

Abstract
Two novel and related C2H2 zinc finger proteins that are highly expressed in the brain, CTIP1 and CTIP2 (COUP TF-interacting proteins 1 and 2, respectively), were isolated and shown to interact with all members of the chicken ovalbumin upstream promoter transcription factor (COUP-TF) subfamily of orphan nuclear receptors. The interaction of CTIP1 with ARP1 was studied in detail, and CTIP1 was found to harbor two independent ARP1 interaction domains, ID1 and ID2, whereas the putative AF-2 of ARP1 was required for interaction with CTIP1. CTIP1, which exhibited a punctate staining pattern within the nucleus of transfected cells, recruited cotransfected ARP1 to these foci and potentiated ARP1-mediated transcriptional repression of a reporter construct. However, transcriptional repression mediated by ARP1 acting through CTIP1 did not appear to involve recruitment of a trichostatin A-sensitive histone deacetylase(s) to the template, suggesting that this repression pathway may be distinct from that utilized by several other nuclear receptors.
COUP-TFI,1 ARP1/COUP-TFII, and Ear2/COUP-TFIII have been grouped in the same subfamily of orphan nuclear receptors based on sequence similarity (1-3), evolutionary analysis (4), and a common capacity to repress ligand-dependent transcriptional activation of target genes mediated by other nuclear receptors, such as retinoic acid (5-10), thyroid hormone (8), estrogen (11-14), and vitamin D3 (9) receptors as well as peroxisome proliferator-activated receptor α (PPARα; Ref. 15).
COUP-TFs play important roles in pattern formation in the developing nervous systems of Xenopus (16) and Drosophila (17). Deletion of the COUP-TFI gene in the mouse results in defects in axonal guidance and aberrant neuronal arborization (18). Such defects involving the glossopharyngeal ganglion and IXth cranial nerve appear to be causally associated with perinatal lethality in COUP-TFI null animals (18).
ARP1 is highly expressed in mesenchymal cells during organogenesis (19), and it is believed to play an important role in mesenchymal-endothelial signaling (20). Deletion of the ARP1 gene results in embryonic lethality at day 10 of mouse embryogenesis, possibly due to defects in angiogenesis and embryonic heart development (20).
The function of the ubiquitously expressed Ear2 (3) during embryonic development or in the adult organism is unknown. However, Ear2 heterodimerizes with both COUP-TFI and ARP1 in solution and on various, directly repeated DNA response elements (21), suggesting that Ear2 may be implicated in both COUP-TFI and ARP1 signaling pathways.
COUP-TF family members are generally considered to be repressors of transcription, and several mechanisms have been proposed to underlie this activity (22). Among these, active transcriptional repression mediated by COUP-TFs may involve recruitment of nuclear receptor co-repressor (NCoR) and/or silencing mediator for retinoid and thyroid hormone receptor (SMRT) to the template (23, 24) in a manner similar to that of other unliganded nuclear receptors (25-28). NCoR and SMRT are components of a larger repressor complex that minimally includes mSin3A/B and a trichostatin-sensitive histone deacetylase (29, 30). Histone deacetylation has been proposed to account for transcriptional repression mediated by nuclear receptors and several other classes of transcriptional repressors (reviewed in Ref. 31).
Toward the goal of elucidating potential mechanisms of COUP-TF signaling, we employed a yeast two-hybrid screen to identify proteins expressed in brain that interact with and may be implicated in transcriptional repression mediated by ARP1. Here we show that CTIP1, a member of a novel family of C2H2 zinc finger proteins that was isolated as an ARP1 interaction partner, harbors autonomous transcriptional repression domains and potentiates ARP1-mediated transcriptional repression independently of trichostatin A-sensitive histone deacetylation. Both CTIP1 and the related CTIP2 are highly expressed in the brain, a tissue also known to express COUP-TF family members abundantly (3, 21, 32), suggesting that this novel family of C2H2 zinc finger proteins may play a role in COUP-TF signaling.
MATERIALS AND METHODS
Yeast Two-hybrid Screening and cDNA Cloning
Yeast two-hybrid screening was conducted as described previously (21) using the hinge region and putative ligand binding domain of ARP1 (amino acids 144–414) as a bait. Fragments corresponding to CTIP1 and CTIP2 (see Fig. 1A) were used to screen mouse cDNA libraries obtained from CLON-TECH and from Dr. René Hen (Columbia University), yielding several overlapping clones. These overlapping clones were then used to prepare a full-length CTIP1 construct that was inserted into the eukaryotic expression vector, pCDNA3+ (Invitrogen).
Fig. 1. Interaction of COUP-TFs with CTIP proteins in yeast and in vitro.
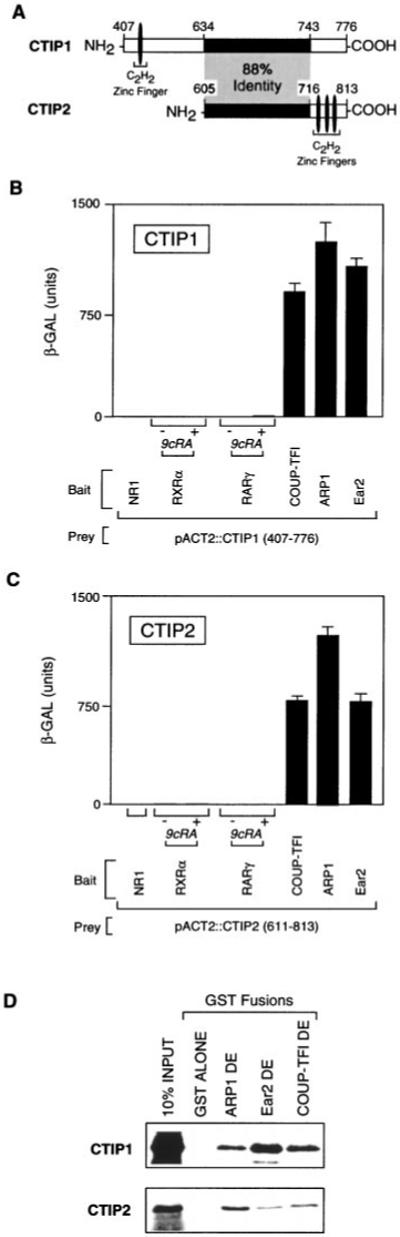
A, diagram of CTIP1 and CTIP2 clones isolated in the yeast two-hybrid screen. The shaded region represents a domain exhibiting 88% identity between the two proteins. B and C, interaction of CTIP1 and CTIP2, respectively, with COUP-TFs, RXRα, RARγ, and the NR1 subunit of the N-methyl-d-aspartate receptor (36). All baits were cloned in pBTM116 as described previously (21) and expressed in S. cerevisiae L40 together with GAL4 activation domain fusions of CTIP1-(407–776) (B) or CTIP2-(611–813) (C). Ligand-dependent interactions were examined in the presence of 1 μm 9-cis-retinoic acid (9cRA). The results shown represent the means ± S.D. of three independent experiments. D, in vitro interaction between COUP-TFs and CTIP1-(602–776) and CTIP2-(611–813). GST/COUP-TFs were bound to glutathione-Sepharose and used as affinity matrices to examine the interaction with the in vitro translated [35S]methionine-labeled CTIPs as described previously (21).
