotinamide-sensitive HDAC(s) to the template and deacetylation of template-associated histones. This hypothesis was tested by conducting ChIP studies in transiently transfected cells. Although the nature and/or extent of chromatin formation on transiently transfected templates likely differs from that of chromosomal genes, Dean and colleagues (36) have previously validated ChIP studies in transiently transfected cells and used this approach to demonstrate HDAC-dependent and -independent transcriptional repression mediated by the Rb protein. Thus, a similar approach was employed in the present study toward the goal of determining if histone deacetylation may underlie CTIP2-mediated transcriptional repression in mammalian cells. Alterations in the levels of acetylated histone H3/H4 associated with the template (a multimerized 17-mer-tk-CAT construct) were not observed at the lowest amount of Gal4-CTIP2 transfected (upper panel of Fig. 1B, compare lanes 10 with 12). However, transfection of increasing amounts of Gal4-CTIP2 dramatically reduced the level of acetylated histone H3/H4 associated with the template (upper panel of Fig. 1B, compare lanes 10, 12, 14, and 16), and this was unaffected by treatment of the transfected cells with TSA (lanes 10–14 of the middle panel of Fig. 1B). However, the deacetylation of template-associated histones observed in CTIP2-transfected cells was partially reversed by treatment of the cells with nicotinamide (compare lanes 10–14 of the top and bottom panels of Fig. 1B). Although inhibition of the deacetylation of promoter-associated histone H3 and/or H4 by treatment of the CTIP2-transfected cells with 10 mm nicotinamide was obvious, complete reversal was not observed (compare lanes 10 and 16 of the bottom panel of Fig. 1B). This may be because of the relative lack of efficacy of nicotinamide as an inhibitor of NAD+-dependent deacetylases and/or insufficient intracellular levels of nicotinamide in treated cells. Concentrations of nicotinamide above 10 mm appeared to result in cellular toxicity, thus, precluding the use of very high levels of nicotinamide. Nonetheless, these findings, which are consistent with transcriptional repression studies (Fig. 1A), further demonstrate that nicotinamide-sensitive histone deacetylation may underlie the mechanistic basis of CTIP2-mediated transcriptional repression in transiently transfected HEK293 cells.
CTIP2 Interacts with and Recruits SIRT1 to the Promoter Template in Mammalian Cells
The above results indicate that a member(s) of class III HDACs may be associated with and mediates the transcriptional repression activity of CTIP2 in mammalian cells. To investigate this, co-immunoprecipitation experiments were conducted using extracts prepared from HEK293 cells transiently transfected with an expression vector encoding FLAG epitope-tagged CTIP2 (FLAG-CTIP2). FLAG-CTIP2 was immunoprecipitated with endogenous SIRT1 by anti-SIRT1 antibody (Fig. 2A, lane 6) but not by preimmune serum (lane 5). This finding demonstrates that transfected CTIP2 directly or indirectly interacts with endogenous SIRT1 in HEK293 cells.
Fig. 2. CTIP2 interacts with and recruits SIRT1 to the promoter template in mammalian cells.
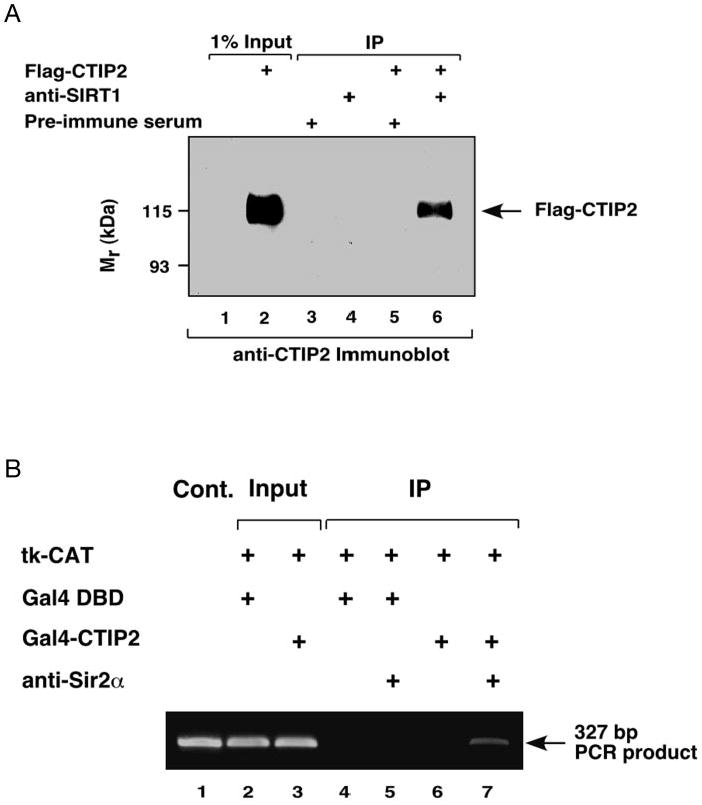
A, FLAG-CTIP2 coimmunoprecipitates (IP) with endogenous SIRT1 from HEK293 cell lysates. Whole cell extracts from HEK293 cells, untransfected and transiently transfected with expression vectors encoding FLAG-CTIP2, were immunoprecipitated with either anti-SIRT1 or preimmune sera, and the immunocomplexes were analyzed by immunoblotting with anti-CTIP2 antibody. The position of FLAG-CTIP2 is indicated. B, endogenous SIRT1 is recruited to promoter template of the reporter gene upon expression of Gal4-CTIP2. HEK293 cells were transfected with 5 μg of (17-mer)4-tk-CAT reporter and 10 μg of expression vectors encoding either Gal4-CTIP2 or Gal4 DBD. Transfection efficiency was normalized as described in the legend of Fig. 1B (input lanes 2 and 3; 2.5% of total). Lanes 4–7 represent template amplification reactions from samples immunoprecipitated with or without antibody directed against SIRT1. Results are representative of three independent experiments.
