

Abstract
Certain human subpopulations are metabolically healthy but obese, or metabolically obese but normal weight; such mutations uncouple obesity from glucose intolerance, revealing pathways implicated in Type 2 diabetes. Current searches for relevant genes consume significant effort. We have reported previously a novel double bromodomain protein called Brd2, which is a transcriptional co-activator/co-repressor with SWI/SNF (switch mating type/sucrose non-fermenting)-like functions that regulates chromatin. In the present study, we show that whole-body disruption of Brd2, an unusual MHC gene, causes lifelong severe obesity in mice with pancreatic islet expansion, hyperinsulinaemia, hepatosteatosis and elevated pro-inflammatory cytokines, but, surprisingly, enhanced glucose tolerance, elevated adiponectin, increased weight of brown adipose tissue, heat production and expression of mitochondrial uncoupling proteins in brown adipose tissue, reduced macrophage infiltration in white adipose tissue, and lowered blood glucose, leading to an improved metabolic profile and avoiding eventual Type 2 diabetes. Brd2 is highly expressed in pancreatic β-cells, where it normally inhibits β-cell mitosis and insulin transcription. In 3T3-L1 pre-adipocytes, Brd2 normally co-represses PPAR-γ (peroxisome-proliferator-activated receptor-γ) and inhibits adipogenesis. Brd2 knockdown protects 3T3-L1 adipocytes from TNF-α (tumour necrosis factor-α)-induced insulin resistance, thereby decoupling inflammation from insulin resistance. Thus hypomorphic Brd2 shifts energy balance toward storage without causing glucose intolerance and may provide a novel model for obese metabolically healthy humans.
Keywords: adipogenesis, β-cell, Brd2, bromodomain, energy balance, obesity, Type 2 diabetes
INTRODUCTION
The problem of human obesity worldwide has reached a crisis in scope and is expected to result in dramatic increases in obesity-related morbidity and early mortality [1]. Much of the projected global incidence of 366 million diabetic individuals by 2030 is expected to be obesity-driven [2]. Thus a deeper molecular understanding of the origins of diabetes is urgently required, particularly signal transduction systems that couple obesity to Type 2 diabetes. Recently, attention has focused on human subpopulations that appear to harbour mutations that uncouple obesity from insulin resistance, because of the opportunity they provide to increase our understanding of the aetiology of obesity-driven diabetes; the molecular mechanisms that drive these processes remain incompletely understood. Such individuals may be either MHO (metabolically healthy but obese) or MONW (metabolically obese but normal weight) [3]. Evidence we provide in the present study suggests that a recently described double bromodomain protein called Brd2 is functionally required in vivo to couple obesity to eventual Type 2 diabetes.
Brd2, formerly ‘RING3’, was first identified as a close homologue of TAF (TATA-box-binding protein-associated factor)II250, a basal transcription co-activator [4]. The gene resides on human chromosome 6p within the MHC class II locus and is found in syntenic regions of other organisms [5]. In mammals, the MHC class II genes encode molecules that participate in processing and presentation of antigenic peptides to T-cells, except BRD2, which encodes a double bromodomain protein that shares no significant sequence similarity to or function with other MHC class II molecules and has been suggested to be involved in signal transduction [6]. Brd2 is one of the protein factors that regulate gene transcription by controlling the activity of transcription complexes, interpreting the histone code and remodelling chromatin. Brd2 belongs to the BET (bromodomain and extraterminal domain) subfamily of proteins that harbour two bromodomains [7]. The bromodomain is a conserved protein motif comprised of approx. 110 amino acids that form four α-helices connected by two loops; the motif binds acetyl-lysine residues of nucleosomal histones [8]. Bromodomain proteins serve as adaptor or scaffolding modules that recruit transcription regulatory factors to chromatin to form protein complexes that regulate gene transcription in response to signal transduction [9,10]. Bromodomains are found in the DNA helicase superfamily members [11] brahma/Brm/Snf2α or Brg1/Snf2β, which are the core catalytic ATPase subunits of the SWI/SNF (switch mating type/sucrose non-fermenting) nucleosome remodelling complex [12]. The bromodomain structure itself does not possess Hat (histone acetyltransferase) activity or other catalytic functions, but it is found in many transcriptional and developmental regulators that function through histone modification and nucleosome remodelling [7–10].
Brd2 is a signal transducer [13] and a nuclear-localized serine/threonine kinase [13,14], and is ubiquitously expressed in mammalian tissues [15]. Brd2 binds to ε-aminoacetyl groups of nucleosomal histone lysine residues with its two bromodomains, particularly acetyl-histone H4 [16], and participates in multiprotein transcription complexes such as Mediator [17]. We have shown previously that Brd2 binds not only transcriptional activators like E2F proteins, but also co-activator TAFs, members of the SWI/SNF complex, Hats and Hdacs (histone deacetylases) [18] to regulate the expression of diverse genes [19]. As with the SWI/SNF complex with which it associates, Brd2 can function as either a transcriptional co-activator or co-repressor, depending on the specific promoter and cellular context [20,21].
One or the other of the Brd2 yeast homologues BDF1 and BDF2 are essential for yeast viability; double mutants are lethal [22]. Bdf1 and yTaf145 together serve a functional role similar to TAFII250 in mammals [23]. Bdf1 has been suggested to play a role in chromatin restructuring [24]. The Drosophila homologue [4,25] of Brd2, fsh [female sterile (1) homeotic] is a maternal effect gene that encodes a double bromodomain-containing developmental factor that restructures chromatin as a co-activator or co-repressor. During Drosophila development, fsh acts as an upstream activator of Hox genes such as trithorax [26] and the Ultrabithorax complex [25], which counter Polycomb repression. Mutation of fsh causes severe homoeotic defects that resemble trithorax mutations [26] and homozygous mutation is lethal [27].
To study Brd2 function in vivo, we expressed it constitutively in the B-cells of transgenic mice, where it causes B-cell lymphoma and leukaemia [28]. A related protein, called Brd4, is also a chromatin regulator and transcriptional adapter [29]. Brd4 binds acetylated histones [30] and is necessary for the G2-to-M transition of the cell cycle [31]. As with fsh-deficient flies, brd4(+/−) mice have severe developmental defects; brd4(−/−) is lethal [32]. Our next step in the characterization of the in vivo function of Brd2 was to engineer a knockout mouse. Given the above considerations, we expected brd2(−/−) to be lethal in mice. Consistent with this expectation, we could not obtain brd2(−/−)-null mice or ES (embryonic stem) cells, and in support of this result, recent reports have established that Brd2 is essential because brd2(−/−)-null mice have neural tube defects and embryonic lethality [33,34]. However, we found, surprisingly, that targeted disruption of the Brd2 gene in the promoter region, leading to the reduced expression of Brd2 (‘brd2 lo’; w/lo), produces a hypomorphic phenotype without gross developmental abnormalities. These mice have extreme obesity with hyperinsulinaemia, but enhanced glucose tolerance and low blood glucose, which is quite distinct from other animal models of obesity.
EXPERIMENTAL
ES cell lines and mice
RRT234 and RRE050 murine ES cell lines were purchased from BayGenomics and were used for microinjection. Homozygous brd2(−/−) ES cells (234/DK; see Figure 1D) were derived from the RRT234 parental line by culturing cells in medium with 8 mg/ml G418. C57BL/6J mice were from the Jackson Laboratory. The regular chow (2918 formulation; Harlan Teklad) was irradiated and contained 57.3 % carbohydrate. Animals were handled humanely in accordance with Boston University IACUC, State and Federal regulatory requirements. Please refer to the Supplementary Experimental section available at http://www.BiochemJ.org/bj/425/bj4250071add.htm for additional details.
Figure 1. Engineering of obese w/lo mice.
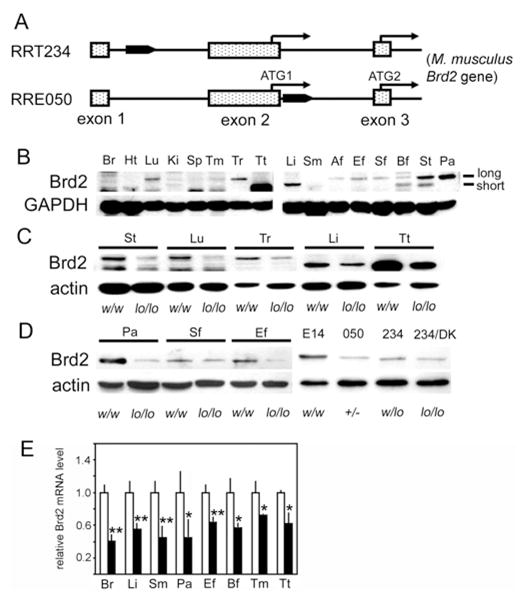
(A) Brd2 gene trap events in RRT234 and RRE050 ES cell lines. The filled pointed rectangular box indicates the gene trap vector. (B) Brd2 expression in w/w mice detected by immunoblotting with a Brd2 antibody. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. (C) Brd2 protein expression in genetically engineered mice. (D) Brd2 protein expression in tissues and ES cell lines. E14, parental E14Tg2A ES cell line; 050, RRE 050 ES cell line; 234, RRT234 ES cell line; 234/DK, brd2 double-mutant RRT234 ES cell line; +/−, heterozygous brd2 knockout from RRE050 ES cells. (E) Quantitative real-time PCR analysis of Brd2 mRNA expression in tissues from heterozygous w/lo mice from RRT234 ES cells (closed bars) and control w/w male mice (open bars) age-matched at 6 months (n = 3; *P < 0.05 and **P < 0.01). Af, abdominal fat; Bf, BAT; Br, brain; Ef, epididymal fat pad; Ht, heart; Ki, kidney; Lu, lung; Li, liver; Pa, pancreas; Sf, subcutaneous fat; Sm, skeletal muscle; Sp, spleen; St, stomach; Tm, thymus; Tr, thyroid; Tt, testis.