Yeast β-Galactosidase Assays and GST Pull-down Experiments
Protein-protein interaction studies in yeast and in vitro have been described previously (21).
Cell Culture
HEK293 cells (ATCC CRL 1573) were cultured and transfected as described previously (21). Culture of CATH.a cells (33) and preparation of mRNA from these cells have been described previously (34).
Reverse Transcription-Polymerase Chain Reaction
First strand cDNA was synthesized using 1 μg of RNA (Ambion, Austin, TX) and 100 ng of random hexamer primers (Promega) as described previously (21). One μl of each reverse transcription reaction was subjected to PCR amplification using primers corresponding to carboxyl termini of CTIP1 and CTIP2. Amplification of 36B4 (35) was used as a control for the quantity of cDNA present in each sample as well as in normalization of gel loading and Southern blotting (21). The following primers were used: CTIP1 forward, 5′-GGAGCTGACGGAGAGCGAGA-3′; CTIP1 reverse, 5′-TCAGCGAGCTGGGGCTACCCA-3′; CTIP1 internal, 5′-GGCTTCGGGCTGAGCCTGGAGGCTGC-3′; CTIP2 forward, 5′-GGTCTTCAAGAACTGTAGCAA-3′; CTIP2 reverse, 5′-CCGTGCCACTTTTTCATGTGT-3′; CTIP2 internal, 5′-CTGACGGTGCACCGGAAGAACAACCACAC-3′; 36B4 forward, 5′-GAGGTCACTGTGCCAGCTCA-3′; 36B4 reverse, 5′-TGATGATGGAGTGAGGCACC-3′; 36B4 internal, 5′-CTGGAGACAAGGTGGGAGCCAGCGAGG-3′.
Northern Blot Analysis
CATH.a cell RNA was isolated using TRI-REAGENT (Molecular Research Center, Inc., Cincinnati, OH) and poly(A)+ RNA was purified on oligo(dT)-cellulose (Amersham Pharmacia Biotech). Brain poly(A)+ RNA was obtained from Ambion. Northern blot analyses were performed following standard procedures, using probes corresponding to the yeast two-hybrid clones of CTIP1 and CTIP2.
Indirect Immunofluorescence and Confocal Microscopy
Forty-eight hours following transfection, HEK293 cells growing on coverslips were fixed and permeabilized in 4% paraformaldehyde and 0.1% Tween. Antibody incubations were performed using standard techniques with the anti-Myc monoclonal antibody (Invitrogen) or anti-HA rabbit polyclonal antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). Myc-ARP1 immune complexes were detected using tetramethylrhodamine isothiocyanate-conjugated goat anti-mouse antibodies (Southern Biotechnology Associates Inc.). HA-CTIP1 immune complexes were detected using fluorescein isothiocyanate-conjugated goat anti-rabbit antibodies (Southern Biotechnology Associates Inc.). Samples were washed in phosphate-buffered saline and counterstained with Hoechst 33258 (1 μg/ml) for 10 min. Images were captured using a Leica inverted confocal microscope model TCS4D and processed using Photoshop 5.0 (Adobe Systems, Inc.).
RESULTS
CTIP1 and CTIP2 Interact Specifically with COUP-TF Family Members
A bait corresponding to the hinge region and putative ligand binding domain of ARP1 (22) was used in a yeast two-hybrid screen (21) to isolate overlapping fragments of CTIP1 and CTIP2 (Fig. 1A). The protein fragments encoded by these novel clones share 88% identity over a 110-amino acid region located at the carboxyl terminus of both proteins (Fig. 1A). CTIP1 (Fig. 1B) and CTIP2 (Fig. 1C) interacted strongly in yeast with all members of the COUP-TF family, COUP-TF1, ARP1, and Ear2 (4). However, neither CTIP1 nor CTIP2 interacted with other nuclear receptors examined, including retinoic acid receptor γ (RARγ), retinoid X receptor α (RXRα), and PPARα, all of which were tested in the presence and absence of activating ligands (Fig. 1, B and C; data not shown). In addition, CTIP1 and CTIP2 did not interact with an unrelated bait corresponding to the carboxyl tail (amino acids 835–938) of the NR1 subunit of the N-methyl-d-aspartate receptor (36). In vitro protein-protein interaction studies confirmed the results of experiments carried out in yeast in that both CTIPs were observed to interact directly and specifically with all COUP-TF family members (Fig. 1D).
CTIP1 and CTIP2 Are Novel C2H2 Zinc Finger Proteins That Are Highly Expressed in Brain
The CTIP1 and CTIP2 fragments isolated from the yeast two-hybrid library were used as probes to obtain full-length cDNAs from mouse brain cDNA libraries. A 2.7-kb CTIP1 fragment (GenBank™ accession number AF186018), which was constructed from several overlapping clones (data not shown), contains an open reading frame encoding a protein comprised of 776 amino acids with a predicted mass of 84,116 Da (Fig. 2A). Several overlapping fragments totaling nearly 3.0 kb of CTIP2 coding sequence (GenBank™ accession number AF186019) have been isolated (Fig. 2B); however, the reading frame of this clone remains open at the 5′-end (data not shown). CTIP1 and CTIP2 are highly related over much of the amino acid sequence of each (Fig. 2C). The predicted amino acid sequences of both proteins are characterized by two centrally located C2H2 zinc fingers that share extensive homology (95% identity; see Fig. 2C). CTIP2 harbors three additional C2H2 zinc fingers at the carboxyl terminus of the protein, two of which are related to the central zinc fingers of both CTIP1 and CTIP2 (71% identity). The amino termini of CTIP1 and CTIP2 are also related over large blocks of amino acid residues, including a conserved C2HC putative zinc finger motif (Fig. 2, A-C), suggesting a commonality of function.
Fig. 2. Amino acid sequence, diagram of amino acid sequence alignment, and expression of CTIP1 and CTIP2 in several mouse tissues.

A and B, amino acid sequence of CTIP1 and CTIP2, respectively. The conserved cysteine and histidine amino acids contributing the C2H2 and C2HC zinc finger motifs of both proteins are underlined. C, schematic diagram of CTIP1 and CTIP2 amino acid alignment. The homologous regions are represented by black boxes and the percentage of identity between each region of CTIP1 and CTIP2 is indicated. The alignment was performed using Clustal X (version 1.63b). The overall characteristics of each domain are indicated. D, Northern blot analysis of CTIP1 and CTIP2 expression in brain and CATH.a cells. Approximately 5 μg of brain and 10 μg of CATH.a poly(A)+ RNA were loaded per lane, and the blots were hybridized with probes derived from the 3′-end of both CTIP1 and CTIP2. E, Southern blot of reverse transcription-polymerase chain reaction analysis of RNA from various tissues of adult mouse and embryo (10–12.5 days postcoitum. Note that partial EST clones of both CTIP1 and CTIP2 have been reported (mi17e04.r1 and mv64 h01.r1, respectively, from the Washington University-HHMI Mouse EST Project).
