

Abstract
This article reviews recent advances in nanomaterial labels in electrochemical immunosensors and immunoassays. Various nanomaterial labels are discussed, including colloidal gold/silver, semiconductor nanoparticles, and markers loaded nanocarriers (carbon nanotubes, apoferritin, silica nanoparticles, and liposome beads). The enormous signal enhancement associated with the use of nanomaterial labels and with the formation of nanomaterial–antibody-antigen assemblies provides the basis for ultrasensitive electrochemical detection of disease-related protein biomarkers, biothreat agents, or infectious agents. In general, all endeavors cited here are geared to achieve one or more of the following goals: signal amplification by several orders of magnitude, lower detection limits, and detecting multiple targets.
Keywords: Nanomaterials, nanoparticles, immunosensors, immunoassays, carbon nanotubes, biomarkers
1. Introduction
Electrochemical immunosensors and immunoassays (EII) have recently attracted considerable interest because of their high sensitivity, low cost, and inherent miniaturization [1–6]. The principle of EII is based on a specific reaction of the antibody and antigen. For immunosensors, the immunologic materials are immobilized on an electrochemical transducer. For immunoassay, the immunological materials are immobilized on a solid supporting materials (microplate wells, magnetic beads, and polystyrene beads). After a sandwich or competitive immunoreaction, nanomaterial labels are attached to the transducer surface or supporting-material surface. Quantification is generally achieved by measuring the specific activity of a nanomaterial label after a releasing step or adding a substrate, i.e., its redox activities and enzyme activities. The novel characteristics of EII make them excellent candidates for clinical and point-of-care diagnosis of disease-related protein biomarkers. Disease biomarkers and biological agents are often present at ultra-low levels and require ultrasensitive methods for detection. Different strategies have been employed for amplifying the transducing signals of antibody-antigen interactions. Most conventional amplification strategies have relied on the use of labels, such as enzymes, electroactive molecules, redox complexes, and metal ions [1]. The emergence of nanotechnology is opening new horizons for the use of nanomaterial labels for signal amplification in EII [7–14]. The power and scope of such nanomaterials can be greatly enhanced by coupling them with immunoreactions and electrical processes (i.e., nanobioelectronics). The applications of nanomaterials in EII can be classified into two categories according to their functions: 1) nanomaterial-modified electrochemical transducers to facilitate antibody immobilization or improve electrochemical properties of transducers, such as low-background current, high signal-to noise ratio, and fast electron transfer; 2) nanomaterial-bimolecular conjugates as labels for EII. In particular, nanomaterial labels are showing the greatest promise for developing ultrasensitive EII [7, 14]. Antibodies (antigens) labeled with nanomaterials can retain their bioactivity and interact with their counterparts, and based on the electrochemical detection of those nanomaterials, the amount or concentration of analytes can be determined. The enormous signal enhancement associated with the use of nanomaterial amplifying labels and with the formation of nanomaterial–antibody-antigen assemblies provides the basis for ultrasensitive EII [7].
This article provides a review of the major advances and milestones in EII development based upon nanomaterial labels and their roles for biomarker detection. Particular attention will be given to new signal amplification and new emerging nanomaterial labels in EII. Such developments provide the pathway for diverse and exciting applications.
2. Nanomaterials used as EII labels
Many kinds of nanomaterials, including metal nanoparticles (gold, silver), semiconductor nanoparticles, enzyme-loaded carbon nanotubes (CNTs) and electroactive component-loaded nanovehicles (silica nanoparticle, polymer beads, and liposome beads), have been widely used in EII (Figure 1). The common characteristics of these nanomaterial labels in EII are providing signal amplifications comparing to the traditional metal ion labels, enzyme labels and redox probe labels.
Figure 1.

Nanomaterial labels in electrochemical immunosensors and immunoassays.