Metabolic measurements
V̇O2 (oxygen consumption) and V̇CO2 (carbon dioxide production) were measured by indirect calorimetry and was used to calculate RER (respiratory exchange ratio) and heat production (CLAMS; Columbus Instruments). Glucose was determined in fresh whole-blood samples using a Glucometer Elite (Bayer). Plasma hormone, cytokine and lipid measurements were performed at the Specialized Assay Core, Joslin Diabetes Center, Boston, MA, U.S.A.
Immunohistochemistry
Brd2 protein was detected with a rabbit polyclonal antibody as described previously [14].
Flow cytometry
Unless otherwise indicated, all reagents were from eBioscience. Standard flow protocols were used and are described in detail in the Supplementary Experimental section.
3T3-L1 pre-adipocyte differentiation and assays
Standard differentiation protocols were used and are described in detail in the Supplementary Experimental section.
Protein immunoblotting and co-immunoprecipitation
Immunoblotting and co-immunoprecipitation were performed according to the manufacturer’s instructions (Santa Cruz Biotechnology) and as described previously [14].
Luciferase assay
Briefly, 3T3-L1 pre-adipocytes were transfected in 24-well plates using Lipofectamine™ 2000 (Invitrogen). Transfections typically consisted of 0.1 μg of tk-PPRE3X-luc (Addgene), 0.015 μg of pRL (Promega), 0.18 μg of pFlag. PPAR-γ (Addgene) and 0.5 μg of effector (pSiBrd2, pmBrd2 or control vectors). Cell lysates were harvested 48 h after transfection and luciferase activity was measured using a Dual Luciferase Reporter Assay System (Promega) on a TD-20/20 luminometer (Turner Biosystems).
Statistical analysis
Values are shown as means ± S.E.M. Paired Student’s t tests were used to compare means, and P < 0.05 was considered significant. Details on the analyses used are described in the Supplementary Experimental section.
RESULTS
Generation and phenotype of w/lo mice
We attempted to generate Brd2-knockout mice using two Brd2 gene-trapped murine ES cell lines from BayGenomics (RRE050 and RRT234). Gene trap vectors were inserted into intron 1, 5′ to the first ATG codon of the Brd2 gene in RRT234 cells, or into intron 2, 3′ to the first ATG codon in RRE050 cells (Figure 1A). The RRE050 disruption (the ES cells used by Shang et al. [33]) reduced Brd2 expression levels more drastically than did the RRT234 disruption (Figures 1C and 1D). Germline transfer succeeded with blastocyst microinjection of RRT234, but not with RRE050, suggesting that Brd2 is critical for embryogenesis, similar to Brd4 [32]. Male chimaeras from RRT234 microinjection were crossed with C57BL/6J female mice to produce heterozygous (F1) Brd2-mutant animals on a 129P2 and C57BL/6J mixed background. The male F1 heterozygote mice were then back-crossed to female wild-type C57BL/6J mice for five generations (F5), then the F5 male and female heterozygotes were mated to produce wild-type (w/w), heterozygous (w/lo; ‘brd2 lo’) and homozygous (lo/lo) animals for study.
To determine whether gene trap insertion into the first intron of Brd2 in RRT234 ES cells disrupts Brd2 expression in vivo and in vitro, first we measured Brd2 protein expression in different tissues of w/w mice. There are at least two forms of Brd2 protein in mice due to alternative mRNA splicing and/or alternative start codons for translation. The long form is comprised of 798 amino acids [NCBI (National Center for Biotechnology Information) accession number NP_034368] and the short form of 679 amino acids (NCBI accession number NP_001020558). Immunoblot analysis showed that, in adult w/w mice, the long form is mainly expressed in lung, thyroid, stomach and pancreas, with low expression in adipose tissues. The short form is mainly expressed in testis, liver, BAT (brown adipose tissue) and stomach (Figure 1B). Unexpectedly, the RRT234 mutation reduced rather than eliminated the expression of both forms of Brd2, as shown by immunoblot analysis of Brd2 for all of the tested tissues from homozygous mice (lo/lo; Figures 1C and 1D) and a homozygous RRT234 ES cell line (double knockout; Figure 1D). Heterozygous mice also had reduced expression (Figure 1E). Live births of homozygous mice were extremely rare and did not yield sufficient animals for study (see Supplementary Table S1 available at http://www.BiochemJ.org/bj/425/bj4250071add.htm). Thus, with RRT234 ES cells, we generated a Brd2 hypomorph (w/lo; ‘brd2 lo’) mouse line.
The w/lo mice were viable and appeared normal at birth, but, surprisingly, they gradually became extremely obese on a diet of regular chow ad libitum (Figure 2A). The penetrance of this phenotype was 100 %. Body weights of w/lo mice diverged from control mice at around 2 months of age, reached 60 g by 4 months of age and continually increased to 90 g by 14 months of age (Figure 2B). Interestingly, the difference in body weight between w/lo mice and w/w mice held steady at 40–50 g at 4 months and thereafter, suggesting a higher ‘set point’ for the body weight of w/lo mice. Body fat deposition was abundant and widespread. Epididymal fat pads were three times heavier than w/w controls of the same age [1.57 ± 0.16 g in w/w mice compared with 6.32 ± 0.50 g in w/lo mice (n = 3; P < 0.01)]. Most dramatically, skeletal muscle was encased in thick layers of fat with adipose deposits throughout. Histopathologically, obese w/lo mice shared certain features with other obese mouse models. Small adipocytes (≤ 2000 μm2 in area) were present in equal numbers in epididymal fat pads of w/w mice and obese w/lo mice (Mann–Whitney rank sum, P = 0.256), but enlarged adipocytes (>2000 μm2) typified the fat depots of obese w/lo mice (P < 0.001); 1.1 % of w/w mice and 62.9 % of w/lo mice epididymal adipocytes were larger than 10000 μm2, and 3.6 % of w/w mice and 48.1 % of w/lo mice subcutaneous adipocytes were larger than 10000 μm2 (Figures 2C–2E), consistent with greatly increased storage in w/lo mice. The w/w mice and w/lo mice shared the same relationship between pancreatic islet area and β-cell number (Figure 2H); pancreatic islets were increased both in number and area in obese w/lo mice (Figures 2F and 2G) and some islets from w/lo mice were extremely large (Figures 2G and 2H). Increased islet number and size is consistent with other murine models of obesity, such as ob/ob, during development of insulin resistance and before the onset of Type 2 diabetes [35]. Islets of obese w/lo mice had high integrity (Figure 2F); β-cell failure, necrosis or declines in islet area or number were never observed in obese w/lo mice, at any age. Severe hepatosteatosis was also apparent in obese w/lo mice (Figure 2I).
Figure 2. Whole-body reduction of Brd2 causes severe obesity in mice.

(A) F6 generation of heterozygous w/lo and w/w control mice at 6 months of age. (B) Body weight curves of w/lo mice (◆) and w/w mice (◇) over time (n = 4–6; *P<0.05 and **P<0.01). (C) Histopathological changes in the epididymal fat pad of obese w/lo mice and w/w mice at 6 months of age. Image analysis of epididymal (D) and subcutaneous (E) adipocytes. Distribution curve fit by non-linear regression. w/w mice, open bars; w/lo mice, closed bars. (F) Histopathological changes in the pancreas of obese w/lo mice and w/w mice at 6 months of age. (G) Box plot analysis of β-cell number per islet from the pancreas of w/w mice and w/lo mice. (H) Regression of β-cell number/islet against islet area of the pancreas from w/w mice (○) and w/lo mice (●). (I) Histopathological changes in the liver of obese w/lo mice and w/w mice at 6 months of age. Sections were visualized with haematoxylin and eosin stain. Scale bar, 100 μm.