A CTIP1 probe corresponding to the yeast two-hybrid clone hybridized to transcripts of approximately 1.2 and 2.7 kb on Northern blots of mRNA isolated from mouse brain and from CATH.a cells (33), a catecholaminergic cell line of neuronal origin (Fig. 2D). The ~1.2-kb CTIP1 transcript corresponds to a splice variant that encodes a form of CTIP1 lacking the central core of the protein.2 The function of this short form of CTIP1 is presently unknown. In contrast, a CTIP2 probe corresponding to the yeast two-hybrid clone hybridized to a single transcript of 5.7 kb (Fig. 2D).
CTIP1 expression was detected at high levels in brain and at lower levels in embryo (10–12.5 days postcoitum), heart, and liver (Fig. 2E). Note, however, that the CTIP1 PCR primers and Southern probe used in these analyses only detect the long form of CTIP1. In addition to embryo and brain, CTIP2 transcripts were detected in lung but not in liver or heart (Fig. 2E), indicating that the tissue expression patterns of the two CTIP genes are only partially overlapping.
The Putative ARP1 AF-2 Core Is Required for in Vitro Interaction with CTIP1
GST pull-down experiments, employing full-length proteins and truncation mutants thereof, were conducted to verify observations made in yeast and to map the respective interaction domains of ARP1 and CTIP1. Full-length ARP1 (residues 1–414) interacted with full-length CTIP1 (residues 1–776) in vitro (Fig. 3A), and this interaction did not require the ARP1 DNA binding domain or the amino-terminal (A/B) or hinge (D) regions (Fig. 3A). However, the putative ARP1 AF-2 core, amino acids 395–399 (37), was required for this interaction inasmuch as deletion of this region completely abolished in vitro interaction with CTIP1 (ARP1-(1–394); Fig. 3A).
Fig. 3. Definition of the in vitro interaction domains of both ARP1 (A) and CTIP1 (B) by deletion mutagenesis.
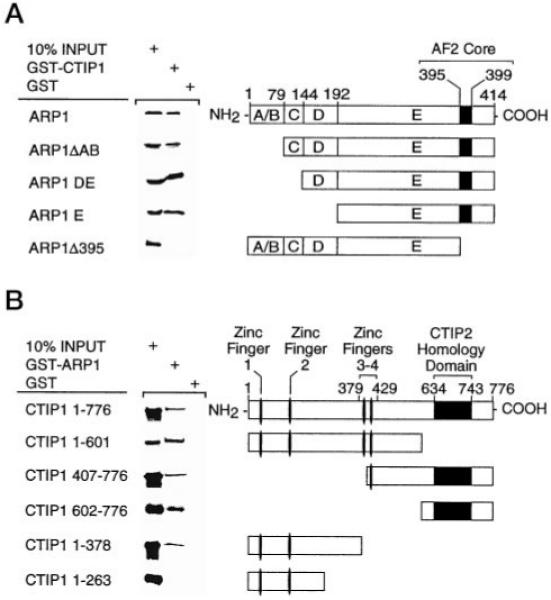
A, CTIP1 interaction domains of ARP1. The ARP1 deletion mutants indicated in the schematic diagram were cloned in pCDNA3 and translated in vitro using the TNT Coupled Reticulocyte Lysate System (Promega). CTIP1-(1–176) was cloned in pGEX-2T. GST/CTIP1 was bound to glutathione-Sepharose and used as affinity matrix to examine the interaction with the in vitro translated [35S]methionine-labeled ARP1 mutants as described previously (21). B, ARP1 interaction interfaces of CTIP1. ARP1-(1–414) was cloned in pGEX-2T, and GST/ARP1 was bound to glutathione-Sepharose and used as affinity matrix to examine the interaction with the in vitro translated [35S]methionine-labeled CTIP1 mutants. CTIP1 deletion mutants indicated in the schematic diagram were cloned in pCDNA3 and in vitro translated using the TNT Coupled Reticulocyte Lysate System (Promega).
CTIP1 Harbors Two ARP1 Interaction Interfaces
Full-length CTIP1 interacted with full-length ARP1 in vitro (Fig. 3B; see also Fig. 3A). Surprisingly, in the context of the full-length protein, deletion of the carboxyl terminus of CTIP1, which is sufficient to mediate interaction with ARP1 in vitro (Fig. 1D) and harbors the CTIP2 homology domain (see Fig. 2C), did not appear to disrupt interaction with full-length ARP1 (Fig. 3B). This unexpected finding suggests that CTIP1 may contain an additional ARP1 interaction domain(s), and, to address this possibility, additional CTIP1 truncation mutants were prepared and tested for interaction with full-length ARP1. Deletion of the CTIP1 carboxyl terminus up to but not including zinc finger 4 did not alter interaction with ARP1 (data not shown), and further deletion of zinc fingers 3 and 4 similarly did not affect the ability of the protein to interact with full-length ARP1 in vitro (CTIP1-(1–378); Fig. 3B). However, deletion of an additional 115 amino acids abolished interaction of CTIP1 with ARP1 (CTIP1-(1–263); Fig. 3B). These findings suggest that a second ARP1 interaction interface, hereafter referred to as ID2, is localized, at least in part, between CTIP1 amino acids 264 and 378. Therefore, the carboxyl-terminal ARP1 interaction domain, which is localized between CTIP1 amino acids 602 and 776 (Fig. 3B) and contains the CTIP2 homology region, is hereafter referred to as ID1. Deletion of the amino-terminal region of CTIP1, which includes ID2, did not disrupt interaction with full-length ARP1 (CTIP1-(407–776); Fig. 3B), presumably because this mutant utilizes CTIP1 ID1 in a manner similar to that of CTIP1-(602–776). Thus, the mechanistic basis of the interaction between ARP1 and CTIP1 is reasonably complex in that at least two CTIP1 interaction interfaces are implicated, ID2 (amino acids 264–378) and ID1 (amino acids 602–776). The bipartite ARP1 interaction domain of CTIP1 is reminiscent of NCoR, which also harbors two nuclear receptor interaction domains (38).
Taken together, these results clearly demonstrate that full-length ARP1 and CTIP1 interact in vitro. This interaction requires the putative AF-2 core of ARP1 and one of two regions of CTIP1, ID1 or ID2.