ChIP experiments were conducted to determine whether endogenous SIRT1 was recruited to the promoter template in cells transfected with a Gal4-CTIP2 expression vector. SIRT1 was not associated with the template in cells transfected with Gal4 DBD (Fig. 2B, lane 5). However, recruitment of endogenous SIRT1 to the template was apparent in cells expressing Gal4-CTIP2 (Fig. 2B, lane 7). These data demonstrate CTIP2-dependent recruitment of endogenous SIRT1 to the promoter template in HEK293 cells, and further suggest a role for this histone deacetylase in the transcriptional repression mechanism of CTIP2.
SIRT1 Enhances CTIP2-mediated Transcriptional Repression
Reporter gene assays were carried out in transiently transfected HEK293 cells to determine whether SIRT1 enhances CTIP2-mediated transcriptional repression. First, the interaction of the co-overexpressed proteins was verified by co-immunoprecipitation experiments using FLAG-CTIP2 and Myc-SIRT1. FLAG-CTIP2 was immunoprecipitated by the anti-Myc antibody but only when Myc-SIRT1 was co-expressed (compare lanes 5 and 6 of Fig. 3A). Similarly, FLAG-CTIP2 was immunoprecipitated with a Myc-tagged, catalytically inactive SIRT1 point mutant, Myc-SIRT1 H363Y (31, 33), to an extent similar to that of the wild-type protein (lane 6 of Fig. 3B). These findings, which are consistent with results presented in Fig. 2, A and B, confirm that transfected CTIP2 interacts with both wild-type SIRT1 and a catalytically inactive form, SIRT1 H363Y, in mammalian cells. The functional consequence of these interactions was investigated in cells transiently cotransfected with expression vectors encoding Gal4-CTIP2, the (17-mer)4-tk-CAT reporter, and either Myc-SIRT1 WT or SIRT1 H363Y. Both wild-type SIRT1 and SIRT1 H363Y repressed reporter gene expression in a concentration-dependent manner in the absence of Gal4-CTIP2 (Fig. 3C, lanes 3–5 and 7-9, respectively). Although the mechanistic basis for recruitment of SIRT1 to the template under these conditions is unknown, these findings, which are consistent with previous reports (33), demonstrate that the catalytic activity of the enzyme is not required for repression of the basal level of reporter gene expression. In contrast, the catalytic activity of SIRT1 was required for enhancement of CTIP2-mediated transcriptional repression. Cotransfection of wild-type SIRT1 stimulated transcriptional repression mediated by Gal4-CTIP2 in a concentration-dependent (Fig. 3C, lanes 10–13) but TSA-insensitive (data not shown) manner, whereas SIRT1 H363Y did not enhance CTIP2-mediated repression (lanes 14–17). These findings demonstrate that the cotransfected SIRT1 stimulates the transcriptional repression activity of CTIP2 and this requires the catalytic activity of the enzyme.
Fig. 3. SIRT1 enhances CTIP2-mediated transcriptional repression.

A, FLAG-CTIP2 coimmunoprecipitates with Myc-SIRT1. Whole cell extracts from HEK293 cells, untransfected and transiently transfected with the indicated expression vectors, were immunoprecipitated with anti-Myc monoclonal antibody. Immunoprecipitates (IP) were resolved by SDS-PAGE and analyzed by Western blotting with anti-FLAG monoclonal antibody that detects FLAG-CTIP2. The position of FLAG-CTIP2 is indicated. B, FLAG-CTIP2 coimmunoprecipitates with the catalytically inactive Myc-SIRT1 H363Y. Transfections, immunoprecipitations, and immunoblots were conducted as described under A. C, wild-type SIRT1, but not SIRT1 H363Y, stimulates CTIP2-mediated transcriptional repression. HEK293 cells were transiently transfected with 5 μg of the (17-mer)4-tk-CAT reporter along with 1.5 μg of expression vectors encoding either Gal4-CTIP2 or Gal4 DBD, and increasing amounts (0.125, 0.25, and 0.5 μg) of expression vectors encoding either SIRT1 WT or SIRT1 H363Y, as indicated. Transfection efficiency was normalized as described in the legend of Fig. 1A. The activity of the CAT reporter in the presence of Gal4 DBD alone (lane 2) was taken to be maximal and that against which all other determined CAT activities were compared. The results presented represent the mean (±S.E.) of three independent experimental determinations. D, SIRT1 stimulates deacetylation of template-associated histones H3 and/or H4 in CTIP2-transfected cells. HEK293 cells were transfected with 5 μg of the (17-mer)4-tk-CAT reporter along with expression vectors encoding Gal4-CTIP2 (1.5 μg) and SIRT1 WT or SIRT1 H363Y (0.5 μg) as indicated. Acetylated histones H3 and H4 were determined by a ChIP assay as described under “Experimental Procedures.” Transfection efficiency was normalized as described in the legend of Fig. 1B. Input lanes (3–6) correspond to amplification reactions conducted using 3.75 (upper panel) and 1.0% (lower panel) of the lysates used for immunoprecipitation reactions. Lanes 7–16 represent template amplification reactions from samples immunoprecipitated with or without anti-acetylated histone H3/H4 antibodies as indicated. Results are representative of three independent experiments.