Obesity is caused by increased energy storage and/or decreased energy expenditure. In general, obese animals are hyperphagic compared with wild-type; hyperphagia was also observed in obese w/lo mice. At 4 weeks of age, the w/lo mice had no statistically significant difference in food intake compared with w/w mice (3.9 ± 0.5 g in w/w mice compared with 4.0 ± 0.4 g in w/lo mice; n = 3), but at 8 weeks began to increase their food intake compared with w/w mice (Figure 3A), and they maintained this pattern as they aged. The obese w/lo mice were hyperinsulinaemic (Figure 3B), but, surprisingly, had low blood glucose (Figure 3C). We noticed the first signs of fed-state low blood glucose in w/lo mice as early as 4 weeks after birth (Figure 3C). Remarkably, fed-state glucose levels held near fasting-state thereafter (Figure 3C). To measure metabolic parameters, we housed obese w/lo mice and w/w mice individually in metabolic cages with regular chow ad libitum and measured food intake, heat production by calorimetry and RER. Photocells aligned with the x-, y- and z- axes of the cages established that habitual activity was not significantly different from w/w mice (results not shown). The w/lo mice produced approximately twice as much heat than controls (Figure 3D). In further support of these data, obese w/lo mice carry twice as much interscapular BAT (257 ± 14 mg) as w/w mice (129 ± 52 mg) or w/w mice rendered obese by 4 months on a HFD (high-fat diet) (138 ± 20 mg) (Figure 3E), and had significant transcriptional up-regulation of Ucps 1, 2 and 3 (uncoupling proteins 1, 2 and 3) in BAT (Figure 3F). Strikingly, RER values of obese w/lo mice held at a fasting level regardless of actual feeding state (Figure 3G), suggesting they constantly catabolize fat stores as their major energy source. Thus w/lo obesity is not explained by reduced energy expenditure, but is more likely to be attributable to increased energy consumption and storage.
Figure 3. Metabolic changes in w/lo mice.

(A) Hyperphagia of w/lo mice at 8 weeks compared with w/w mice at the same age (n = 6; *P = 0.0003). (B) Hyperinsulinaemia in w/lo mice at 40 weeks (closed bars) compared with w/w mice at the same age (open bars) (n = 4; *P < 0.05). (C) Obese w/lo mice develop fed-state low blood glucose as they age. Plasma glucose at 4 and 40 weeks of age for w/lo mice (closed bars) and w/w mice (open bars) (n = 6; *P < 0.05). (D) Heat production at 40 weeks of age for obese w/lo mice (closed bars) and w/w mice (open bars) (n = 4; *P < 0.05). (E) Weight of interscapular BAT depots of age-matched w/w and lean mice (w/w lean), w/w and obese mice after 4 months on an HFD (w/w HFD) or obese w/lo mice (n = 3; *P = 0.015 and **P = 0.0017). (F) Real-time RT–PCR assay of mRNA levels of Ucp1, 2 and 3 compared with β-actin in interscapular BAT from (E); (open bars) obese w/w mice; (closed bars) obese w/lo mice (n = 3; *P < 0.05). (G) A representative RER of w/w mice (◆) and obese w/lo mice (◇) in fed and fasted states at 40 weeks.
Obesity is a chronic disease and is usually accompanied by serological features such as hyperinsulinaemia, hyperleptinaemia, hypoadiponectinaemia, dyslipidaemia and elevated pro-inflammatory cytokines. Certain serological parameters of the obese w/lo mice reflected chronic obesity. Plasma levels of hormones, such as insulin, glucagon and leptin, pro-inflammatory cytokines, such as IL-1β (interleukin-1β) and TNF-α (tumour necrosis factor-α), and lipids, including cholesterol and triacylglycerol (triglyceride), were all elevated in obese w/lo mice (Table 1), but the level of NEFAs (non-esterified fatty acids; ‘free fatty acids’) remained unchanged, suggesting obese w/lo mice have hyperinsulinaemia and moderate dyslipidaemia. However, unlike obese animal models, plasma adiponectin in obese w/lo mice was elevated.
Table 1. Blood chemistry and endocrine characteristics of w/lo and w/w mice at 9 months of age.
P values were determined using Student’s t test.
| Parameter | w/w | w/lo | P value | n |
|---|---|---|---|---|
| Random plasma glucose (mg/dl) | 161.8 ± 11.0 | 110.3 ± 20.0 | 2.1×10−5 | 8 |
| Fasting plasma glucose (mg/dl) | 109.7 ± 18.5 | 88.1 ± 12.6 | 5.4×10−4 | 11 |
| Random plasma insulin (ng/ml) | 1.5 ± 0.3 | 4.2 ± 0.7 | 3.2×10−5 | 7 |
| Fasting plasma insulin (ng/ml) | 1.1 ± 0.4 | 3.2 ± 0.5 | 2.6×10−4 | 7 |
| Adiponectin (μg/ml) | 20.9 ± 7.2 | 30.4 ± 5.1 | 8.8×10−3 | 9 |
| Leptin (ng/ml) | 44.7 ± 18.7 | 132.0 ± 56.2 | 3.2×10−3 | 9 |
| Glucagon (pg/ml) | 77.4 ± 24.5 | 108.2 ± 21.2 | 3.6×10−4 | 9 |
| NEFAs (mmol/l) | 1.2 ± 0.2 | 1.4 ± 0.1 | 0.098 | 8 |
| Cholesterol (mg/ml) | 1.4 ± 0.4 | 2.0 ± 0.4 | 0.007 | 8 |
| Triacylglycerol (mg/ml) | 0.9 ± 0.2 | 1.2 ± 0.3 | 0.045 | 8 |
| IL-1β (pg/ml) | 30.0 ± 27.2 | 234.4 ± 164.3 | 0.047 | 4 |
| TNF-α (pg/ml) | 0.1 ± 0.1 | 10.8 ± 10.5 | 0.01 | 8 |
Akin to insulin-resistant obese humans, obese w/lo mice are hyperinsulinaemic, yet they have lower random blood glucose than w/w controls (Figure 3C), suggesting their metabolism is unbalanced toward energy storage and that they remain insulin-responsive. Obese w/lo mice resemble MHO humans [3] in having slightly, yet significantly, elevated triacylglycerol and elevated leptin, but NEFAs and fasting glucose were not significantly elevated above normals. As with MHO and normal humans, but unlike insulin-resistant obese humans, w/lo mice retain a robust insulin-stimulated ability to dispose of glucose. The w/lo mice differ from MHO, obese and normal humans: none of these phenotypes have lower blood glucose. The lower ‘random blood glucose’ probably mimics a permanently fasting state, which may explain why the RER value of w/lo mice holds at a fasting level. It is widely accepted that low blood glucose is a strong hunger signal, which in w/lo mice probably provides a driving force for chronic hyperphagia. Therefore reduced Brd2 caused hyperinsulinaemia, but unalleviated low blood glucose in the fed state, and hyperadiponectinaemia, which are exceptionally rare in obese subjects.
In obese animals, blood glucose tends to be above normal due to obesity-induced insulin resistance, which is a major cause of eventual β-cell failure and Type 2 diabetes. To explore glucose usage in obese w/lo mice, we performed IGTTs (intraperitoneal glucose tolerance tests): 1.5 g of D-glucose/kg of body weight was injected to challenge young (4 week) w/lo mice before obesity onset, older (40 week) w/lo mice with severe obesity and w/w controls. Interestingly, plasma glucose in both young and old w/lo mice only reached ~ 250 mg/dl and fell quickly to baseline in young mice. Conversely, plasma glucose in w/w mice rose to ~ 500 mg/dl and fell more slowly to baseline (Figures 4A and 4B). These results indicate that w/lo mice are more efficient at clearing glucose from the blood and more tolerant than w/w mice to glucose challenge during chronic obesity and even before the onset of obesity (Figure 2B). Insulin secretion during IGTT is much higher in w/lo mice than in w/w mice (Figure 4C). Taken together, these data make it clear that obese w/lo mice, although hyperinsulinaemic, do not progress to glucose intolerance and β-cell failure despite months of chronic obesity.
Figure 4. Reduced Brd2 enhances glucose tolerance in vivo and protects 3T3-L1 adipocytes from TNF-α-induced insulin resistance in vitro.

IGTT (1.5 g of D-glucose/kg of body weight) in w/w mice (◇) and w/lo mice (■) at 4 weeks (A) and 40 weeks (B) of age. (C) Plasma insulin production during IGTT in w/w mice (◇) and w/lo mice (■). (D) Flow cytometry of F4/80+ macrophages in the SVF isolated from the epididymal fat pad of obese w/lo mice (solid grey line) compared with obese w/w mice on a HFD [w/w (HFD)] as a positive control (solid black line) and isotype control (broken line). (E) Quantification of F4/80+ macrophages. Open bar, obese w/w mice on a HFD [w/w (HFD)]; closed bar, obese w/lo mice. (F) Normal expression of MHC class II molecules [PE (phycoerythrin)-Cy5] on the surface of antigen-presenting B-cells (B220) from the spleens of w/w mice and w/lo mice; percentages of the gated lymphocytes are shown for each quadrant of the contour plot, as described previously [57]. (G) Immunoblot for Brd2 in 3T3-L1 pre-adipocytes stably transfected with Brd2 shRNA (SiBrd2) or control (SiC) plasmids. (H) Baseline (open bars) and insulin-stimulated (closed bars) glucose uptake by 3T3-L1 adipocytes. (I) Glucose uptake of 3T3-L1 adipocytes stably transfected with Brd2 or control shRNA plasmids and treated with 4 ng/ml TNF-α for 4 days to induce insulin resistance. Open bars, baseline; grey-shaded bars, TNF-α alone; closed bars, TNF-α+insulin. n = 7 in (A), n = 4 in (B, C, H and I), and n = 3 in (E); *P<0.05.