CTIP1 Potentiates ARP1-mediated Transcriptional Repression in a Trichostatin-insensitive Manner
Cotransfection experiments were conducted in human embryonic kidney 293 (HEK293) cells to determine the functional significance of the interaction observed between CTIP1 and ARP1 in yeast and in vitro. ARP1 is known to bind strongly as a homodimeric complex to a direct repeat of the hexanucleotide, AGGTCA, spaced by 1 base pair (DR1; Refs. 8 and 21). However, cotransfection of ARP1 with a reporter gene driven by a DR1-containing promoter had only a negligible effect on expression of the reporter gene in HEK293 cells (Fig. 4A, lanes 1 and 2). Cotransfection with increasing amounts of full-length CTIP1 resulted in a strong repression of this reporter in the presence (Fig. 4A, lanes 3–5) but not in the absence (Fig. 4B, lanes 3–5) of ARP1, indicating that recruitment of CTIP1 to the DR1-bound ARP1 complex resulted in transcriptional repression. CTIP1-(1–601), which lacks the CTIP1 ID1, only weakly potentiated ARP1-mediated repression of the reporter (Fig. 4A, compare lanes 3–5 and 6–8). This finding suggested that ID1 and/or additional residues localized within or around the CTIP1 carboxyl terminus may play an important role(s) in transcriptional repression mediated by ARP1·CTIP1 complexes in cells. CTIP1-(407–776), which contains ID1 but not ID2 and interacted with ARP1 in vitro (Fig. 2B), did not potentiate ARP1-mediated transcriptional repression (Fig. 4A, lanes 9–11). However, this mutant also lacks the major CTIP1 repression domain contained within the amino terminus of the protein (see below). CTIP1-(1–378), which contains ID2, but not ID1, did not potentiate the transcriptional repression activity of ARP1 (Fig. 4A, lanes 12–14), nor did the amino-terminal mutant, CTIP1-(1–263), which lacks both ARP1 interaction domains (Fig. 4A, lanes 15–17).
Fig. 4. CTIP1 potentiates the ARP1-mediated repression in a TSA-insensitive manner.
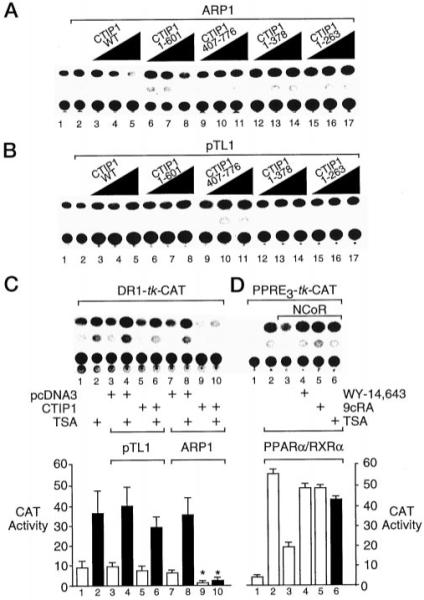
A and B, HEK 293 cells were cotransfected with 5 μg of the DR1-TK-CAT reporter, 5 μg of Myc-ARP1 expression vector (A) or empty vector (pTL1; B) and increasing amounts (0.2, 0.66, and 2.22 μg) of expression vectors encoding full-length HA-CTIP1 or HA-CTIP1 deletion mutants as indicated. CAT reporter activity in cell extracts was determined as described previously (21). C, HEK 293 cells were cotransfected with 5 μg of DR1-TK-CAT reporter, 5 μg of Myc-ARP1 expression vector or empty vector (pTL1), and 2.2 μg of HA-CTIP1 or empty vector (pcDNA3). Cells were treated with TSA (100 ng/ml; solid bars) as indicated for 24 h prior to harvesting and CAT assays. D, cotransfection of HEK 293 cells with 2 μg of PPRE-TK-CAT reporter and expression vectors for PPARα/RXRα (0.5 μg each) and NCoR (2 μg) as indicated. Cells were treated with either vehicle (0.1% Me2SO; lane 3); the RXR agonist, 9-cis-retinoic acid (9cRA; 1 μm); or the PPARα agonist, WY-14,643 (10 μm), or TSA as noted for 24 h prior to harvesting and determination of CAT activity. The quantifications shown below B and C represent mean CAT activities ± S.D. derived from three independent experiments. The CAT activity values in lanes 9 and 10 are statistically different from those shown in lanes 7 and 8, respectively, as determined by Student’s t test (p < 0.05, indicated by asterisks).
Transcriptional repression mediated by ARP1·CTIP1 complexes was only minimally sensitive to reversal by the histone deacetylase inhibitor, trichostatin A (TSA, Ref. 39; Fig. 4B, compare lanes 9 and 10), whereas this compound nearly completely reversed NCoR-mediated repression of PPARα·RXRα complexes (Ref. 28; Fig. 4C, lanes 3–6). These findings suggest that CTIP1-mediated transcriptional repression probably does not involve recruitment of a TSA-sensitive histone deacetylase(s) to the template and, therefore, appears to be mechanistically distinct from that mediated by NCoR (or SMRT) acting through the histone deacetylase complex (29, 30).
CTIP1 Exhibits a Punctate Distribution in the Nucleus and Recruits Cotransfected ARP1 to These Foci
To confirm the association of CTIP1 and ARP1 within the nucleus, cotransfected cells were examined by indirect immunofluorescence confocal microscopy. HA-CTIP1 presented a distinct punctate distribution in interphase nuclei in approximately 80% of transfected cells examined, with the remaining 20% being composed of a combination of focal and diffuse staining (Fig. 5A, see also Fig. 6A and Table I). In contrast, Myc-ARP1 exhibited a diffuse nuclear staining pattern in 100% of 120 transfected cells examined when transfected independently (Fig. 5C). However, when cotransfected with HA-CTIP1, Myc-ARP1 was recruited to the punctate structures defined by HA-CTIP1 staining (Fig. 6B). Overlaying the corresponding images revealed that the nuclear localization of the two proteins was entirely coincident in all transfected cells examined (Fig. 6C). This finding strongly suggests that HA-CTIP1 and Myc-ARP1 interact directly within the nuclei of cotransfected HEK293 cells as evidenced by the ability of the former protein to disrupt the nuclear localization of the latter protein. HA-CTIP1-(1–601), a mutant that interacts in vitro with (Fig. 3B) and weakly potentiates the transcriptional activity of ARP1 in cotransfected cells (Fig. 4A), formed less well defined punctate structures and appeared to be localized in larger blocks within interphase nuclei (Fig. 6E). These blocks appeared to be a mixture of foci superimposed upon a partially diffuse staining pattern that was clearly distinct from the highly punctate bodies in which HA-CTIP1 was found (Fig. 6A; see also Table I). However, HA-CTIP1 1–601, which contains ID2 but not ID1, interacted with and recruited Myc-ARP1 to these loci (Fig. 6, F and G), albeit less efficiently than full-length CTIP1 (see Fig. 6, B and C). The carboxyl-terminal mutant, HA-CTIP1-(407–776), which interacted in vitro with ARP1 (Fig. 3B) but did not potentiate ARP1-mediated repression in cells (Fig. 4A), was found to exhibit a completely diffuse nuclear staining in all cells examined (Fig. 6I; see also Table I). Although the staining pattern observed for HA-CTIP1-(407–776), which contains ID1 but not ID2, and Myc-ARP1 appeared coincident within the nucleus, this amino-terminal truncation mutant was clearly incapable of localizing in or redistributing Myc-ARP1 to punctate bodies (Fig. 6, J and K). Two amino-terminal CTIP1 fragments (HA-CTIP1-(1–378) and HA-CTIP1-(1–263)) also did not localize in (Fig. 6, M and Q, respectively) or recruit Myc-ARP1 to (Fig. 6, panels N and O and panels R and S, respectively) punctate bodies. These mutants also exhibited extensive cytoplasmic staining (Fig. 6, M and Q, respectively), suggesting that the putative CTIP1 NLS located at codons 569–572, and not those present at codons 186–189 and 631–637 (data not shown), is critical for proper nuclear localization of CTIP1.