ChIP assays were conducted to determine whether histone deacetylation may underlie the molecular basis for the observed stimulation of CTIP2-mediated transcriptional repression by SIRT1. The amounts of transfected expression vectors used in these studies were titrated downward to levels at which neither CTIP2 (upper panel of Fig. 3D, lane 12; 1.5 μg transfected) nor SIRT1 (upper panel, lane 14; 0.5 μg transfected) independently affected the acetylation level of promoter-associated histone H3/H4 to an appreciable degree. This was necessary as both Gal4-CTIP2 (Fig. 1B) and SIRT1 (data not shown) individually decreased acetylation of histone H3/H4 when transfected at higher levels. Under the experimental conditions employed, cotransfection of Gal4-CTIP2 and SIRT1 resulted in a large decrease in the level of acetylated histone H3/H4 that was associated with the reporter gene template and this effect was clearly greater than that observed by transfection of either expression vector individually (compare lanes 10, 12, 14, and 16 of the upper panel of Fig. 3D). The catalytically inactive point mutant, SIRT1 H363Y, did not enhance deacetylation of promoter-associated histones H3/H4 in cells transfected with Gal4-CTIP2 (compare lanes 10, 12, 14, and 16 of the bottom panel of Fig. 3D). These findings suggest that SIRT1-catalyzed histone deacetylation may underlie, at least in part, the ability of the enzyme to stimulate CTIP2-mediated transcriptional repression.
CTIP2 Interacts Directly with SIRT1 in Vitro
The co-immunoprecipitation experiments presented above (Figs. 2A, 3A, and 3B) indicated that CTIP2 and SIRT1 associate with similar complexes in transfected cells. However, these studies are incapable of distinguishing between direct physical interaction and indirect association resulting from interaction with a common, intermediary protein(s) within a complex of proteins. Thus, in vitro GST pull-down experiments were conducted to determine whether CTIP2 interacts directly with SIRT1. Full-length CTIP2 (CTIP2-(1–813)) was found to interact with full-length SIRT1 fused to GST (Fig. 4A, lane 3), but not with GST alone (lane 2). These results demonstrate that CTIP2 and SIRT1 participate in a direct, physical interaction. The use of a series of CTIP2 deletion mutants (see Fig. 4A) revealed that the SIRT1 interaction interface is contained within CTIP2 amino acids 171–350 (Fig. 4A, lane 3 of panel E). All CTIP2 deletion mutants containing this region strongly interacted with GST-SIRT1 (Fig. 4A, lane 3 of panels A, B, D, and E) but mutants lacking it interacted weakly or not at all (Fig. 4A, lane 3 of panels C and F–I). Thus, CTIP2-(171–350), a region that is relatively rich in proline, but devoid of C2H2 zinc finger motifs (1), appears to be primarily responsible for interaction with SIRT1 in vitro.
Fig. 4. CTIP2 interacts directly with SIRT1 in vitro.
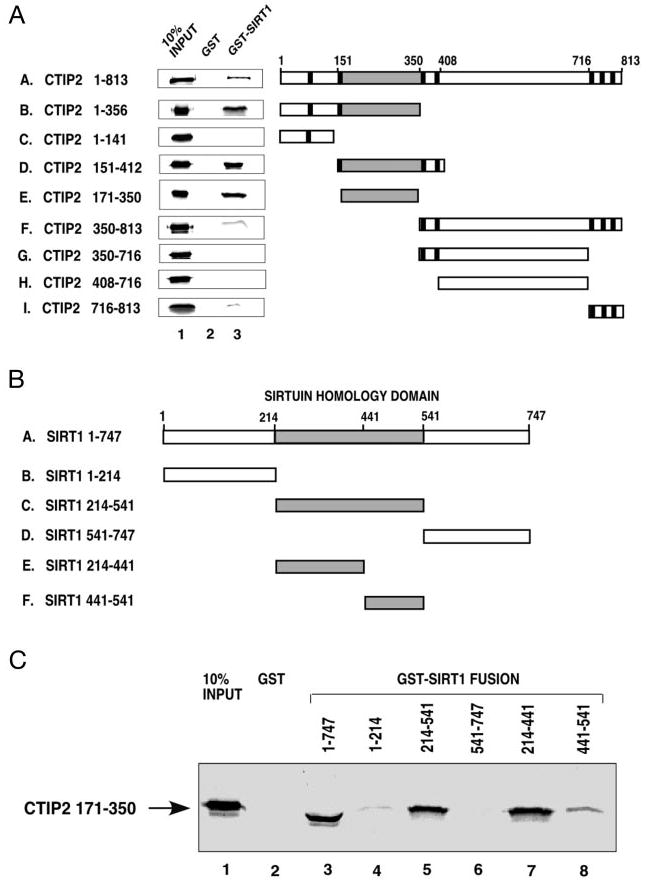
A, in vitro translated and [35S]Met-labeled full-length CTIP2 and truncation CTIP2 mutants were incubated with equivalent amounts of bacterially expressed GST (lane 2) or GST-SIRT1 fusion protein (lane 3). After extensive washing, [35S]Met-labeled CTIP2 associated with the affinity resin was determined by SDS-PAGE and auto-radiography. Input [35S]Met-labeled proteins are shown in lane 1. CTIP2 truncation mutants used in these studies (panels B–I) are schematically represented on the right with zinc finger motifs are denoted by vertical bars. B, a schematic representation of full-length SIRT1 and SIRT1 truncation mutants used to generate GST-SIRT1 fusion proteins for in vitro pull-down experiments (see below). C, [35S]Met-labeled CTIP2-(171– 350) (the minimal SIRT1-interaction domain) was incubated with equivalent amounts of GST (lane 2) or GST-SIRT1 fusion proteins (lanes 3–8). The position of bound [35S]Met-labeled CTIP2-(171– 350) is indicated by an arrow on the left. Lane 1 corresponds to 10% of the [35S]Met-labeled CTIP2-(171–350) that was incubated with GST or GST-SIRT1 fusion proteins. Shown in A and C are representative experiments that were replicated 3–5 times.