Reduced Brd2 levels block macrophage infiltration in vivo and insulin resistance in vitro
It is well established that chronic inflammation due to increased production of pro-inflammatory cytokines and adipose deposition in liver and muscle (two major targets of insulin action) cause insulin resistance in obesity and Type 2 diabetes [36]. Unexpectedly, despite their severe obesity, glucose intolerance was not observed in w/lo mice, although they have severe hepatosteatosis (Figure 2I) and elevated pro-inflammatory cytokines (Table 1). Furthermore, obese w/lo mice are more tolerant than w/w mice to a glucose challenge. These findings suggest inflammatory responses may be deficient in w/lo mice. We undertook additional experiments to explore the possibility that Brd2, as a signal transduction effector [13,14,17,19,28], might be required in normal cells to transduce signals from pro-inflammatory cytokines.
Macrophages and adipocytes release pro-inflammatory cytokines that can induce insulin resistance in vitro and in vivo, an important one of which is TNF-α [36]. Although w/lo mice have elevated serum TNF-α (Table 1), we were surprised to find that F4/80+ (classically activated) inflammatory macrophages that might provide a source of TNF-α were missing from the SVF (stromal vascular fraction) of the epididymal fat depot of obese w/lo mice, although these macrophages were nevertheless abundant in epididymal fat depots of w/w controls rendered obese by an HFD (Figure 4D; quantification is shown in Figure 4E). The lack of infiltration of fat in w/lo mice by these inflammatory cells probably provides another form of protection against insulin resistance in adipose tissues. Given the position of the lacZ disruption of the Brd2 gene in RRT234 ES cells (Figure 1A), within the MHC class II locus, we performed control experiments to confirm that these mice did not have a global defect in antigen presentation that might provide a trivial explanation for immunodeficiency. The surface expression of MHC class II molecules was measured by flow cytometry of splenic B-cells from adult w/lo mice and was verified to be normal (Figure 4F). To test the hypothesis that whole-body reduction in Brd2 protects cells against cytokine-induced insulin resistance, Brd2 expression was reduced in the 3T3-L1 system by stable transfection of the pre-adipocytes with a plasmid encoding Brd2 shRNA (small-hairpin RNA) (pSiBrd2). Then the cells were differentiated to adipocytes with insulin/dexamethasone/isobutylmethylxanthine and pioglitazone. We confirmed that pSiBrd2 significantly reduced Brd2 protein in 3T3-L1 adipocytes (Figure 4G). The adipocytes were then treated with TNF-α and insulin-stimulated glucose uptake was measured. We found that knockdown of Brd2 itself had marginal effects on baseline and insulin-stimulated glucose uptake (Figure 4H). As expected, TNF-α inhibited insulin-stimulated glucose uptake in control cells, but cells with knocked-down Brd2 maintained their insulin sensitivity (Figure 4I). This result indicates that Brd2 is required for TNF-α signalling to induce insulin resistance in vitro.
Brd2 inhibits β-cell growth and insulin transcription
The hyperinsulinaemia and pancreatic islet expansion of obese animals and humans are usually considered to be compensatory responses of the organism to increased demand for insulin due to obesity-induced insulin resistance. Thus an interesting question arose: if obese w/lo mice are more tolerant to glucose challenge and have lower blood glucose than w/w mice, why do they still manifest hyperinsulinaemia and islet expansion? Given the role of Brd2 and its family of related proteins as cell-cycle and developmental regulators, we hypothesized first that β-cell proliferation might be uncoupled from glucose homoeostasis and β-cells might proliferate if Brd2 levels were reduced.
The long form of Brd2 is highly expressed in the pancreas of w/w mice (Figure 1B). We used indirect immunofluorescence to identify the cells of the pancreas that express Brd2 and found that Brd2 is exclusively expressed in islet β-cells, the insulin-producing cells of the pancreas (Figure 5A). This result suggested that β-cell function might be deregulated if Brd2 levels were reduced. We then hypothesized that, rather than a chronic manifestation of increased demand for insulin, the observed islet expansion in obese w/lo mice might be a driving force behind the obesity, if reduced Brd2 exerts a direct positive effect on β-cell mass.
Figure 5. Brd2 regulates pancreatic β-cell biology.

(A) Immunofluorescence images of a pancreatic islet from a 16-week-old w/w mouse stained with anti-Brd2 (αBrd2) and anti-insulin (αinsulin) antibodies, and DAPI (4′,6-diamidino-2-phenylindole) to visualize nuclei, showing β-cell-restricted expression. Scale bar, 50 μm. (B) Growth curves of β-TC-6 cells stably transfected with plasmids for overexpression of the long form of Brd2 (broken line), shRNA (dotted line) or control (solid line) as in Figure 4 (n = 3; *P<0.05). (C) Representative BrdU assay of β-TC-6 cells stably transfected with plasmids as above. Percentages of cells in S-phase of the cell cycle are shown. (D) Quantification of flow cytometry results (n = 3; *P<0.05). Open bar, mock; light-grey bar, pSiC; solid black bar, pSiBrd2 knockdown; coarse hatched bar, pcDNA control; fine hatched bar, pmBrd2 overexpression. (E) Real-time RT–PCR of insulin mRNA levels compared with β-actin in stably transfected β-TC-6 cells cultured in 0.5 or 25 mM glucose medium (n = 3; *P<0.05 and **P<0.01). Open bar, mock; light-grey bar, pSiC; closed bar, pSiBrd2 knockdown; vertical striped bar, pcDNA control; horizontal striped bar, pmBrd2 overexpression. (F) Pancreatic islets of w/lo mice and w/w mice at 8 weeks of age, before onset of obesity. Scale bar, 100 μm.
Therefore we manipulated Brd2 expression in β-TC-6 cells, a murine insulinoma cell line, to model Brd2 function in β-cells. We stably transfected the cells with plasmids that express the long form of mouse Brd2 or Brd2 shRNA and counted cells over time (Figure 5B). Compared with controls, knockdown of Brd2 accelerated cell proliferation, but overexpression of Brd2 reduced cell proliferation and increased the number of dead cells (Figure 5D). Specifically, BrdU (bromodeoxyuridine) incorporation studies showed that knockdown of Brd2 promoted the entry into S-phase (Figure 5C; quantification is shown in Figure 5D). Conversely, overexpression of Brd2 reduced the number of cells entering S-phase and increased the number of sub- G0/G1 (presumably apoptotic) events (Figure 5D). These results suggest that the long form of Brd2 normally functions in β-cells to restrict β-cell proliferation, offering an interesting contrast with lymphoid cells and fibroblasts, in which ectopic expression of the short form of Brd2 promotes proliferation [14] and drives malignancy in Eμ-Brd2 transgenic mice [28].
Given the function of Brd2 as a transcriptional co-regulator, its high expression in β-cells and the hyperinsulinaemia of w/lo mice, we next hypothesized that Brd2 is a co-repressor of insulin transcription [21]. Therefore we measured insulin mRNA levels in the transfected β-TC-6 cells with real-time RT (reverse transcription)–PCR. Knockdown of Brd2 increased insulin transcription in low-glucose medium (0.5 mM), but did not further increase insulin transcription stimulated by high-glucose medium (25 mM; Figure 5E). Conversely, overexpression of the long form of Brd2 (pmBrd2) inhibited insulin transcription in β-cells cultured in both low and high glucose (Figure 5E), which supports the hypothesis. Taken together, these results suggested that pancreatic islet expansion and hyperinsulinaemia in obese w/lo mice are not an adaptive response to glucose intolerance, but are direct consequences of reduced Brd2 levels in pancreatic β-cells. We therefore returned to young w/lo mice and gender-matched w/w mice at 8 weeks of age and examined their islets, before the onset of obesity. We found significant islet expansion (Figure 5F). The median number of β-cells per field of view of young w/w mouse islets was 50 cells (with 25 and 75 %percentiles of 33.5 and 83.5 cells; n = 5), but for young w/lo mouse islets the number was 222 cells (with 25 and 75 % percentiles of 191.2 and 579.0 cells; n = 5), which supports the hypothesis that islet expansion is a primary event in w/lo mice.
Brd2 knockdown promotes adipogenic differentiation in vitro
In obesity, adipocytes expand both in size and number to accommodate increased energy storage. Adipogenesis, the process by which adipocyte precursors develop into adipocytes, is regulated by a group of specific transcription factors. Because we found above that Brd2 probably functions as a co-repressor of insulin transcription, we wondered whether a similar co-repression mechanism might be at work in adipocytes. Thus we returned to the 3T3-L1 system. Pre-adipocytes were stably transfected with the same plasmids to overexpress or knockdown Brd2 (Figures 4 and 5), then adipogenic differentiation was induced with insulin/dexamethasone/isobutylmethylxanthine. We found that Brd2 knockdown promoted adipogenic differentiation, as shown qualitatively (Figure 6B) and quantitatively (Figure 6C); conversely, Brd2 overexpression strongly suppressed adipogenic differentiation (Figures 6B and 6C). In addition, Brd2 overexpression inhibited the induction of PPAR-γ (peroxisome-proliferator-activated receptor-γ) and ACC (acetyl-CoA carboxylase), two markers of adipogenesis (Figure 6E). These results suggest that Brd2 normally functions to inhibit adipogenic differentiation. Consistent with this interpretation, endogenous Brd2 expression in 3T3-L1 pre-adipocytes declines during adipogenic differentiation (Figure 6F). In control experiments, we confirmed that, in the presence of a PPAR-γ agonist (pioglitazone), wild-type 3T3-L1 pre-adipocytes differentiate as fully as pre-adipocytes in which Brd2 has been knocked down (see Supplementary Figure S1A available at http://www.BiochemJ.org/bj/425/bj4250071add.htm).