2.1 Colloidal gold/silver
The applications of colloidal gold/silver in immunoassays date back to the early 1970s when 5- to 50-nm colloidal gold particles were first used as electron-dense probes in electron microscopy and thus enabled sensitive, high-resolution immunocytochemistry [15]. The subsequent development of silver-enhanced methods then allowed gold labels to provide very specific and sensitive immunocytochemistry. Because of the intrinsic electrochemical characteristics of gold and silver, colloidal gold/silver was introduced into the research fields of EII. The use of colloidal gold as an electrochemical label for voltammetric monitoring of protein interactions was pioneered in 2000 by Gonzalez-Garcia et al. [16] and Dequaire et al. [17]. Costa-Garcia’s group thus demonstrated the monitoring of streptavidin-biotin interactions down to the 2.5-nM streptavidin level with adsorptive voltammetric measurements of the colloidal gold tags at a carbon-paste electrode [16]. Dequaire et al. reported a sensitive electrochemical immunoassay based on a colloidal gold label that was indirectly determined by anodic stripping voltammetry (ASV) at a single-use, carbon-based, screen-printed electrode (SPE) after oxidative gold metal dissolution in an acidic solution (Figure 2) [17]. The method was evaluated for a noncompetitive heterogeneous immunoassay of an immunoglobulin G (IgG), and a concentration as low as 3×10−12 M was determined, which was competitive with a colorimetric enzyme-linked immunosorbent assay (ELISA) or with immunoassays based on fluorescent europium chelated labels. To avoid the acid-dissolution step, Liu et al. developed an electrochemical magnetic immunosensor based on magnetic beads and gold nanoparticle labels [18]. The captured gold nanoparticle labels on the immunosensor surface were directly quantified by electrochemical stripping analysis. The stripping signal of gold nanoparticles is related to the concentration of target IgG in the sample solution. The detection limit of 0.02 μg mL−1 of IgG was obtained under optimum experimental conditions. Ambrosi et al. reported double-codified gold nanolabels for simultaneous electrochemical and optical immunoassays [19]. A built-in magnet graphite-epoxy-composite electrode allowed a sensibly enhanced adsorption and electrochemical quantification of the specifically captured gold nanoparticle labels on the paramagnetic bead surface. The detection limits for this double-codified nanoparticle-based assay were 52 and 260 pg of human IgG/mL for the spectrophotometric (horseradish peroxidase [HRP]-based) and electrochemical (AuNP-based) detections, respectively, much lower than those typically achieved by ELISA tests.
Figure 2.

Schematic representation of the noncompetitive heterogeneous electrochemical immunoassay based on a colloidal gold label. (Based on Ref. 17 with permission).
To further enhance the sensitivity of gold nanoparticle-label-based EII, various methods and technologies have been developed. For example, the nanoparticle-promoted precipitation of sliver on gold nanoparticle labels was used to amplify the transduction of antibody-antigen biorecognition events [20–23]. Chu et al. reported an electrochemical immunoassay based on silver on colloidal gold labels, which, after silver metal dissolution in an acidic solution, was indirectly determined by ASV at a glassy-carbon electrode [20]. The method was evaluated for a noncompetitive heterogeneous immunoassay of using IgG as a model analyte. The anodic stripping peak current of silver depended linearly on the IgG concentration over the range of 1.66 ng mL−1 to 27.25 ng mL−1 in a logarithmic plot. A detection limit as low as 1 ng mL−1 (i.e., 6×10−12 M) human IgG was achieved. The method was applied to detect schistosoma japonicum antibodies (SjAb) in rabbit serum [21]. A detection limit as low as 3.0 ng/mL SjAb was achieved. A potentiometric immunosensor based on gold nanoparticle labels and an ion-selective microelectrode was reported by Chumbimuni-Torres et al. [22]. This immunosensor is based on a sandwich immunoassay where the target mouse IgG antigen is captured by the primary anti-mouse gold substrate modified by IgG antibodies, followed by adding a secondary anti-mouse IgG antibody conjugated to gold nanoparticle tags and by catalytic silver enlargement onto the gold labels. The precipitated silver is oxidatively dissolved with hydrogen peroxide to yield dilute electrolyte backgrounds for the potentiometric detection of the released silver ions with a polymer-membrane, silver, ion-selective microelectrode. Silver-enhanced gold nanoparticle labels were also used to develop conductive immunosensors. For example, Velev and Kaler reported a conductive immunosensor using antibody-functionalized latex spheres and a microelectrode gap [23]. A sandwich immunoassay led to the binding of a secondary gold-nanoparticle-labeled antibody on the latex spheres that were located in the gap, followed by catalytic deposition of a silver layer “bridging” the two electrodes. Such a formation of conductive paths across interdigitated electrodes led to a measurable conductive signal and enabled the ultrasensitive detection of human IgG down to the 0.2-pM level. The method holds promise for creating miniaturized on-chip protein arrays.