Fig. 5. Localization of HA-CTIP1 (A) and Myc-ARP1 (C) in HEK293 cell nuclei.
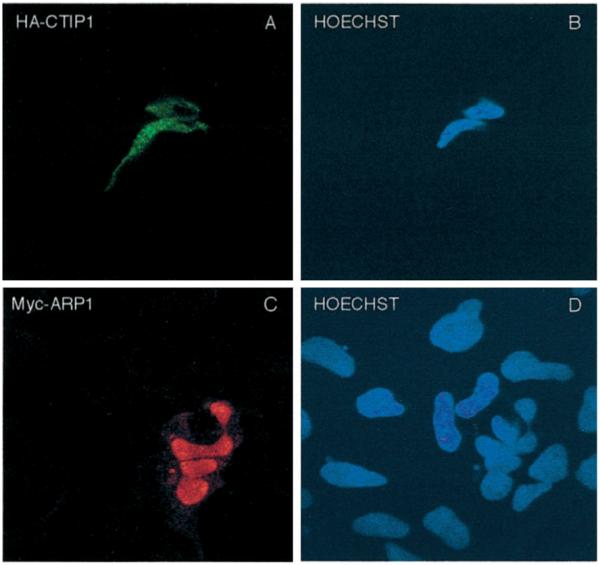
HEK293 cells growing on coverslips were transiently transfected with expression vectors encoding HA-CTIP1 (A) and Myc-ARP1 (C). Forty-eight hours after transfection, the cells were fixed and incubated with anti-HA (A) or anti-Myc (C) antibodies and stained with appropriate fluorescein isothiocyanate- or tetramethylrhodamine isothiocyanate-conjugated secondary antibodies detecting HA-CTIP1 and Myc-ARP1 immune complexes, respectively. B and D, Hoechst-counterstained cells shown in A and C, respectively. HA-CTIP1 exhibited the punctate staining pattern depicted in A in 80% of transfected cells examined, with the balance displaying a combination of focal and diffuse staining (see Table I). In contrast, Myc-ARP1 exhibited the diffuse staining pattern in 100% of 120 transfected cells examined by a naive observer. Images were obtained on a Leica inverted confocal microscope model TCS4D using a × 100 objective. The images shown are derived from a representative experiment that was replicated several times.
Fig. 6. ARP1 is redistributed in the nucleus when cotransfected with CTIP1.
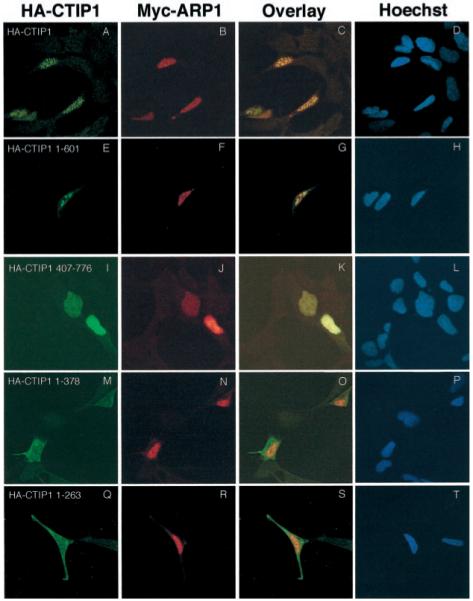
HEK293 cells were transiently cotransfected with expression vectors encoding Myc-ARP1 and either HA-CTIP1 or HA-CTIP1 mutants as indicated. The corresponding proteins were localized 48 h after transfection by indirect immunofluorescence confocal microscopy. Cells were stained for HA-CTIP1 (first column) and Myc-ARP1 (second column) as described in the legend of Fig. 5. An overlay of the images presented in the first two columns is shown in the third column, and the fourth column represents counterstaining of the cells with Hoechst as indicated. Shown are representative experiments that were replicated 3–7 times. Each micrograph was prepared using a × 100 objective on a Leica inverted confocal microscope model TCS4D, and overlays were prepared using Photoshop 5.0.
Table I. Summary of the activities and subcellular distribution of CTIP1 mutants.
Refer to Fig. 3, A and B, for a schematic representation of each mutant. Note that CTIP1-(1–378) and -(1–263) both exhibited disrupted nuclear localization (see Fig. 6, M–P and Q–T, respectively) relative to wild-type CTIP1-(1–776) and the two other truncation mutants (1–601) and 407–776). HEK293 cells were transfected with HA-CTIP1 expression vectors (in the absence of cotransfected Myc-ARP1), stained with appropriate antibodies, and qualitatively scored for the observed distribution pattern (focal, focal and diffuse, diffuse) by a naive observer. The numbers are derived from three independent transfection experiments
| CTIP1 mutant | Interaction with ARP1-(1–414) in vitro |
Potentiation of ARP1-mediated repression |
Subcellular distribution (indirect immunofluorescence/confocal microscopy) |
|||
|---|---|---|---|---|---|---|
| Foci | Foci/Diffuse | Diffuse | Comments | |||
| observed frequency/number of cells counted | ||||||
| 1–776 | + | ++ | 96/120 | 24/120 | 0/120 | Nuclear only |
| 1–601 | + | + | 0/90 | 76/90 | 14/90 | Nuclear only |
| 407–776 | + | − | 0/120 | 0/120 | 120/120 | Nuclear only |
| 1–378 | + | − | 0/90 | 0/90 | 90/90 | Cytoplasmic and nuclear |
| 1–263 | − | − | 0/90 | 0/90 | 90/90 | Cytoplasmic and nuclear |
Considered together, the results of in vitro GST pull-down experiments and colocalization studies in transfected cells suggest that the enhancement of ARP1-mediated repression by CTIP1 correlates with the capacity of CTIP1 to localize in punctate nuclear structures and to recruit ARP1 to these structures. Even if capable of interaction with ARP1 in vitro, CTIP1 mutants that were unable to localize in these punctate structures also lacked the ability to enhance ARP1-mediated repression. Thus, we conclude that these punctate, nuclear bodies are implicated in ARP1·CTIP1-mediated transcriptional repression.
CTIP1 Harbors Autonomous Transcriptional Regulatory Functions
To investigate the possibility that CTIP1 may harbor an autonomous transcriptional regulatory function(s), full-length CTIP1 and three isolated regions of the protein were fused to the GAL4 DNA binding domain (DBD; Fig. 7A) and tested in transient transfection experiments. Although full-length CTIP1 did not appear to modulate expression of a GAL4-dependent reporter when fused to the GAL4 DBD (Fig. 7B, lanes 4–6), both the isolated amino-terminal region of CTIP1 (amino acids 1–171) and the central core (amino acids 172–434) repressed basal transcription significantly in this context (Fig. 7B, lanes 7–9 and 13–15, respectively). The extent of transcriptional repression mediated by both GAL4 DBD-CTIP1-(1–171) and GAL4 DBD-CTIP1-(172–434) was unaffected by treatment of the cells with TSA, suggesting that neither involved recruitment of a TSA-sensitive histone deacetylase to the template (not shown). Unexpectedly, the carboxyl-terminal region of CTIP1 (amino acids 428–776) appeared to stimulate basal transcription weakly but significantly in these experiments (Fig. 5B, lanes 10–12).