The CTIP2 interaction interface of SIRT1 was similarly mapped by deletion mutagenesis (Fig. 4B). Based on the crystal structure of SIRT2 (41), we generated three deletion mutants of SIRT1 (see Fig. 4B): the amino-terminal region, the centrally located sirtuin homology domain, and the carboxyl terminus, all of which were fused to GST for use in in vitro pull-down experiments. CTIP2-(171–350) weakly interacted with the amino-terminal region of SIRT1 (residues 1–214; Fig. 4C, lane 4) but did not interact with the carboxyl terminus (residues 541– 747; lane 6). However, CTIP2-(171–350) interacted strongly with the SIRT1 sirtuin homology domain (residues 214–541; Fig. 4C, lane 5). Two additional deletion mutants within the sirtuin homology domain were constructed to map the CTIP2 interaction interface more precisely, SIRT1-(214–441) and SIRT1-(441–541) (see Fig. 4B). CTIP2-(171–350) was found to interact primarily with SIRT1-(214–441) (Fig. 4C, lane 7), and less so with SIRT1-(441–541) (lane 8). Thus, these results suggest that CTIP2 interacts primarily with a portion of the SIRT1 sirtuin homology domain that overlaps, at least partially, with the catalytic domain of the enzyme. Collectively, these results suggest that CTIP2 and SIRT1 interact directly in vitro and this interaction requires the proline-rich region of CTIP2 (amino acids 171–350) and residues within the amino-terminal part of the sirtuin homology domain of SIRT1.
CTIP2 and SIRT1 Are Components of a Large Complex in Jurkat Cell Nuclear Extracts
The above co-immunoprecipitation studies were performed using transiently transfected cells overexpressing CTIP2. However, it is important to verify that endogenous CTIP2 and SIRT1 interact in the cellular context, i.e. in the absence of overexpression. To test this possibility, nuclear extracts were prepared from untransfected Jurkat cells, which express both CTIP2 (Fig. 5A, lane 1) and SIRT1 (data not shown) endogenously. Endogenous human CTIP2 was co-immunoprecipitated with SIRT1 by both the purified anti-Sir2α antibody (Fig. 5A, lane 3), and anti-SIRT1 antiserum (data not shown), but not by an irrelevant antibody (anti-HA, lane 4) or control IgG (lane 5). Similar results were obtained when the precipitating and detecting antibodies were reversed (data not shown). These findings demonstrate that endogenous CTIP2 and SIRT1 physically associate with similar complexes in the nuclei of mammalian cells when expressed at physiological levels.
Fig. 5. CTIP2 and SIRT1 are components of a large complex in Jurkat cell nuclear extracts.
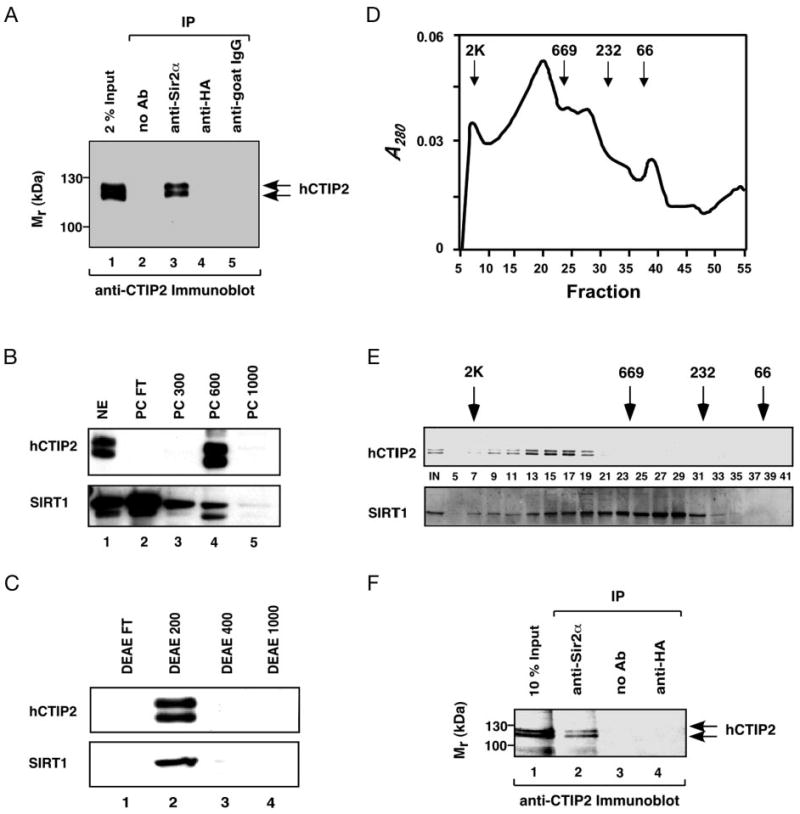
A, endogenous CTIP2 and SIRT1 interact in Jurkat cell nuclear extracts. Nuclear extracts prepared from Jurkat cells were immunoprecipitated (IP) with no antibody, anti-Sir2α, or irrelevant antibodies (anti-HA or goat IgG). The immunocomplexes were then analyzed by Western blotting with anti-CTIP2 antibody. The positions of two forms of endogenous human CTIP2 are indicated, which may correspond to two CTIP2 transcripts present in Jurkat cells as previously reported (5). B, fractionation of CTIP2 and SIRT1 immunoreactivity on a P11 phosphocellulose column. Jurkat cell nuclear extract was applied to a phosphocellulose column, eluted, and eluates from this column were analyzed by immunoblotting as described under “Experimental Procedures.” Each lane of the gel contained 10 μg of protein except lane 5 in which 2.5 μg of protein was loaded. Shown is an autoradiograph from the chemiluminescent detection of CTIP2 (upper panel) and SIRT1 (lower panel). C, fractionation of PC600 on a DEAE column. The 600 mm step off the phosphocellulose column (PC600) was applied to a DEAE Bio-Gel A column and eluted stepwise with increasing salt. Steps (6–8 μg of protein per lane) were analyzed for the presence of CTIP2 and SIRT1 immunoreactivity as described above. D–E, fractionation of DEAE200 on Superose 6 size exclusion column. The 200 mm step off the DEAE column (DEAE200) was applied to a Superose 6 column and the column was eluted isocratically in Buffer 100. The chromatographic elution profile of this column is shown in D, whereas the upper and lower panels of E, respectively, correspond to immunodetection of CTIP2 and SIRT1 in the fractions (fraction numbers are indicated between the CTIP2 and SIRT1 immunoblots; equal volumes from each fraction were analyzed). The positions of elution of calibrating proteins are indicated by downward arrows in both D and E. F, co-immunoprecipitation of SIRT1 and CTIP2 in the Superose 6 purified fractions. Fractions 14–18 from the Superose 6 column shown in D–E were pooled, immunoprecipitated with an antibody that recognizes SIRT1, and the immunoprecipitate was analyzed for the presence of CTIP2 as described under A.