A novel dendritic amplification procedure has been developed for quartz crystal microbalance (QCM) immunosensing by applying antibody-functionalized Au nanoparticles as the primary amplifying probe and a dendritic-type immunocomplex of protein A- and antibody-modified Au nanoparticles as the secondary amplifying probe [24]. The primary amplification of the recognition process is implemented via the interaction of the sensing interface with the antibody-functionalized Au nanoparticles, and the secondary dendritic amplification is performed through interaction with the immunocomplex of protein A- and antibody-modified Au nanoparticles. The frequency decreases of the primary-amplified and the secondary-amplified sensing process are observed to be linearly dependent upon the IgG concentration in the range of 10.9 ng mL−1 to 10.9 μg mL−1 with a detection limit of 3.5 ng mL−1, while in the absence of the amplification processes, the antigen-antibody recognition event can only be detected for an IgG concentration as high as 10.9 μg mL−1.
Recently, Mao et al. presented a new method based on cyclic accumulation of gold nanoparticles for detecting human immunoglobulin G (IgG) by ASV [25]. The dissociation reaction between dethiobiotin and avidin in the presence of biotin provides an efficient means for the cyclic accumulation of gold nanoparticles used for the final analytical quantification. The anodic peak current increases gradually with the increasing accumulation cycles. Five cycles of accumulation are sufficient for the assay. The low background of the proposed method is a distinct advantage, providing a possibility for determining at least 0.1 ng/mL human IgG.
An electrochemical immunoassay method based on Au nanoparticle-labeled immunocomplex enlargement was reported by Zhou et al. [26]. When the aggregates formed from nano-Au labeled goat-anti-human C-3 and nano-Au labeled rabbit-anti-goat IgG were immobilized on the electrode surface by the sandwich method (antibody/antigen/aggregate), the electrochemical signal of the electrode was enlarged greatly. The reported immunosensor could quantitatively determine complement C-3 in the range of 0.12, similar to 117.3 ng mL−1, and the detection limit was 0.02 ng mL−1.
Liao et al. reported an amplified electrochemical immunoassay by autocatalytic deposition of Au3+ onto gold nanoparticle labels [27]. By coupling the autocatalytic deposition with square-wave stripping voltammetry, enlarged gold nanoparticles were used as labels on goat anti-rabbit immunoglobulin G (GaRIgG-Au) and, thus, the rabbit immunoglobulin G (RIgG) analyte could be determined quantitatively. The detection limit was 0.25 pg mL−1 (1.6 fM), which is three orders of magnitude lower than that obtained by a conventional immunoassay using the same gold nanoparticle labels.
Recently Das et al. reported an ultrasensitive electrochemical immunosensor using the gold nanoparticle labels as electrocatalyts [28]. In this case, the gold nanoparticle labels were attached to the immmunosensor surface (indium tin oxide as substrate electrode) by sandwich immunoreaction; the signal amplification was achieved by catalytic reduction of p-nitrophenol (NP) to paminophenol (AP) and chemical reduction of p-quinone imine (QI) to AP by NaBH4 (Figure 3). Such dual amplification events gave a 1-fg mL−1 detection limit, and its linear range of measurement ranged from 1 fg mL−1 to 10 μg mL−1, which covered a 10-order concentration range.
Figure 3.
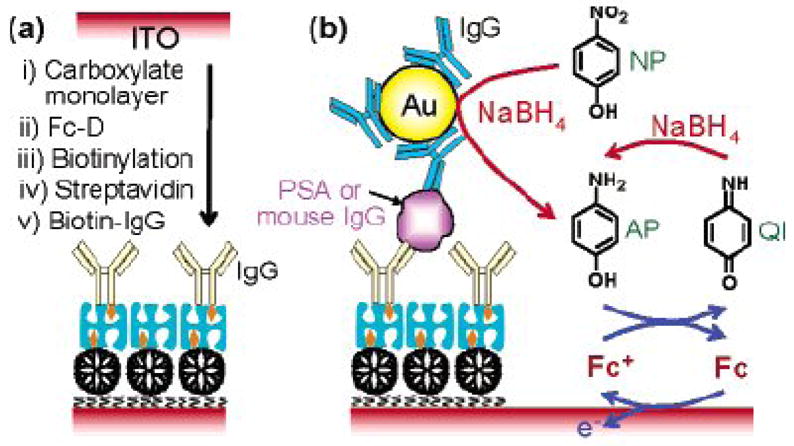
(a) Schematic representation of the preparation of an immunosensing layer. (b) Schematic view of electrochemical detection of mouse IgG or PSA. (Reproduced from Ref. 28 with permission)
2.2 Semiconductor nanoparticles