Fig. 7. Autonomous transcriptional modulatory activity of CTIP1.
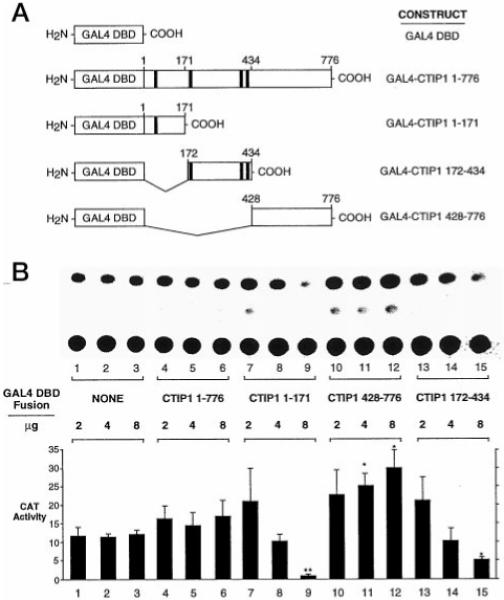
A, diagram of GAL4 DBD-CTIP1 fusion constructs used in the transfection experiments illustrated in B. The black boxes represent the zinc finger motifs of CTIP1. B, CAT assays using extracts of HEK293 cells transiently transfected with 7.5 μg of the (17-mer)5 reporter (pG5CAT, CLONTECH) and the indicated amounts of expression vectors for GAL4 DBD (pM; CLONTECH) or GAL4 DBD-CTIP1 fusion proteins. Statistically significant effects are indicated by asterisks (p < 0.05, Student’s t test) or double asterisks (p < 0.01) when comparing GAL4-CTIP fusions to the corresponding amount of transfected GAL4 DBD. Note that the quantitation shown represents the mean ± S.D. of CAT activities from six independent experiments, whereas the CAT assay shown in B is from a single, representative experiment.
DISCUSSION
The findings described herein identify a potential point of convergence between two large families of transcription factors, the nuclear receptor superfamily and proteins containing C2H2 zinc fingers. Both CTIP1 and CTIP2, novel C2H2 zinc finger proteins, were found to interact strongly with members of the COUP-TF family of orphan nuclear receptors in yeast and in vitro, and at least one of these proteins (CTIP1) potentiated the transcriptional repression activity of ARP1 in HEK293 cells independently of trichostatin-sensitive histone deacetylation. Potentiation of ARP1-mediated repression by CTIP1 correlated with the capacity of CTIP1 to form punctate nuclear structures and to recruit ARP1 to these structures. These studies suggest a novel mechanism for nuclear receptormediated repression that, rather than acting through recruitment of histone deacetylase(s), favors transcriptional silencing by redistributing nuclear receptors to distinct nuclear structures, possibly associated with heterochromatic regions, which may afford a transcriptionally nonpermissive environment. A similar mechanism of repression was suggested in the case of another C2H2 zinc finger protein, RP58 (40). Pipaon and colleagues recently demonstrated that COUP-TF I positively regulates NGF-IA expression through a putative Sp1 binding site (41). Sp1, like both CTIPs, is a C2H2 zinc finger protein. Thus, it is conceivable that complexes composed of a COUP-TF family member and CTIP1 (or CTIP2) may activate transcription in some promoter contexts.
Interaction with CTIP1 was found to require the putative AF-2 of ARP1. However, it is unknown if the ARP1 AF-2 core is sufficient to mediate interaction with CTIP1 or, more likely, this region simply enucleates the interaction interface that is formed by the juxtaposition of ARP1 α-helical regions within the putative ligand binding domain, such as has been described for the coactivator interaction interface of other nuclear receptors (42–44). CTIP1 appeared to harbor two separable ARP1 interaction domains, ID1 (amino acids 602–776) and ID2 (amino acids 264–378), and deletion mutants lacking one of these, but not both, were found to interact with ARP1 in vitro in a manner indistinguishable from that of full-length protein. Again, the structural basis for CTIP1-ARP1 interactions is not known; however, it is of interest that NCoR, another protein that couples nuclear receptors to the transcriptional repression machinery, similarly harbors two receptor interaction domains, both of which are capable of direct and independent interaction with nuclear receptors (38).
Interaction with ARP1 in HEK293 cells appeared to expose transcriptional repression domains of CTIP1 that were not functional when the full-length protein was fused to a heterologous DNA binding domain (Fig. 7B, lanes 4–6). However, fusion of the isolated regions of CTIP1 to the GAL4 DNA binding domain allowed delineation of two separable transcription repression functions. Similar results, suggesting a highly modular domain organization characteristic of other C2H2 zinc finger proteins, such as Ikaros (45) and YY1 (46), have been previously reported.
Transcriptional repression mediated by CTIP1 did not appear to be sensitive to inhibition by TSA, suggesting that histone deacetylation, which has been implicated in transcriptional repression mediated by many other classes of transcription factors, including nuclear receptors (29, 30) and some C2H2 zinc finger proteins such as YY1 (47), may not underlie the action of CTIP1. The mechanism(s) underlying transcriptional repression mediated by CTIP1 in mammalian cells is unknown but may involve interaction with and disruption of the function of the general transcription machinery (48, 49), interaction with chromatin components, or remodeling factors resulting in stabilization of inactive chromatin structure (50), titration of transcriptional coactivators such as p300 and CBP (46), interaction with general corepressor proteins such as Groucho (51) or CtBP2 (52), recruitment of general repressors such as NC2 (53), inhibition of the assembly of the transcriptional preinitiation complex through interaction with TATA binding protein (reviewed in Ref. 54), and/or a novel mechanism.
Both CTIP1 and CTIP2, like the COUP-TF proteins, are highly expressed in the central nervous system and in the developing embryo (3, 19, 21, 32), suggesting that CTIPs may mediate, at least in part, the activity of these orphan receptors during mammalian neurogenesis and organogenesis in tissues such as heart and liver. However, it is possible that CTIP proteins may also function independently of COUP-TF family members. Both CTIP1 and CTIP2 contain a highly conserved and repeated C2H2 zinc finger motif that is related to other such motifs implicated in site-specific DNA recognition, such as those present in PRD-1 (55, 56) and YY1 (57, 58). Similarly, the COUP-TF proteins may regulate transcription independently of CTIP1 and CTIP2 through interaction with SMRT (23), NCoR (23), and/or splice variants of NCoR (24), possibly suggesting a role for histone deacetylation in the COUP-TF signaling pathways leading to transcriptional repression. The relative contribution and importance of CTIP1 and histone deacetylation-dependent pathways to transcriptional repression mediated by COUP-TF family members remains to be established.