Figure 6. Brd2 inhibits adipogenic differentiation and co-represses PPAR-γ transcription.

(A) Schematic of Brd2 structure [4,14] and reported interacting proteins [18,19]. BD, bromodomain; NLS, nuclear localization signal; ET, extraterminal domain; E2F, E2F-complex association domain [19]; ++, basic domain; − −, acidic domain. Human and mouse Brd2 proteins are approx. 91 % identical. Associated proteins are discussed in the Introduction. CAF, chromatin assembly factor; NAP, nucleosome assembly protein. (B) Manipulation of Brd2 in 3T3-L1 adipocytes with Brd2 shRNA, Brd2 overexpression or controls. Oil Red O staining visualizes lipid content in cells that have undergone adipogenic differentiation. Adipogenic differentiation of 3T3-L1 adipocytes from pre-adipocytes was performed as described previously [58]. UD, undifferentiated (see the legend to Figure 4 for plasmid abbreviations). (C) Quantification of lipid content in transfected 3T3-L1 cells after adipogenic differentiation (n = 3; *P< 0.05). Lipid content using Oil Red O stain was determined as described previously [59]. (D) Immunoblot analysis verifies increased Brd2 expression in 3T3-L1 pre-adipocytes transfected with pmBrd2 or controls. (E) Overexpression of Brd2 with pmBrd2 suppresses normal expression of two adipogenic markers, PPAR-γ and ACC, as detected by RT–PCR at different time points during adipogenic differentiation. (F) Kinetics of Brd2 and PPAR-γ expression in 3T3-L1 pre-adipocytes during adipogenesis. (G) Co-immunoprecipitation (IP) of Brd2 and PPAR-γ in 3T3-L1 cells 3 days after induction of differentiation. IgG, non-specific control. (H) Brd2 inhibits PPAR-γ activity, as assessed by transcriptional activity of a PPAR-γ-driven luciferase reporter in 3T3-L1 pre-adipocytes, in response to manipulation of Brd2 (n = 3; *P< 0.05).
Brd2 co-represses PPAR-γ
PPAR-γ is the master regulator of adipogenesis and is regulated in part by multiprotein transcriptional complexes composed of co-regulators. The activity of PPAR-γ is suppressed by a co-repressor protein complex before being activated. Upon activation through ligand binding, a co-activator complex replaces the inhibitory co-repressor complex. We have previously characterized Brd2-associated transcriptional protein complexes and reported that, among other factors, such complexes contain Hats/Hdacs and nucleosome remodelling factors, including members of the SWI/SNF complex [18]. These transcriptional co-factors are also found in the protein complexes that co-regulate PPAR-γ transcriptional activity [37]. Thus we hypothesized that Brd2-dependent complexes and PPAR-γ-dependent complexes share overlapping composition and activities (Figure 6A) and that Brd2 opposes PPAR-γ action as a direct co-repressor. It follows first that Brd2 and PPAR-γ should associate with each other. Therefore we performed protein co-immunoprecipitation of Brd2 and PPAR-γ with cell lysates from 3T3-L1 adipocytes 3 days after induction of differentiation, when Brd2 and PPAR-γ are co-expressed (Figure 6F). We found that Brd2 associates with PPAR-γ in vitro in a ligand-independent fashion (Figure 6G). To determine whether the binding of Brd2 and PPAR-γ regulates PPAR-γ transcriptional activity, 3T3-L1 pre-adipocytes were transiently transfected with a luciferase reporter plasmid that contains a triplicated PPAR-responsive element (PPRE-3X-Luc), pPPAR-γ, Renilla luciferase and effector plasmids (pSiBrd2, pmBrd2 and controls); then luciferase activity was measured. We found that reduced Brd2 expression increased PPAR-γ transcriptional activity, whereas increased Brd2 expression reduced PPAR-γ transcriptional activity (Figure 6H). These results suggest that Brd2 acts as a PPAR-γ co-repressor; Brd2 knockdown increases PPAR-γ activity and facilitates adipogenesis, consistent with the dramatically increased adipose tissue burden of w/lo mice.
DISCUSSION
The newly described bromodomain protein Brd2 performs profound, diverse and non-redundant functions in the life of specialized cells. Although elevated expression of Brd2 has been linked to cancer, the broader normal functions of the ubiquitous double bromodomain-containing BET class of transcriptional co-regulators are not well understood. In mice, knockout of the closely related gene Brd4 is lethal, and haploinsufficiency poses severe problems for cell cycle and organ development [32]. Clues from studies of the yeast single bromodomain homologues BDF1 and BDF2, and the Drosophila homologue fsh, imply that these proteins function as chromatin-directed developmental regulators with maternal effects in animals [24,27]. Bromodomain-containing protein complexes respond to signal transduction to mediate protein–protein interactions among TAFs, histone-modification enzymes and the chromatin remodelling machinery at specific promoters during development and in differentiated cells of the adult, thereby establishing repressive or transcriptionally active chromatin. Such a broad mechanism makes it difficult to establish specific paradigms for action on target genes or to predict how mutations cause specific diseases; we suspect that brd2 mutations functionally resemble swi/snf mutations in their networked pleiotropic effects [20]. Our attempts to generate a Brd2-knockout mouse line with RRE050, a Brd2 gene-trapped murine ES cell-line failed, consistent with Brd4 knockout attempts [32]. Shang et al. [33] found that brd2(−/−)null embryos derived from RRE050 ES cells showed a phenotype different from brd4(−/−)-null embryos; they hypothesized a deficiency in neuronal differentiation and brain development. This phenotype has been confirmed very recently [34]. However, our gene disruption in the 5′ region of the Brd2 gene in the ES cell line RRT234, which reduced rather than eliminated Brd2 gene expression, overcame problems of early lethality to give w/lo mice. The unexpected severely obese phenotype of the w/lo mice indicates that, among other functions, Brd2 in mammals is also a regulator of energy balance. The body weights that adult w/lo mice attain (Figure 2B) are among the highest reported for obese mouse models and rival adiponectin transgenic mice on the ob/ob background [38]. Surprisingly, w/lo mice have long lives (~ 20 months) despite their obesity and do not appear to die of tumours, haematological or immunological abnormalities, opportunistic infections, pulmonary disease or musculoskeletal disorders. The discovery of these mice was completely serendipitous and could not have been anticipated from the ES cells we used.
The w/lo mice pose two problems. First, why do they gain weight with a well-defined mutation located in a gene not previously thought to play any role in energy metabolism? Secondly, upon development of severe obesity, why does the same mutation protect them from the development of Type 2 diabetes, which marks a major departure from numerous other mouse models of obesity?
In animals, insulin is the major regulator of anabolism. Maintenance of proper β-cell mass is crucial for energy homoeostasis [39], and failure of β-cell production of insulin is a characteristic defect in both Type 1 and Type 2 diabetes. In most obese animals, hyperinsulinaemia and β-cell expansion are compensatory for insulin resistance. Pancreatic β-cell mass is regulated by at least four independent mechanisms: β-cell apoptosis, β-cell replication by mitosis, β-cell size and β-cell neogenesis from pancreatic progenitors. The issue of whether the reduction in human β-cell mass in diabetes results from increased β-cell apoptosis, reduced β-cell neogenesis/replication, or both factors, remains unresolved [40]. However, recent evidence indicates that maintenance of β-cell mass in young adult mice is primarily due to β-cell replication [41]; proper control of proliferation is critical to maintain sufficient β-cells to produce enough insulin. The transition from G1- to S-phase of the cell cycle is controlled by the D-type cyclins in conjunction with E2Fs, cyclin-dependent kinases and their inhibitors, which are important for β-cell growth; if D-type cyclin activity falls below a critical level, β-cell mitogenesis is almost totally ablated [42].
The expression of different forms of Brd2 varies significantly among mouse tissues (Figure 1B), with the long form of Brd2 highly expressed in β-cells (Figure 5A). We have established previously that the short form of Brd2 is a pro-mitotic factor in lymphoid cells [28], through its transcriptional activation of E2F-dependent cyclin genes, including cyclin D1 [19]. β-Cell expansion and hyperinsulinaemia in obese w/lo mice appear to be uncoupled from hyperglycaemia, which suggests Brd2 plays a direct non-sensing role in the regulation of β-cell mass and insulin production. We tested this hypothesis in vitro in β-TC-6 cells, where knockdown of Brd2 forces more cells into S-phase and promotes proliferation, which suggests that the long form of Brd2 normally acts as a negative regulator of the G1 checkpoint, to oppose β-cell mitogenesis and thus restrict β-cell mass. Hyperinsulinaemia in the absence of glucose intolerance creates chronic lower blood glucose; glucose is rapidly cleared from the blood and stored as fat. Thus we interpret w/lo mice: chronic hyperphagia addresses their low blood glucose, but in the absence of increased physical activity to balance increased storage they become obese.