Owing to their unique (size-tunable fluorescent) properties, semiconductor (quantum dot, QD) nanoparticles have generated considerable interest for optical biodetection [29]. The intrinsic redox properties and the sensitive electrochemical stripping analysis of the metal components of semiconductor nanoparticles (CdS, PbS, and ZnS) cause the labels in the electrochemical biosensors to be very sensitive [7, 14]. The concept was first demonstrated by Wang’s group using semiconductor nanoparticle labels for the electrochemical DNA hybridization assay [30, 31], and then it was extended in EII [32]. Liu et al. [29] reported an electrochemical immunoassay protocol for the simultaneous measurements of multiple protein targets based on the use of different semiconductor nanoparticle tracers (CdS, ZnS and PbS). Carbamate linkage is used for conjugating the hydroxyl-terminated semiconductor nanoparticles with the secondary antibodies. The multianalyte electrical sandwich immunoassay involves a dual binding event, based on antibodies linked to the nanocrystal tags and magnetic beads (Figure 4). Each biorecognition event yields a distinct voltammetric peak whose position and size reflects the identity and level, respectively, of the corresponding antigen. The multi-protein electrical detection capability is coupled to the amplification feature of electrochemical stripping transduction (to yield fmol detection limits) and with an efficient magnetic separation (to minimize nonspecific adsorption effects). The concept has been demonstrated for a simultaneous immunoassay of α2-microglobulin, IgG, bovine serum albumin, and C-reactive protein in connection with ZnS, CdS, PbS, and CuS colloidal crystals, respectively. Hong et al. reported a similar approach using commercial QD (ZnS@CdS) labels for the electrochemical immunoassay of a protein biomarker, interleukin-1α (IL-1α) [33]. After sandwich immunoreaction, QD labels were attached to the IL-1α antibody-coated magnetic beads by the formed antibody-antigen immunocomplex. The concentration of IL-1α was determined by electrochemical stripping analysis of the cadmium component of the captured QD labels after an acid-dissolution step. The voltammetric response is highly linear over the range of 0.5 to 50 ng mL−1 IL-1α, and the limit of detection is estimated to be 0.3 ng mL−1 (18 pM).
Figure 4.

Multiprotein electrical detection protocol based on different metallic sulfide nanoparticle labels. (Based on Ref. 29 with permission)
Recently, Hansen et al. reported using a QD/Aptamer-based ultrasensitive electrochemical biosensor to detect multiple protein targets [34]. The protocol is based on a simple single-step displacement assay involving the co-immobilization of several thiolated aptamers, along with binding of the corresponding QD-tagged proteins on a gold surface, adding the protein sample, and monitoring the displacement through electrochemical detection of the remaining nanocrystals. Such electronic transduction of aptamer-protein interactions is extremely attractive for meeting the low power, size, and cost requirements of decentralized diagnostic systems. Unlike two-step sandwich assays used in the above QD-based electrochemical immunoassays, the new aptamer biosensor protocol relies on a single-step displacement protocol. This biosensor allows assays of samples with target concentrations that are 3 to 4 orders of magnitude with a detection limit of 20 ng L−1 (0.5 pM). Such a detection limit corresponds to 54.5 attomole (2 pg) in the 100-μL sample.
QD has also been used as a label for potentiometric immunoassays [35]. Thurer reported the potentiometric bioanalysis of proteins in a microtiter plate format with semiconductor nanocrystal labels. After sandwich immunoreaction on the microtiter plate, the captured CdSe QDs were found to be easily dissolved/oxidized in a matter of minutes with hydrogen peroxide. The released Cd2+ ions were measured by Cd2+ selective micropipette electrodes. The potentiometric protein immunoassay exhibits a log-linear response ranging from 0.15 to 4.0 pmol of IgG, with a detection limit of <10 fmol in 150-μL sample wells.