Jurkat cell nuclear extracts were then fractionated to characterize the nature of CTIP2 complexes. First, Jurkat cell nuclear extract was applied to a P11 phosphocellulose column, which was then eluted in a stepwise fashion with increasing salt. CTIP2 immunoreactivity was eluted in the 600 mm step (Fig. 5B, lane 4 of the upper panel), although lesser amounts of CTIP2 could be detected in the 1000 mm step with longer exposures of the blot (data not shown). In contrast, the majority of the SIRT1 immunoreactivity appeared in the flow-through of the phosphocellulose column (Fig. 5B, lane 2 of the lower panel). These findings suggest that most of SIRT1 in Jurkat cell nuclei does not exist within CTIP2 complexes, at least when subjected to the chromatographic conditions utilized in this study. This would be consistent with the notion that SIRT1 plays multiple cellular roles, many of which appear to be independent of CTIP2. Nonetheless, a small amount of SIRT1 did co-chromatograph with CTIP2 within the 600 mm step off the phosphocellulose fraction and this material was then dialyzed and applied to a DEAE column to fractionate the CTIP2 immunoreactivity further. Both CTIP2 (upper panel) and SIRT1 (lower panel) immunoreactivity eluted in the 200 mm step off the DEAE column (Fig. 5C, lane 2). The DEAE-purified material was then applied to a Superose 6 sizing column to determine the relative molecular masses of CTIP2 and SIRT1 complexes. CTIP2 was eluted from the Superose 6 column as a sharp peak with a relative mass between approximately 1 and 2 mDa (Fig. 5D, and upper panel of E, fractions 7–19). In contrast, SIRT1 was eluted as a broad peak beginning at 2 mDa and ending at ∼232 kDa (lower panel of Fig. 5E, fractions 7–31). These size-exclusion chromatography results demonstrate that: 1) native CTIP2 migrated at least 10-fold larger than the predicted size of the monomeric protein (95.5 and 88.5 kDa, respectively, for the CTIP2 splice variants); 2) SIRT1 appeared to be present in heterogenous complex(es), some of which corresponded to CTIP2 immunoreactivity (lower panel of Fig. 5E, fractions 7–19), others that clearly did not (fractions 21–31), and all of which migrated much larger than the predicted size of the monomeric SIRT1 (81.7 kDa). Fractions containing both CTIP2 and SIRT1 immunoreactivity were pooled and immunoprecipitated with the SIRT1 antibody. The immunoprecipitate was analyzed by immunoblotting with the CTIP2 antibody to verify that CTIP2 and SIRT1 co-existed within a similar complex in this partially purified preparation. Indeed, CTIP2 was found to co-immunoprecipitate with the SIRT1 (Fig. 5F, lane 2), indicating the CTIP2-SIRT1 interaction that was observed in Jurkat cell nuclear extracts (Fig. 5A) was maintained through a high-salt extraction and three sequential chromatographic steps.
Discussion
The combinatorial, covalent modification of histones, including acetylation, methylation, phosphorylation, and ubiquitylation has been proposed to underlie the mechanistic basis of dynamic transcriptional regulation from yeast to man. In general, histone acetylation, phosphorylation, and ubiquitylation are believed to promote decondensation of chromatin and have been implicated in the mode of action of transcriptional activators (42). The transcriptional outcome of histone methylation, including mono-, di-, and trimethylation of lysine residues located primarily in the tails of histones H3 and H4, is more complex and can either promote activation (43) or repression (44, 45) in a context-dependent manner. In contrast, many transcriptional repressors, including unliganded nuclear receptors and members of numerous other transcription factor families, have been shown to recruit TSA-sensitive HDACs to the template, resulting in condensation of chromatin and transcriptional silencing (16). In most of these cases, the HDACs recruited to the template of RNA polymerase II-transcribed genes by transcriptional repressors have been shown or suspected to be either class I or II HDACs (46).
We have previously observed that CTIP1 (1), a member of a novel family of C2H2 zinc finger proteins, repressed transcription of a reporter gene in a manner that was only minimally sensitive to reversal by TSA. Similar findings were reported herein for CTIP2 (Fig. 1A). However, CTIP2-mediated transcriptional repression was found to be inhibited, at least partially, by nicotinamide, an inhibitor of the NAD+-dependent, class III HDAC of the SIRT family. Consistent with these findings, expression of CTIP2 in HEK293 cells resulted in recruitment of SIRT1 to the promoter template, and deacetylation of template-associated histones H3/H4 in a manner that was unaffected by TSA but reversed by nicotinamide. Moreover, co-expression of SIRT1, but not a catalytically inactive point mutant, was found to enhance CTIP2-mediated transcriptional repression in transiently transfected cells. The possibility that SIRT1 plays a role in CTIP2-mediated transcriptional repression was given strong support by the finding that the two proteins interacted directly and co-immunoprecipitated from extracts prepared from both co-transfected and singly transfected HEK293 cells, as well as non-transfected Jurkat cells. Finally, SIRT1 partially co-purified with CTIP2 over three chromatographic steps, and the apparent mass of this SIRT1-containing, CTIP2 complex was estimated to be between 1 and 2 mDa, indicating the possible existence of several other component proteins.