Adipogenesis is also crucial for energy balance. Deregulated adipogenesis can cause lipoatrophy or obesity; both are harmful. A group of transcription factors, such as PPAR-γ and C/EPBα (CCAAT enhancer-binding protein-α), play critical roles in this process, especially PPAR-γ: its function is necessary and probably sufficient for adipogenesis [43]. Interestingly, Brd2 protein expression is high in pre-adipocytes and falls to barely detectable amounts in differentiated adipocytes, which suggested to us that Brd2 might normally function as a negative regulator of the adipogenic differentiation programme, possibly as a transcriptional co-repressor of PPAR-γ. The activity of PPAR-γ is regulated by ligands and transcriptional co-regulators. We found that Brd2 binds to PPAR-γ in vitro and this binding cannot be reversed by the PPAR-γ agonist pioglitazone. Furthermore, the long form of Brd2 inhibits PPAR-γ transcriptional activity, which we interpret as an explanation for the increased adipogenic differentiation in 3T3-L1 cells with knocked-down Brd2. In this respect, Brd2 functionally resembles another chromatin-directed transcriptional co-regulator, Adp, which has been reported to bind histones and Hdac3, thus inhibiting PPAR-γ activity and adipogenesis [44]. We speculate that the increased energy storage brought about by elevated insulin works together with reduced co-repression of PPAR-γ-driven adipogenesis to promote severe obesity in w/lo mice.
Key factors also clearly protect obese w/lo mice from glucose intolerance. The RER results suggest constant lipid oxidation reduces fat accumulation, but this pathway is insufficient to prevent severe obesity. Normally, chronic hyperglycaemia and increased blood NEFAs in obesity impair β-cell function through nutrient-induced β-cell apoptosis. The reduced feeding blood glucose and normal NEFA levels in obese w/lo mice probably preserve β-cells in these mice. Interestingly, obese w/lo phenocopy mice that harbour a β-cell-restricted deletion of Pten (encoding phosphatase and tensin homologue deleted on chromosome 10), which clear injected glucose better than wild-type mice and have increased islet numbers and total mass of islets, as well as having lower fasting blood glucose and resistance to β-cell apoptosis [45], suggesting that Brd2 may be a transcriptional effector of the Pten/Akt pathway in β-cells.
Furthermore, adiponectin, an important adipokine, is probably relevant in this model. Adiponectin is a protective insulin-sensitizing factor that can temper obesity-induced insulin resistance [46]. But a paradox has been noted: in most obese animals, including humans, an increased number of adipocytes ought to produce more adiponectin; however, observed levels of adiponectin are in fact lower. It is likely that insulin resistance plays a role in this phenomenon, reducing blood adiponectin and vice versa, thus forming a ‘vicious circle’ of diminished adiponectin [47]. Interestingly, unlike in other animal models of obesity, the plasma adiponectin level in obese w/lo mice is increased (31.3 ± 3.0 μg/ml), thus it is reasonable to attribute superior glucose sensitivity in obese w/lo mice partly to their elevated adiponectin; hyperadiponectinaemia is associated with improved metabolic profiles of obese patients [48]. This serum adiponectin concentration matches the hyperadiponectinaemia of male adiponectin transgenic mice [49]; approximately half this concentration is sufficient to rescue ob/ob mice from Type 2 diabetes [38]. It will be important to determine whether Brd2 normally co-represses adiponectin transcription, similar to its action on insulin transcription and the PPAR-γ-dependent promoter we tested. Scherer and co-workers have noted an interesting ‘feed-forward’ relationship between PPAR-γ agonist action and hyperadiponectinaemia [38]. Given the evidence that Brd2 reduction phenocopies thiazolidinedione action, promoting adipogenesis, it is possible that the transcriptional activity of Brd2 provides a functional link between the two processes.
Finally, it is well established that production of pro-inflammatory cytokines from adipose cells accumulated in liver and muscle during obesity promotes insulin resistance [50]. Unexpectedly, despite their severe obesity, increased serum levels of pro-inflammatory cytokines, such as TNF-α and IL-1β, hepatosteatosis and highly elevated transcription of these cytokine genes in WAT (white adipose tissue; see Supplementary Figure S1B), obese w/lo mice are more tolerant than w/w mice to glucose challenge; blood glucose levels in both the fed state and during an IGTT fall short of the levels attained in w/w mice. Interestingly, aP2 (adipocyte fatty acid-binding protein 4)-deficient mice appear to uncouple dietary obesity from insulin resistance in part through failure of TNF-α production [51], yet we encountered a surprise while investigating fat depots: adipose tissue of obese w/lo mice lack infiltration by F4/80+ inflammatory macrophages (Figure 4D), a phenotype previously reported for adiponectin transgenic mice [38]. It is currently unclear whether this lack of macrophage recruitment is a specific consequence of elevated serum adiponectin or a lack of adipocyte necrosis; unlike adiponectin transgenic mice, obese w/lo mice have adipocyte hypertrophy but not hyperplasia. Furthermore, in vitro glucose uptake experiments with 3T3-L1 adipocytes reveal that Brd2 is required for TNF-α-induced insulin resistance. Thus (i) elevated adiponectin, (ii) increased BAT and elevated uncoupling proteins, (iii) high quality adipose tissue without infiltration by inflammatory cells, and (iv) molecular uncoupling of elevated TNF-α signalling from insulin resistance work together to protect obese w/lo mice from eventual Type 2 diabetes.
Certain naturally occurring genetic environments, such as MHO or MONW human subpopulations [3], uncouple obesity from the insulin resistance and Type 2 diabetes commonly found in the general population, thus a substantial fraction of obese patients avoid insulin resistance. There is intense interest in obtaining greater mechanistic understanding of the genes involved, first to deepen our understanding of the basic biology of insulin resistance and secondly to identify possible therapeutic targets. The location of the BRD2 gene within the MHC class II locus of both humans and mice, situated among genes devoted to antigen presentation but itself unrelated to them, nevertheless suggests intriguing links to the immune system, particularly to inflammation. The lack of development of inflammation-associated glucose intolerance in these mice is unlikely to be due to a global deficiency in antigen presentation, because lacZ gene disruption of the Brd2 gene in RRT234 ES cells did not impair expression of MHC class II molecules in the periphery. Immune and metabolic systems are closely related, developmentally and functionally, and disorders in the immune system are associated with obesity and diabetes [52]. Although Brd2 expression in the adult mouse brain is low, it remains possible that Brd2 in the brain plays a role in the regulation of energy metabolism; however, given the chromosomal location of the BRD2 gene at 6p21.3, we nevertheless favour a role in immunity and inflammation. Our present data suggest that lowered levels of Brd2 reduce inflammatory signal transduction, which lead us to speculate that the reduced inflammatory profile of MHO humans might be related to polymorphism at the BRD2 locus. There are no published studies of such polymorphisms at the time of writing.
We speculate further that the strong influence of Brd2 levels on insulin production and action suggest Brd2 is probably a promising target for diabetes treatment, but also imply that overactive Brd2 might cause diabetes. Interestingly, the MHC class II in humans is the most strongly linked locus to Type 1 diabetes [53]. The molecular mechanisms of this phenomenon are still not fully understood. ‘Islet insult’ or β-cell death is probably required for the aetiology of Type 1 diabetes. Notably, Brd2 not only regulates β-cell growth and insulin transcription in vitro, but overexpression of Brd2 also increases the number of apoptotic cells (Figure 5D), which, together with the potential role of Brd2 in the immune system, suggests further studies to investigate a possible role of Brd2 in the aetiology of Type 1 diabetes.
Body energy balance is regulated by a complex network of signals from both inside and outside of the body. More than 100 genes have been implicated in body weight regulation [54]. Emerging evidence reveals critical roles for transcriptional co-regulators in energy balance, such as PGC-1, a PPAR-γ co-activator, which controls energy storage and expenditure by regulating the development and function of adipose tissue, skeletal muscle and liver [55]. Similarly, PRDM16 (PR-domain-containing lb) interacts with the transcriptional co-regulators PGC-1α and PGC-1β to control an important differentiation switch between BAT and skeletal muscle [56]. Thus specific MDa complexes of transcription-factor-associated co-regulators are essential for proper control of energy metabolism. We suggest Brd2 and its associated protein complexes should be included in this group of transcriptional co-regulators that control energy balance.