2.3 Carbon nanotubes
CNTs have been explored extensively in electroanalytical chemistry research fields [see reviews, 36–40]. Because of their big surface area-to-weight ratio, excellent mechanical properties, and fast electron-transfer capabilities, CNTs have been widely used to prepare electrochemical sensors and biosensors and study the electron-transfer characteristics of proteins. Recently, Wang and coworkers investigated their novel applications by using CNTs as labels for ultrasensitive electrochemical immunoassays [41]. In this case, CNTs were used as a “carrier” to load numerous enzyme tracers by covalent functionalization of their surface (Figure 5). A coverage of around 9600 enzyme molecules per CNT (1 μm length) was estimated. The numerous enzyme-loaded CNT super labels were applied for the magnetic beads-based electrochemical immunoassay of IgG, and a detection limit of 500 fg mL−1 (160 zmol in 25 μL samples) was obtained. A 100-fold enhancement in the resulting electrochemical signal was observed from the enzyme-CNT label when compared to a system consisting of a single-enzyme label. Further sensitivity enhancement was achieved with a layer-by-layer (LBL) assembly of multilayer enzyme films on the CNT template [42]. Such coupling of CNT labels and protein multiple architectures was shown to maximize the ratio of enzyme tracers per binding event, to offer a remarkably high amplification factor, and hence to achieve an extremely low detection limit down to 2000 protein molecules (67 aM). Rustling’s team reported on CNT “forest” amperometric immunosensor platforms with multi-labeled secondary antibody-CNT bioconjugates for highly sensitive detection of a cancer biomarker in serum and tissue lysates [43]. Greatly amplified sensitivity was attained by using bioconjugates featuring HRP labels and secondary antibodies (Ab2) linked to CNTs at a high HRP/Ab2 ratio. This approach provided a detection limit of 4 pg mL−1 (100 amol mL−1), for prostate-specific antigen (PSA) in 10 μL of undiluted calf serum, a mass detection limit of 40 fg.
Figure 5.

Ultrasensitive electrochemical immunoassay of protein based on the numerous enzymes loaded with CNT labels. (Based on Ref. 41 with permission)
2.4 Apoferritin nanovehicles
Recently, we used apoferritin as a template to prepare nanoparticle labels for a highly sensitive electrochemical immunoassay of protein [44–47]. Apoferritin consists of a spherical protein shell (around 12.5 nm in diameter) composed of 24 subunits surrounding an aqueous cavity with a diameter of about 8 nm that is capable of accommodating around 4500 iron atoms [48]. The protein cage of apoferritin can be disassociated into 24 subunits at low pH (2.0) and reconstituted at a high pH (8.5) environment. There are 14 channels connecting the outside of apoferritin with its interior, among which 6 are hydrophobic channels, and 8 are hydrophilic channels. Based on its unique properties, two types of apoferritin nanoparticle labels, including the redox probe-loaded apoferritin and metallic phosphate-loaded apoferritin, were developed (Figure 6). The concept was first demonstrated by loading hexacyanoferrate in the cavity of apoferritin for the electrochemical immunoassay of an IgG model analyte [45]. The preparation of hexacyanoferrate-loaded apoferritin is illustrated in Figure 6A. Briefly, apoferritin was dissociated into subunits at pH 2 and then reconstituted at pH 8.5, thereby trapping hexacyanoferrate in solution within its interior. Around 150 hexacyanoferrates were loaded into an apoferritin. The hexacyanoferrate-loaded apoferritin was thus modified with biotin and used as a label for a magnetic bead-based sandwich immunoassay of IgG. The electrochemical response of the target was obtained by square-wave voltammetric measuring of the released hexacyanoferrate tracers from the captured nanoparticle labels. A detection limit of 0.08 ng mL−1 (0.52 pM) was obtained in connection with the 60-min immunoreaction time. This detection limit corresponds to 26 attomols in the 50-μL sample solution.
Figure 6.

(A) Preparation of K3Fe(CN)6-loaded apoferritin nanoparticle labels (based on Ref. 45 with permission); (B) apoferritin template synthesis metallic phosphate nanoparticle tag (based on Ref. 46 and 47 with permission).
Inspired by the use of semiconductor nanoparticle labels in the electrochemical bioassay, we developed a simple and facile templated synthesis strategy based on an apoferritin template to prepare uniform-size, metal phosphate nanoparticle labels for a highly sensitive electrochemical immunoassay (Figure 6B) [46]. Comparing the semiconductor nanoparticle tags, new metallic phosphate nanoparticle tag is easy to prepare (simply by diffusion or adjusting the pH of the solution) and functionalize (protein shell). Releasing metal components from the nanoparticle was performed in the mild condition, pH 4.6 acetate buffer, instead of a strong acid solution (1 M HNO3 to dissolve the semiconductor nanoparticle tags). New nanoparticle was used as a label for the electrochemical immunoassay of the tumor necrosis factor a (TNF-α) protein biomarker with a detection limit of 2 pg mL−1 (77 fM), that is, 3.9 attomols or 2.33×106 TNF-α biomarker molecules in a 50-μL sample, which is lower than that of the semiconductor nanoparticle tag. The ability to simultaneously measure multiple proteins in a single assay was also demonstrated by detecting TNF-α and macrophage chemotactic protein-1 protein biomarkers with cadmium phosphate and lead phosphate nanoparticle tags.