To our knowledge, the results of ChIP assays conducted herein provide the first cellular evidence that SIRT1 is capable of deacetylating histones H3 and H4, histones that play a key role in transcriptional regulatory events (17, 47). Although the chromatin organization of transiently transfected DNA templates may differ from that of their chromosomal counterparts (48), the effect of TSA on basal CAT activity (Fig. 1A), and the observed decrease in template-associated, acetylated histone H3/H4 as determined by ChIP analyses (Figs. 1B and 3B) indicated that the transiently transfected reporter template adopted some form of chromatin structure. This finding is also supported by previous reports (36, 49, 50).
The proline-rich region of CTIP2, which harbors an autonomous transcriptional repression activity,5 was found to interact with the centrally located sirtuin homology domain of SIRT1 (see Figs. 4, B and C). Because the sirtuin homology domain is highly conserved among all SIRT proteins, it is conceivable that other members of the sirtuin family may also serve as an alternative CTIP2-interacting partner(s). If this is true, one may envision that other SIRT proteins may play a role(s) in CTIP2-mediated transcriptional repression, perhaps in different cellular and/or promoter contexts. Similarly, the proline-rich region of CTIP2 is also present and highly conserved in CTIP1 (69% identity over 138 amino acids) and may also be expected to interact with SIRT proteins. Indeed, we have also observed that CTIP1 interacts directly with SIRT1 in vitro and in transfected cells.4
Other C2H2 zinc finger proteins, such as the Kruppel-associated box (KRAB) protein Kox1, have also been reported to repress transcription in a TSA-insensitive manner (51, 52). Transcriptional repression by KRAB zinc finger proteins requires interaction with the KRAB domain-binding protein, KAP-1, which in turn may recruit members of heterochromatin protein 1 (HP1) family to the template (53, 54). These findings suggest a role for HP1 proteins and heterochromatin formation in transcriptional repression mediated by KRAB·KAP-1 complexes. Whereas none of these studies addressed the potential role of TSA-insensitive histone deacetylation, possibly catalyzed by a SIRT family member, in transcriptional repression and/or heterochromatinization mediated by KRAB·KAP-1 complexes, deacetylation of H3–K9 is required at the initial step of heterochromatin formation (17). This is believed to be followed by SUV39H-catalyzed methylation of H3–K9, which provides a binding surface for HP1 proteins that bind and self-assemble into a supramolecular, heterochromatinized template (45). Therefore, it is of interest that we have also observed a potential role for HP1 proteins in CTIP-mediated transcriptional repression (55). In this context, CTIPs may serve to recruit SIRT1 (or other SIRT family members) to a particular genomic locus, either by direct DNA binding or via tethering to a COUP-TF family member. Once recruited to the template, SIRT1 may catalyze histone deacetylation, ultimately leading to HP1 binding, heterochromatin formation, and gene silencing. Verification of this model will require identification of all of the component proteins of the CTIP repressor complex(es) bound to the promoter regions of target genes.
In summary, the present results strongly suggest that CTIP2-mediated transcriptional repression involves the recruitment of SIRT1 to the template, at which the TSA-insensitive, NAD+-dependent histone deacetylase catalyzes deacetylation of promoter-associated histones H3 and/or H4. One outcome of this deacetylation may simply be chromatin condensation and short term silencing. Alternatively or additionally, these deacetylation events may ultimately result in formation of heterochromatin, contributing to a persistent, silenced state. In either case, recruitment of SIRT1 to the DNA template by CTIP2 would be expected to result in gene silencing. This feature might be helpful for identification and characterization of the target genes under the control of CTIP proteins in cells of the hematopoietic system and/or the developing central nervous system.
Acknowledgments
We thank Drs. T. Kouzarides and M.-j. Tsai for providing constructs, M. Marusich for raising the anti-CTIP2 monoclonal antibody, and C. Kiossi and L. Tora for advice with the ChIP assay. We are grateful to M. Beilstein for expert technical advice.
Footnotes
This work was supported in part by National Institutes of Health Grant GM60852 (to M. L.) and NIEHS Center National Institutes of Health Grant ES00210 to the Oregon State University Environmental Health Sciences Center.
The abbreviations used are: CTIP 1 and 2, COUP-TF-interacting proteins 1 and 2; COUP-TF, chicken ovalbumin upstream promoter transcription factor; BCL, B cell leukemia; CAT, chloramphenicol acetyltransferase; GST, glutathione S-transferase; HA, hemagglutinin; HDAC, histone deacetylase; HEK293, human embryonic kidney 293 cells; mDa, megadalton; Sir2, silent information regulator 2; SIRT1, Sir2-like protein 1 or sirtuin 1; TSA, trichostatin A; ChIP, chromatin immunoprecipitation; DBD, DNA binding domain; KRAB, Kruppel-associated box; HP1, heterochromatin protein 1.