Supplementary Material
Acknowledgments
We note the National Institutes of Health sponsorship of both the National Heart Lung and Blood Institute –BayGenomics, and the National Center for Research Resources – Mutant Mouse Regional Resource Center, and we thank the Murine Targeted Genomics Laboratory at the University of California, Davis, CA, U.S.A. We also thank H. Li (Joslin Diabetes Center, Boston, MA, U.S.A.) for serological assays; Z. Arany (Brigham and Women’s Hospital, Boston, MA, U.S.A); and the Transgenic Core Facility, Flow Cytometry Core Facility and M. Kelly of the Section of Endocrinology, Diabetes and Nutrition (all at Boston University School of Medicine) for joint projects, helpful discussion and reagents. We thank B. Corkey, S. Farmer, S. Fried, P. Pilch, N. Ruderman and other members of the Boston Obesity Nutrition Research Center (P30 DK046200) for useful discussions. We thank D. Faller, A. Sinha and other members of the Cancer Research Center for invaluable help and support. This paper is dedicated to the memory of Edward M. Kennedy, United States Senator for Massachusetts, who was an ardent supporter of Boston Medical Center and its mission.
FUNDING
This work was supported by grants from the American Cancer Society; the National Institutes of Health [grant number T32 AI007309]; and pilot funds awarded to G.V.D. from the Boston Area Diabetes Endocrinology Research Center [grant number P30 DK057521].
Abbreviations used
- ACC
acetyl-CoA carboxylase
- BAT
brown adipose tissue
- BET
bromodomain and extraterminal domain
- BrdU
bromodeoxyuridine
- ES
embryonic stem
- fsh
female sterile (1) homeotic
- Hat
histone acetyltransferase
- Hdac
histone deacetylase
- HFD
high-fat diet
- IGTT
intraperitoneal glucose tolerance test
- IL-1β
interleukin-1β
- MHO
metabolically healthy but obese
- MONW
metabolically obese but normal weight
- NCBI
National Center for Biotechnology Information
- NEFA
non-esterified fatty acid (‘free fatty acid’)
- PPAR-γ
peroxisome-proliferator-activated receptor-γ
- PGC
PPAR-γ co-activator
- Pten
phosphatase and tensin homologue deleted on chromosome 10
- RER
respiratory exchange ratio
- RT
reverse transcription
- shRNA
small-hairpin RNA
- SVF
stromal vascular fraction
- SWI/SNF
switch mating type/sucrose non-fermenting
- TAF
TATA-box-binding protein-associated factor
- TNF-α
tumour necrosis factor-α
- Ucp
uncoupling protein
Footnotes
AUTHOR CONTRIBUTION
Fangnian Wang performed most of the experiments with the necessary assistance of Hongsheng Liu, Wanda Blanton and Anna Belkina, who helped Fangnian Wang with the flow cytometry. Nathan LeBrasseur performed body composition, calorimetry and respiratory exchange ratio measurements. Fangnian Wang wrote a first draft of the paper, and all authors consulted on the final version. Gerald Denis conceptualized the project, directed the experiments, contributed to the calculation of the statistics, finalized the paper and wrote the grants that provided funding.
References
- 1.Zimmet P, Alberti KG, Shaw J. Global and societal implications of the diabetes epidemic. Nature. 2001;414:782–787. doi: 10.1038/414782a. [DOI] [PubMed] [Google Scholar]
- 2.Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27:1047–1053. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- 3.Succurro E, Marini MA, Frontoni S, Hribal ML, Andreozzi F, Lauro R, Perticone F, Sesti G. Insulin secretion in metabolically obese, but normal wt, and in metabolically healthy but obese individuals. Obesity. 2008;16:1881–1886. doi: 10.1038/oby.2008.308. [DOI] [PubMed] [Google Scholar]
- 4.Beck S, Hanson I, Kelly A, Pappin DJC, Trowsdale J. A homologue of the Drosophila female sterile homeotic (fsh) gene in the class II region of the human MHC. DNA Seq. 1992;2:203–210. doi: 10.3109/10425179209020804. [DOI] [PubMed] [Google Scholar]
- 5.Salter-Cid L, Pasquier L, Flajnik M. RING3 is linked to the Xenopus major histocompatibility complex. Immunogenetics. 1996;44:397–399. [PubMed] [Google Scholar]
- 6.Ting JP, Trowsdale J. Genetic control of MHC class II expression. Cell. 2002;109:S21–S33. doi: 10.1016/s0092-8674(02)00696-7. [DOI] [PubMed] [Google Scholar]
- 7.Florence B, Faller DV. You bet-cha: a novel family of transcriptional regulators. Front Biosci. 2001;6:D1008–D1018. doi: 10.2741/florence. [DOI] [PubMed] [Google Scholar]
- 8.Zeng L, Zhou MM. Bromodomain: an acetyl-lysine binding domain. FEBS Lett. 2002;513:124–128. doi: 10.1016/s0014-5793(01)03309-9. [DOI] [PubMed] [Google Scholar]
- 9.Denis GV. Bromodomain motifs and “scaffolding”? Front Biosci. 2001;6:D1065–D1068. doi: 10.2741/a668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.LeRoy G, Rickards B, Flint SJ. The double bromodomain proteins Brd2 and Brd3 couple histone acetylation to transcription. Mol Cell. 2008;30:51–60. doi: 10.1016/j.molcel.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tamkun JW, Deuring R, Scott MP, Kissenger M, Pattatucci AM, Kaufman TC, Kennison JA. brahma: a regulator of Drosophila homeotic genes structurally related to the yeast transcriptional activator SWI2/SNF2. Cell. 1992;68:561–572. doi: 10.1016/0092-8674(92)90191-e. [DOI] [PubMed] [Google Scholar]
- 12.Wang W, Côte J, Xue Y, Zhou S, Khavari PA, Biggar SR, Muchardt C, Kalpana GV, Goff SP, Yaniv M, Workman JL, Crabtree GR. Purification and biochemical heterogeneity of the mammalian SWI-SNF complex. EMBO J. 1996;15:5370–5382. [PMC free article] [PubMed] [Google Scholar]
- 13.Denis GV, Green MR. A novel, mitogen-activated nuclear kinase is related to a Drosophila developmental regulator. Genes Dev. 1996;10:261–271. doi: 10.1101/gad.10.3.261. [DOI] [PubMed] [Google Scholar]
- 14.Sinha A, Faller DV, Denis GV. Bromodomain analysis of Brd2-dependent transcriptional activation of cyclin A. Biochem J. 2005;387:257–269. doi: 10.1042/BJ20041793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thorpe KL, Beck S. DNA sequence and structure of the mouse RING3 gene: identification of variant RING3 transcripts. Immunogenetics. 1998;48:82–86. doi: 10.1007/s002510050406. [DOI] [PubMed] [Google Scholar]
- 16.Nakamura Y, Umehara T, Nakano K, Jang MK, Shirouzu M, Morita S, Uda-Tochio H, Hamana H, Terada T, Adachi N, et al. Crystal structure of the human BRD2 bromodomain: insights into dimerization and recognition of acetylated histone H4. J Biol Chem. 2007;282:4193–4201. doi: 10.1074/jbc.M605971200. [DOI] [PubMed] [Google Scholar]
- 17.Kornberg RD. Mediator and the mechanism of transcriptional activation. Trends Biochem Sci. 2005;30:235–239. doi: 10.1016/j.tibs.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 18.Denis GV, McComb ME, Faller DV, Sinha A, Romesser PB, Costello CE. Identification of transcription complexes that contain the double bromodomain protein Brd2 and chromatin remodeling machines. J Proteome Res. 2006;5:502–511. doi: 10.1021/pr050430u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Denis GV, Vaziri C, Guo N, Faller DV. RING3 kinase transactivates promoters of cell cycle regulatory genes through E2F. Cell Growth Differ. 2000;11:417–424. [PMC free article] [PubMed] [Google Scholar]
- 20.Holstege FC, Jennings EG, Wyrick JJ, Lee TI, Hengartner CJ, Green MR, Golub TR, Lander ES, Young RA. Dissecting the regulatory circuitry of a eukaryotic genome. Cell. 1998;95:717–728. doi: 10.1016/s0092-8674(00)81641-4. [DOI] [PubMed] [Google Scholar]
- 21.Denis GV. Duality in bromodomain-containing protein complexes. Front Biosci. 2001;6:D849–D852. doi: 10.2741/denis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chua P, Roeder GS. Bdf1, a yeast chromosomal protein required for sporulation. Mol Cell Biol. 1995;15:3685–3696. doi: 10.1128/mcb.15.7.3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matangkasombut O, Buratowski RM, Swilling NW, Buratowski S. Bromodomain factor 1 corresponds to a missing piece of yeast TFIID. Genes Dev. 2000;14:951–962. [PMC free article] [PubMed] [Google Scholar]
- 24.Matangkasombut O, Buratowski S. Different sensitivities of bromodomain factors 1 and 2 to histone H4 acetylation. Mol Cell. 2003;11:353–363. doi: 10.1016/s1097-2765(03)00033-9. [DOI] [PubMed] [Google Scholar]