Acknowledgments
We gratefully thank Drs. H. Nakshatri, P. Chambon, R. Losson, P. Kastner, A. Krust, T. Lufkin, R. Hen, P. Dowell, and S. Nakanishi for generously sharing plasmid constructs and reagents; Valerie Peterson for expert technical assistance; and the Oregon State University Center for Gene Research and Biotechnology for DNA sequencing.
Footnotes
This work was supported in part by American Heart Association Grant 9640219N; NIEHS, National Institutes of Health, Grants ES00210 and ES00040; the Oregon State University College of Pharmacy; and the Laboratory of Molecular Pharmacology. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
- COUP-TF
- chicken ovalbumin upstream promoter-transcription factor
- RXR
- retinoid X receptor
- DR
- direct repeat
- HEK
- human embryonic kidney
- CAT
- chloramphenicol acetyltransferase
- DBD
- DNA binding domain
- TSA
- trichostatin A
- GST
- glutathione S-transferase
- PPARα
- peroxisome proliferator-activated receptor α
- NCoR
- nuclear receptor co-repressor
- SMRT
- silencing mediator for retinoid and thyroid hormone receptor
- HA
- hemagglutinin
- kb
- kilobase pair(s)
REFERENCES
- 1.Ladias JAA, Hadzopoulou-Cladaras M, Kardassis D, Cardot P, Cheng J, Zannis V, Caldaras C. J. Biol. Chem. 1992;267:15849–15860. [PubMed] [Google Scholar]
- 2.Wang LH, Ing NH, Tsai SY, O’Malley BW, Tsai M-J. Gene Expr. 1991;1:207–216. [PMC free article] [PubMed] [Google Scholar]
- 3.Jonk LJC, de Jonge EJ, Pals CEGM, Wissink S, Vervaart JMA, Schoorlemmer J, Kruijer W. Mech. Dev. 1994;47:81–97. doi: 10.1016/0925-4773(94)90098-1. [DOI] [PubMed] [Google Scholar]
- 4.Escriva H, Safi R, Hanni C, Langlois M-C, Saumitou-Laprade P, Stehelin D, Capron A, Pierce R, Laudet V. Proc. Natl. Acad. Sci. U. S. A. 1997;94:6803–6808. doi: 10.1073/pnas.94.13.6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kliewer SA, Umesono K, Heyman RA, Mangelsdorf DJ, Dyck JA, Evans RM. Proc. Natl. Acad. Sci. U. S. A. 1992;89:1448–1452. doi: 10.1073/pnas.89.4.1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Islam TC, Toftgard R. Biochem. Biophys. Res. Commun. 1994;203:545–552. doi: 10.1006/bbrc.1994.2217. [DOI] [PubMed] [Google Scholar]
- 7.Tran P, Zhang X-K, Salbert G, Hermann T, Lehmann JM, Pfahl M. Mol. Cell. Biol. 1992;12:4666–4676. doi: 10.1128/mcb.12.10.4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cooney AJ, Tsai SY, O’Malley BW, Tsai M-J. Mol. Cell. Biol. 1992;12:4153–4163. doi: 10.1128/mcb.12.9.4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cooney AJ, Leng X, Tsai SY, O’Malley BW, Tsai M-J. J. Biol. Chem. 1993;268:4152–4160. [PubMed] [Google Scholar]
- 10.Leng X, Cooney AJ, Tsai SY, Tsai M-J. Mol. Cell. Biol. 1996;16:2332–2340. doi: 10.1128/mcb.16.5.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shigeta H, Newbold RR, McLachlan JA, Teng C. Mol. Reprod. Dev. 1996;45:21–30. doi: 10.1002/(SICI)1098-2795(199609)45:1<21::AID-MRD3>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 12.Klinge C, Silver BF, Driscoll MD, Sathyan G, Bambara RA, Hilf R. J. Biol. Chem. 1997;272:31465–31474. doi: 10.1074/jbc.272.50.31465. [DOI] [PubMed] [Google Scholar]
- 13.Chu K, Zingg HH. J. Mol. Endocrinol. 1997;19:163–172. doi: 10.1677/jme.0.0190163. [DOI] [PubMed] [Google Scholar]
- 14.Chu K, Boutin J-M, Breton C, Zingg HH. Mol. Cell. Endocrinol. 1998;137:145–154. doi: 10.1016/s0303-7207(97)00241-4. [DOI] [PubMed] [Google Scholar]
- 15.Miyata KS, Zhang B, Marcus SL, Capone JP, Rachubinski RA. J. Biol. Chem. 1993;268:19169–19172. [PubMed] [Google Scholar]
- 16.van der Wees J, Matharu PJ, de Roos K, Destree OH, Godsave SF, Durston AJ, Sweeney GE. Mech. Dev. 1996;54:173–184. doi: 10.1016/0925-4773(95)00471-8. [DOI] [PubMed] [Google Scholar]
- 17.Mlodzik M, Hiromi Y, Weber U, Goodman CS, Rubin GM. Cell. 1990;60:211–224. doi: 10.1016/0092-8674(90)90737-y. [DOI] [PubMed] [Google Scholar]
- 18.Qiu Y, Pereira FA, DeMayo FJ, Lydon JP, Tsai SY, Tsai MJ. Genes Dev. 1997;11:1925–1937. doi: 10.1101/gad.11.15.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pereira FA, Qiu Y, Tsai M-J, Tsai SY. J. Steroid Biochem. Mol. Biol. 1995;53:503–508. doi: 10.1016/0960-0760(95)00097-j. [DOI] [PubMed] [Google Scholar]
- 20.Pereira FA, Qiu Y, Zhou G, Tsai MJ, Tsai SY. Genes Dev. 1999;13:1037–1049. doi: 10.1101/gad.13.8.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Avram D, Ishmael JE, Nevrivy DJ, Peterson VJ, Lee SH, Dowell P, Leid M. J. Biol. Chem. 1999;274:14331–14336. doi: 10.1074/jbc.274.20.14331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsai SY, Tsai M-J. Endocr. Rev. 1997;18:229–240. doi: 10.1210/edrv.18.2.0294. [DOI] [PubMed] [Google Scholar]
- 23.Shibata H, Nawaz Z, Tsai SY, O’Malley B, Tsai M-J. Mol. Endocrinol. 1997;11:714–724. doi: 10.1210/mend.11.6.0002. [DOI] [PubMed] [Google Scholar]
- 24.Bailey PJ, Dowhan DH, Franke K, Burke LJ, Downes M, Muscat GEO. J. Steroid Biochem. Mol. Biol. 1997;63:165–174. doi: 10.1016/s0960-0760(97)00079-4. [DOI] [PubMed] [Google Scholar]
- 25.Horlein AJ, Naar AM, Heinzel T, Torchia J, Gloss B, Kurokawa R, Ryan A, Kamei Y, Soderstrom M, Glass CK, Rosenfeld MG. Nature. 1995;377:397–404. doi: 10.1038/377397a0. [DOI] [PubMed] [Google Scholar]