D. Avram and M. Leid, unpublished results.
D. Shepherd and M. Leid, manuscript in preparation.
M. Beilstein and M. Leid, unpublished results.
D. Avram and M. Leid, unpublished results.
References
- 1.Avram D, Fields A, Pretty On Top K, Nevrivy D, Ishmael JE, Leid M. J Biol Chem. 2000;275:10315–10322. doi: 10.1074/jbc.275.14.10315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Avram D, Fields A, Senawong T, Topark-Ngarm A, Leid M. Biochem J. 2002;368:555–563. doi: 10.1042/BJ20020496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nakamura T, Yamazaki Y, Saiki Y, Moriyama M, Largaespada DA, Jenkins NA, Copeland NG. Mol Cell Biol. 2000;20:3178–3186. doi: 10.1128/mcb.20.9.3178-3186.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Satterwhite E, Sonoki T, Willis TG, Harder L, Nowak R, Arriola EL, Liu H, Price HP, Gesk S, Steinemann D, Schlegelberger B, Oscier DG, Siebert R, Tucker PW, Dyer MJ. Blood. 2001;98:3413–3420. doi: 10.1182/blood.v98.12.3413. [DOI] [PubMed] [Google Scholar]
- 5.Bernard OA, Busson-LeConiat M, Ballerini P, Mauchauffe M, Della Valle V, Monni R, Nguyen Khac F, Mercher T, Penard-Lacronique V, Pasturaud P, Gressin L, Heilig R, Daniel MT, Lessard M, Berger R. Leukemia. 2001;15:1495–1504. doi: 10.1038/sj.leu.2402249. [DOI] [PubMed] [Google Scholar]
- 6.Wakabayashi Y, Inoue J, Takahashi Y, Matsuki A, Kosugi-Okano H, Shinbo T, Mishima Y, Niwa O, Kominami R. Biochem Biophys Res Commun. 2003;301:598–603. doi: 10.1016/s0006-291x(02)03069-3. [DOI] [PubMed] [Google Scholar]
- 7.Liu P, Keller JR, Ortiz M, Tessarollo L, Rachel RA, Nakamura T, Jenkins NA, Copeland NG. Nat Immunol. 2003;4:525–532. doi: 10.1038/ni925. [DOI] [PubMed] [Google Scholar]
- 8.Wakabayashi Y, Watanabe H, Inoue J, Takeda N, Sakata J, Mishima Y, Hitomi J, Yamamoto T, Utsuyama M, Niwa O, Aizawa S, Kominami R. Nat Immunol. 2003;4:533–539. doi: 10.1038/ni927. [DOI] [PubMed] [Google Scholar]
- 9.Saiki Y, Yamazaki Y, Yoshida M, Katoh O, Nakamura T. Genomics. 2000;70:387–391. doi: 10.1006/geno.2000.6385. [DOI] [PubMed] [Google Scholar]
- 10.De Ruijter AJ, Van Gennip AH, Caron HN, Kemp S, Van Kuilenburg AB. Biochem J. 2002;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gray SG, Ekstrom TJ. Exp Cell Res. 2001;262:75–83. doi: 10.1006/excr.2000.5080. [DOI] [PubMed] [Google Scholar]
- 12.Gao L, Cueto MA, Asselbergs F, Atadja P. J Biol Chem. 2002;277:25748–25755. doi: 10.1074/jbc.M111871200. [DOI] [PubMed] [Google Scholar]
- 13.Fischer DD, Cai R, Bhatia U, Asselbergs FA, Song C, Terry R, Trogani N, Widmer R, Atadja P, Cohen D. J Biol Chem. 2002;277:6656–6666. doi: 10.1074/jbc.M108055200. [DOI] [PubMed] [Google Scholar]
- 14.Fischle W, Dequiedt F, Fillion M, Hendzel MJ, Voelter W, Verdin E. J Biol Chem. 2001;276:35826–35835. doi: 10.1074/jbc.M104935200. [DOI] [PubMed] [Google Scholar]
- 15.Zhou X, Marks PA, Rifkind RA, Richon VM. Proc Natl Acad Sci U S A. 2001;98:10572–10577. doi: 10.1073/pnas.191375098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Struhl K. Genes Dev. 1998;12:599–606. doi: 10.1101/gad.12.5.599. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Y, Reinberg D. Genes Dev. 2001;15:2343–2360. doi: 10.1101/gad.927301. [DOI] [PubMed] [Google Scholar]
- 18.Hassig CA, Fleischer TC, Billin AN, Schreiber SL, Ayer DE. Cell. 1997;89:341–347. doi: 10.1016/s0092-8674(00)80214-7. [DOI] [PubMed] [Google Scholar]
- 19.Laherty CD, Yang WM, Sun JM, Davie JR, Seto E, Eisenman RN. Cell. 1997;89:349–356. doi: 10.1016/s0092-8674(00)80215-9. [DOI] [PubMed] [Google Scholar]
- 20.Yang WM, Inouye C, Zeng Y, Bearss D, Seto E. Proc Natl Acad Sci U S A. 1996;93:12845–12850. doi: 10.1073/pnas.93.23.12845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kadosh D, Struhl K. Cell. 1997;89:365–371. doi: 10.1016/s0092-8674(00)80217-2. [DOI] [PubMed] [Google Scholar]
- 22.Roopra A, Sharling L, Wood IC, Briggs T, Bachfischer U, Paquette AJ, Buckley NJ. Mol Cell Biol. 2000;20:2147–2157. doi: 10.1128/mcb.20.6.2147-2157.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaczynski J, Zhang JS, Ellenrieder V, Conley A, Duenes T, Kester H, van Der Burg B, Urrutia R. J Biol Chem. 2001;276:36749–36756. doi: 10.1074/jbc.M105831200. [DOI] [PubMed] [Google Scholar]
- 24.Heinzel T, Lavinsky RM, Mullen TM, Soderstrom M, Laherty CD, Torchia J, Yang WM, Brard G, Ngo SD, Davie JR, Seto E, Eisenman RN, Rose DW, Glass CK, Rosenfeld MG. Nature. 1997;387:43–48. doi: 10.1038/387043a0. [DOI] [PubMed] [Google Scholar]