- 25.Chang YL, King B, Lin SC, Kennison JA, Huang DH. A double-bromodomain protein, FSH-S, activates the homeotic gene Ultrabithorax through a critical promoter-proximal region. Mol Cell Biol. 2007;27:5486–5498. doi: 10.1128/MCB.00692-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mozer BA, Dawid IB. Cloning and molecular characterization of the trithorax locus of Drosophila melanogaster. Proc Natl Acad Sci USA. 1989;86:3738–3742. doi: 10.1073/pnas.86.10.3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Digan ME, Haynes SR, Mozer BA, Dawid IB, Forquignon F, Gans M. Genetic and molecular analysis of fs(1)h, a maternal effect homeotic gene in Drosophila, Dev. Biol. 1986;114:161–169. doi: 10.1016/0012-1606(86)90392-1. [DOI] [PubMed] [Google Scholar]
- 28.Greenwald R, Tumang JR, Sinha A, Currier N, Cardiff RD, Rothstein TL, Faller DV, Denis GV. Eμ-BRD2 transgenic mice develop B cell lymphoma and leukemia. Blood. 2004;103:1475–1484. doi: 10.1182/blood-2003-06-2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu SY, Chiang CM. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J Biol Chem. 2007;282:13141–13145. doi: 10.1074/jbc.R700001200. [DOI] [PubMed] [Google Scholar]
- 30.Dey A, Chitsaz F, Abbasi A, Misteli T, Ozato K. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc Natl Acad Sci USA. 2003;100:8758–8763. doi: 10.1073/pnas.1433065100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dey A, Ellenberg J, Farina A, Coleman AE, Maruyama T, Sciortino S, Lippincott-Schwartz J, Ozato K. A bromodomain protein, MCAP, associates with mitotic chromosomes and affects G2-to-M transition. Mol Cell Biol. 2000;20:6537–6549. doi: 10.1128/mcb.20.17.6537-6549.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Houzelstein D, Bullock SL, Lynch DE, Grigorieva EF, Wilson VA, Beddington RSP. Growth and early postimplantation defects in mice deficient for the bromodomain-containing protein Brd4. Mol Cell Biol. 2002;22:3794–3802. doi: 10.1128/MCB.22.11.3794-3802.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shang E, Wang X, Wen D, Greenberg DA, Wolgemuth DJ. Double bromodomain-containing gene Brd2 is essential for embryonic development in mouse. Dev Dyn. 2009;238:908–917. doi: 10.1002/dvdy.21911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gyuris A, Donovan DJ, Seymour KA, Lovasco LA, Smilowitz NR, Halperin AL, Klysik JE, Freiman RN. The chromatin-targeting protein Brd2 is required for neural tube closure and embryogenesis. Biochim Biophys Acta. 2009;1789:413–421. doi: 10.1016/j.bbagrm.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tomita T, Doull V, Pollock HG, Krizsan D. Pancreatic islets of obese hyperglycemic mice (ob/ob) Pancreas. 1992;7:367–357. doi: 10.1097/00006676-199205000-00015. [DOI] [PubMed] [Google Scholar]
- 36.Kroder G, Bossenmaier B, Kellerer M, Capp E, Stoyanov B, Mühlhöfer A, Berti L, Horikoshi H, Ullrich A, Häring H. Tumor necrosis factor-α- and hyperglycemia-induced insulin resistance. Evidence for different mechanisms and different effects on insulin signaling. J Clin Invest. 1996;97:1471–1477. doi: 10.1172/JCI118569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rosen ED, Walkey CJ, Puigserver P, Spiegelman BM. Transcriptional regulation of adipogenesis. Genes Dev. 2000;14:1293–1307. [PubMed] [Google Scholar]
- 38.Kim JY, van de Wall E, Laplante M, Azzara A, Trujillo ME, Hofmann SM, Schraw T, Durand JL, Li H, Li G, et al. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J Clin Invest. 2007;117:2621–2637. doi: 10.1172/JCI31021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kahn SE. The importance of the β-cell in the pathogenesis of type 2 diabetes mellitus. Am J Med. 2000;108(Suppl 6a):2S–8S. doi: 10.1016/s0002-9343(00)00336-3. [DOI] [PubMed] [Google Scholar]
- 40.Rhodes CJ. Type 2 diabetes: a matter of β-cell life and death? Science. 2005;307:380–384. doi: 10.1126/science.1104345. [DOI] [PubMed] [Google Scholar]
- 41.Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic β-cells are formed by self-duplication rather than stem-cell differentiation. Nature. 2004;429:41–46. doi: 10.1038/nature02520. [DOI] [PubMed] [Google Scholar]
- 42.Kushner JA, Ciemerych MA, Sicinska E, Wartschow LM, Teta M, Long SY, Sicinski P, White MF. Cyclins D2 and D1 are essential for postnatal pancreatic β-cell growth. Mol Cell Biol. 2005;25:3752–3762. doi: 10.1128/MCB.25.9.3752-3762.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rosen ED, Sarraf P, Troy AE, Bradwin G, Moore K, Milstone DS, Spiegelman BM, Mortensen RM. PPARγ is required for the differentiation of adipose tissue in vivo and in vitro. Mol Cell. 1999;4:611–617. doi: 10.1016/s1097-2765(00)80211-7. [DOI] [PubMed] [Google Scholar]
- 44.Suh JM, Zeve D, McKay R, Seo J, Salo Z, Li R, Wang M, Graff JM. Adipose is a conserved dosage-sensitive antiobesity gene. Cell Metab. 2007;6:195–207. doi: 10.1016/j.cmet.2007.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stiles BL, Kuralwalla-Martinez C, Guo W, Gregorian C, Wang Y, Tian J, Magnuson MA, Wu H. Selective deletion of Pten in pancreatic β-cells leads to increased islet mass and resistance to STZ-induced diabetes. Mol Cell Biol. 2006;26:2772–2781. doi: 10.1128/MCB.26.7.2772-2781.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guerre-Millo M. Adiponectin: an update. Diabetes Metab. 2008;34:12–18. doi: 10.1016/j.diabet.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 47.Weyer C, Funahashi T, Tanaka S, Hotta K, Matsuzawa Y, Pratley RE, Tataranni PA. Hypoadiponectinemia in obesity and type 2 diabetes: close association with insulin resistance and hyperinsulinemia. J Clin Endocrinol Metab. 2001;86:1930–1935. doi: 10.1210/jcem.86.5.7463. [DOI] [PubMed] [Google Scholar]
- 48.Côté M, Mauriège P, Bergeron J, Alméras N, Tremblay A, Lemieux I, Després JP. Adiponectinemia in visceral obesity: impact on glucose tolerance and plasma lipoprotein and lipid levels in men. J Clin Endocrinol Metab. 2005;90:1434–1439. doi: 10.1210/jc.2004-1711. [DOI] [PubMed] [Google Scholar]
- 49.Combs TP, Pajvani UB, Berg AH, Lin Y, Jelicks LA, Laplante M, Nawrocki AR, Rajala MW, Parlow AF, Cheeseboro L, et al. A transgenic mouse with a deletion in the collagenous domain of adiponectin displays elevated circulating adiponectin and improved insulin sensitivity. Endocrinology. 2004;145:367–383. doi: 10.1210/en.2003-1068. [DOI] [PubMed] [Google Scholar]
- 50.Shoelson SE, Herrero L, Naaz A. Obesity, inflammation, and insulin resistance. Gastroenterology. 2007;132:2169–2180. doi: 10.1053/j.gastro.2007.03.059. [DOI] [PubMed] [Google Scholar]
- 51.Hotamisligil GS, Johnson RS, Distel RJ, Ellis R, Papaioannou VE, Spiegelman BM. Uncoupling of obesity from insulin resistance through a targeted mutation in aP2, the adipocyte fatty acid binding protein. Science. 1996;274:1377–1379. doi: 10.1126/science.274.5291.1377. [DOI] [PubMed] [Google Scholar]
- 52.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 53.Jahromi MM, Eisenbarth GS. Genetic determinants of type 1 diabetes across populations. Ann NY Acad Sci. 2006;1079:289–299. doi: 10.1196/annals.1375.044. [DOI] [PubMed] [Google Scholar]
- 54.Leibel RL. Energy in, energy out, and the effects of obesity-related genes. N Engl J Med. 2008;359:2603–2604. doi: 10.1056/NEJMe0808660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Handschin C, Spiegelman BM. Peroxisome proliferator-activated receptor gamma co-activator 1 co-activators, energy homeostasis, and metabolism. Endocr Rev. 2006;27:728–735. doi: 10.1210/er.2006-0037. [DOI] [PubMed] [Google Scholar]
- 56.Seale P, Bjork B, Yang W, Kajimura S, Chin S, Kuang S, Scimè A, Devarakonda S, Conroe HM, Erdjument-Bromage H, et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature. 2008;454:961–967. doi: 10.1038/nature07182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Longe H, Romesser PB, Rankin A, Faller DV, Eller MS, Gilchrest BA, Denis GV. Telomere homolog oligonucleotides induce apoptosis in malignant but not in normal lymphoid cells: mechanism and therapeutic potential. Int J Cancer. 2009;124:473–482. doi: 10.1002/ijc.23946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Student AK, Hsu RY, Lane MD. Induction of fatty acid synthetase synthesis in differentiating 3T3-L1 pre-adipocytes. J Biol Chem. 1980;255:4745–4750. [PubMed] [Google Scholar]
- 59.Ramírez-Zacarías JL, Castro-Muñozledo F, Kuri-Harcuch W. Quantitation of adipose conversion and triglycerides by staining intracytoplasmic lipids with Oil red O. Histochemistry. 1992;97:493–497. doi: 10.1007/BF00316069. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.