- 26.Kurokawa R, Soderstrom M, Horlein A, Halachmi S, Brown M, Rosenfeld MG, Glass CK. Nature. 1995;377:451–454. doi: 10.1038/377451a0. [DOI] [PubMed] [Google Scholar]
- 27.Chen JD, Evans RM. Nature. 1995;377:454–457. doi: 10.1038/377454a0. [DOI] [PubMed] [Google Scholar]
- 28.Dowell P, Ishmael JE, Avram D, Peterson VJ, Nevrivy DJ, Leid M. J. Biol. Chem. 1999;274:15901–15907. doi: 10.1074/jbc.274.22.15901. [DOI] [PubMed] [Google Scholar]
- 29.Heinzel T, Lavinsky RM, Mullen TM, Soderstrom M, Laherty CD, Torchia J, Yang WM, Brard G, Ngo SD, Davie JR, Seto E, Eisenman RN, Rose DW, Glass CK, Rosenfeld MG. Nature. 1997;387:43–48. doi: 10.1038/387043a0. [DOI] [PubMed] [Google Scholar]
- 30.Nagy L, Kao HY, Chakravarti D, Lin RJ, Hassig CA, Ayer DE, Schreiber SL, Evans RM. Cell. 1997;89:373–380. doi: 10.1016/s0092-8674(00)80218-4. [DOI] [PubMed] [Google Scholar]
- 31.Struhl K. Genes Dev. 1998;12:599–606. doi: 10.1101/gad.12.5.599. [DOI] [PubMed] [Google Scholar]
- 32.Qiu Y, Cooney A, Kuratani S, DeMayo FJ, Tsai SY, Tsai M-J. Proc. Natl. Acad. Sci. U. S. A. 1994;91:4451–4455. doi: 10.1073/pnas.91.10.4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suri C, Fung BP, Tischler AS, Chikaraishi DM. J. Neurosci. 1993;13:1280–1291. doi: 10.1523/JNEUROSCI.13-03-01280.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bouvier C, Avram D, Peterson VJ, Hettinger B, Soderstrom K, Murray TF, Leid M. Eur. J. Pharmacol. 1998;348:85–93. doi: 10.1016/s0014-2999(98)00132-0. [DOI] [PubMed] [Google Scholar]
- 35.Kronczynska AM, Coutts M, Makrides S, Braweman G. Nucleic Acids Res. 1989;17:6408. doi: 10.1093/nar/17.15.6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moriyoshi K, Masu M, Ishii T, Shigemoto R, Mizuno N, Nakanishi S. Nature. 1991;354:31–37. doi: 10.1038/354031a0. [DOI] [PubMed] [Google Scholar]
- 37.Achatz G, Holzi B, Speckmayer R, Hauser C, Sandhofer F, Paulweber B. Mol. Cell. Biol. 1997;17:4914–4932. doi: 10.1128/mcb.17.9.4914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seol W, Mahon MJ, Lee YK, Moore DD. Mol. Endocrinol. 1996;10:1646–1655. doi: 10.1210/mend.10.12.8961273. [DOI] [PubMed] [Google Scholar]
- 39.Yoshida M, Horinouchi S, Beppu T. BioEssays. 1995;17:423–430. doi: 10.1002/bies.950170510. [DOI] [PubMed] [Google Scholar]
- 40.Aoki K, Meng G, Suzuki K, Takashi T, Kameoka Y, Nakahara K, Ishida R, Kasai M. J. Biol. Chem. 1998;273:26698–26704. doi: 10.1074/jbc.273.41.26698. [DOI] [PubMed] [Google Scholar]
- 41.Pipaon C, Tsai SY, Tsai MJ. Mol. Cell. Biol. 1999;19:2734–2745. doi: 10.1128/mcb.19.4.2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Darimont BD, Wagner RL, Apriletti JW, Stallcup MR, Kushner PJ, Baxter JD, Fletterick RJ, Yamamoto KR. Genes Dev. 1998;12:3343–3356. doi: 10.1101/gad.12.21.3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nolte RT, Wisely GB, Westin S, Cobb JE, Lambert MH, Kurokawa R, Rosenfeld MG, Willson TM, Glass CK, Milburn MV. Nature. 1998;395:137–143. doi: 10.1038/25931. [DOI] [PubMed] [Google Scholar]
- 44.Westin S, Kurokawa R, Nolte RT, Wisely GB, McInerney EM, Rose DW, Milburn MV, Rosenfeld MG, Glass CK. Nature. 1998;395:199–202. doi: 10.1038/26040. [DOI] [PubMed] [Google Scholar]
- 45.Georgopoulos K, Winandy S, Avitahl N. Annu. Rev. Immunol. 1997;15:155–176. doi: 10.1146/annurev.immunol.15.1.155. [DOI] [PubMed] [Google Scholar]
- 46.Austen M, Luscher B, Luscher-Firzlaff JM. J. Biol. Chem. 1997;272:1709–1717. doi: 10.1074/jbc.272.3.1709. [DOI] [PubMed] [Google Scholar]
- 47.Yang WM, Inouye C, Zeng Y, Bearss D, Seto E. Proc. Natl. Acad. Sci. U. S. A. 1996;93:12845–12850. doi: 10.1073/pnas.93.23.12845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Olave I, Reinberg D, Vales LD. Genes Dev. 1998;12:1621–1637. doi: 10.1101/gad.12.11.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Um M, Li C, Manley JL. Mol. Cell. Biol. 1995;15:5007–5016. doi: 10.1128/mcb.15.9.5007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim J, Sif S, Jones B, Jackson A, Koipally J, Heller E, Winandy S, Viel A, Sawyer A, Ikeda T, Kingston R, Georgopoulos K. Immunity. 1999;10:345–355. doi: 10.1016/s1074-7613(00)80034-5. [DOI] [PubMed] [Google Scholar]
- 51.Ren B, Chee KJ, Kim TH, Maniatis T. Genes Dev. 1999;13:125–37. doi: 10.1101/gad.13.1.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Turner J, Crossley M. EMBO J. 1998;17:5129–5140. doi: 10.1093/emboj/17.17.5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ikeda K, Halle JP, Stelzer G, Meisterernst M, Kawakami K. Mol. Cell. Biol. 1998;18:10–18. doi: 10.1128/mcb.18.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maldonado E, Hampsey M, Reinberg D. Cell. 1999;99:455–458. doi: 10.1016/s0092-8674(00)81533-0. [DOI] [PubMed] [Google Scholar]
- 55.Keller AD, Maniatis T. Genes Dev. 1991;5:868–879. doi: 10.1101/gad.5.5.868. [DOI] [PubMed] [Google Scholar]
- 56.Keller AD, Maniatis T. Mol. Cell. Biol. 1992;12:1940–1949. doi: 10.1128/mcb.12.5.1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hariharan N, Kelley DE, Perry RP. Proc. Natl. Acad. Sci. U. S. A. 1991;88:9799–9803. doi: 10.1073/pnas.88.21.9799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Seto E, Shi Y, Shenk T. Nature. 1991;354:241–245. doi: 10.1038/354241a0. [DOI] [PubMed] [Google Scholar]