- 25.Nagy L, Kao HY, Chakravarti D, Lin RJ, Hassig CA, Ayer DE, Schreiber SL, Evans RM. Cell. 1997;89:373–380. doi: 10.1016/s0092-8674(00)80218-4. [DOI] [PubMed] [Google Scholar]
- 26.Alland L, Muhle R, Hou H, Jr, Potes J, Chin L, Schreiber-Agus N, DePinho RA. Nature. 1997;387:49–55. doi: 10.1038/387049a0. [DOI] [PubMed] [Google Scholar]
- 27.Huang EY, Zhang J, Miska EA, Guenther MG, Kouzarides T, Lazar MA. Genes Dev. 2000;14:45–54. [PMC free article] [PubMed] [Google Scholar]
- 28.Imai S, Armstrong CM, Kaeberlein M, Guarente L. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 29.Frye RA. Biochem Biophys Res Commun. 1999;260:273–279. doi: 10.1006/bbrc.1999.0897. [DOI] [PubMed] [Google Scholar]
- 30.Frye RA. Biochem Biophys Res Commun. 2000;273:793–798. doi: 10.1006/bbrc.2000.3000. [DOI] [PubMed] [Google Scholar]
- 31.Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- 32.Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 33.Langley E, Pearson M, Faretta M, Bauer UM, Frye RA, Minucci S, Pelicci PG, Kouzarides T. EMBO J. 2002;21:2383–2396. doi: 10.1093/emboj/21.10.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grozinger CM, Chao ED, Blackwell HE, Moazed D, Schreiber SL. J Biol Chem. 2001;276:38837–38843. doi: 10.1074/jbc.M106779200. [DOI] [PubMed] [Google Scholar]
- 35.Bitterman KJ, Anderson RM, Cohen HY, Latorre-Esteves M, Sinclair DA. J Biol Chem. 2002;277:45099–45107. doi: 10.1074/jbc.M205670200. [DOI] [PubMed] [Google Scholar]
- 36.Luo RX, Postigo AA, Dean DC. Cell. 1998;92:463–473. doi: 10.1016/s0092-8674(00)80940-x. [DOI] [PubMed] [Google Scholar]
- 37.Dowell P, Ishmael JE, Avram D, Peterson VJ, Nevrivy DJ, Leid M. J Biol Chem. 1997;272:33435–33443. doi: 10.1074/jbc.272.52.33435. [DOI] [PubMed] [Google Scholar]
- 38.Nevrivy DJ, Peterson VJ, Avram D, Ishmael JE, Hansen SG, Dowell P, Hruby DE, Dawson MI, Leid M. J Biol Chem. 2000;275:16827–16836. doi: 10.1074/jbc.275.22.16827. [DOI] [PubMed] [Google Scholar]
- 39.Dignam JD, Lebovitz RM, Roeder RG. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dowell P, Peterson VJ, Zabriskie TM, Leid M. J Biol Chem. 1997;272:2013–2020. doi: 10.1074/jbc.272.3.2013. [DOI] [PubMed] [Google Scholar]
- 41.Finnin MS, Donigian JR, Pavletich NP. Nat Struct Biol. 2001;8:621–625. doi: 10.1038/89668. [DOI] [PubMed] [Google Scholar]
- 42.Jenuwein T, Allis CD. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 43.Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NC, Schreiber SL, Mellor J, Kouzarides T. Nature. 2002;419:407–411. doi: 10.1038/nature01080. [DOI] [PubMed] [Google Scholar]
- 44.Lachner M, Jenuwein T. Curr Opin Cell Biol. 2002;14:286–298. doi: 10.1016/s0955-0674(02)00335-6. [DOI] [PubMed] [Google Scholar]
- 45.Lachner M, O'Carroll D, Rea S, Mechtler K, Jenuwein T. Nature. 2001;410:116–120. doi: 10.1038/35065132. [DOI] [PubMed] [Google Scholar]
- 46.Hermanson O, Glass CK, Rosenfeld MG. Trends Endocrinol Metab. 2002;13:55–60. doi: 10.1016/s1043-2760(01)00527-6. [DOI] [PubMed] [Google Scholar]
- 47.Guarente L. Genes Dev. 2000;14:1021–1026. [PubMed] [Google Scholar]
- 48.Smith CL, Hager GL. J Biol Chem. 1997;272:27493–27496. doi: 10.1074/jbc.272.44.27493. [DOI] [PubMed] [Google Scholar]
- 49.Cereghini S, Yaniv M. EMBO J. 1984;3:1243–1253. doi: 10.1002/j.1460-2075.1984.tb01959.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reeves R, Gorman CM, Howard B. Nucleic Acids Res. 1985;13:3599–3615. doi: 10.1093/nar/13.10.3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lorenz P, Koczan D, Thiesen HJ. Biol Chem. 2001;382:637–644. doi: 10.1515/BC.2001.075. [DOI] [PubMed] [Google Scholar]
- 52.de Haan G, Chusacultanachai S, Mao C, Katzenellenbogen BS, Shapiro DJ. J Biol Chem. 2000;275:13493–13501. doi: 10.1074/jbc.275.18.13493. [DOI] [PubMed] [Google Scholar]
- 53.Lechner MS, Begg GE, Speicher DW, Rauscher FJ., 3rd Mol Cell Biol. 2000;20:6449–6465. doi: 10.1128/mcb.20.17.6449-6465.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ryan RF, Schultz DC, Ayyanathan K, Singh PB, Friedman JR, Fredericks WJ, Rauscher FJ., 3rd Mol Cell Biol. 1999;19:4366–4378. doi: 10.1128/mcb.19.6.4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rohr O, Lecestre D, Chasserot-Golaz S, Marban C, Avram D, Aunis D, Leid M, Schaeffer E. J Virol. 2003;77:5415–5427. doi: 10.1128/JVI.77.9.5415-5427.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]