Except for the preparation of single-metal-component, metal phosphate nanoparticle tags, compositionally (multiple metals) encoded nanoparticle tags were also prepared by the apoferritin template [47]. By incorporating different predetermined levels of multiple metal ions, such compositionally encoded nanoparticles can lead to a large number of recognizable voltammetric signatures and hence to a reliable detection of a larger number of protein biomarkers. Each nanoparticle thus yields a characteristic multi-peak voltammogram, whose peak potentials and current intensities reflect the identity of the corresponding biomolecule target. The apoferritin template synthesis of the encoded nanoparticle provides a simpler, faster approach to prepare electrochemical nanoparticle labels for diverse applications.
2.5 Liposome
Liposomes are composed of a lipid bilayer with the hydrophobic chains of the lipids forming the bilayer and the polar headgroups of the lipids oriented towards the extravesicular solution and inner cavity [49]. The sizes of the liposomes vary, ranging from nanometers to several micrometers, which depends on the synthesis conditions [49]. Owing to its high surface area, large internal volume, and capability to conjugate bilayer lipids with a variety of biorecognition elements, liposomes have been widely used as bioassay labels by encapsulating enzymes, fluorescent dyes, electrochemical and chemiluminescent markers, DNA, RNA, ions, and radioactive isotopes [50, 51]. While excellent reviews of the uses of liposomes in immunoassays are available in the literature [49, 50], we focus on the applications of liposomes as labels in EII. Figure 7 displays the typical liposome label-based electrochemical immunoassay. Most of the early works are based on ion-encapsulated liposome labels and ion-selective electrodes for serum protein analysis [52–54]. For example, D’Orazio and Rechnitz used trimethylphenylammonium ion (TMPA+)-loaded sheep red-blood-cell ghosts (which act in the same capacity as liposome) to quantitate the complement enzymes present in serum samples by using a TMPA+ ion selective electrode [52]. They were later able to adapt this technique to indirectly detect antibodies in bovine serum albumin. Similarly, Shiba et al. demonstrated the potentiometric determination of tetrapentylammonium ion (TPA+) with a TPA+ ion-selective electrode upon complement disruption of TPA+ of the TPA+-loaded liposomes [53]. Shiba et al. first used potassium ion-loaded liposomes and a potassium ion-selective electrode for monitoring the complement-mediated immune lysis reaction [54].
Figure 7.
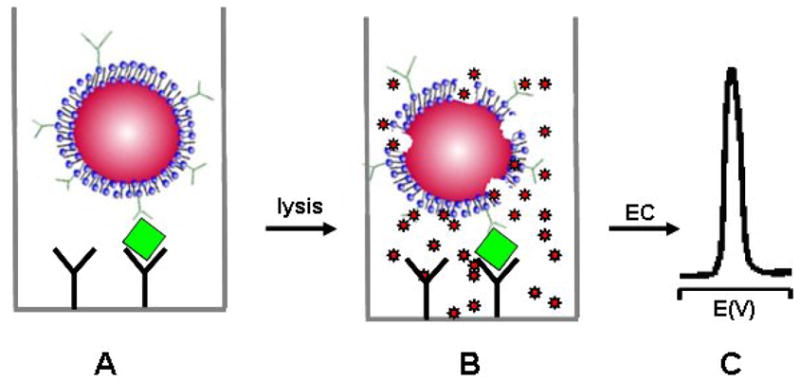
Schematic of an electrochemical immunoassay based on markers loaded with liposome labels: (A) immunocapturing liposome labels; (B) lysis to release the markers; (C) electrochemical detection.
Liposomes encapsulating electroactive molecules, such as ferrocyanide and ascorbic acid, have been used as labels for EII [55, 56]. Kannuck reported ferrocyanide-encapsulated liposome for selective and sensitive (0.1 nM) determination of immuno-agents in serum matrices [55]. Ferrocyanide was encapsulated in the cavity of liposomes at concentrations of approximately 104 molecules/liposome. The ferrocyanides were released from within the liposome by either the addition of surfactant or the complement lysis of the membrane and were monitored by differential pulse voltammetry on a polymer-modified electrode. Liposomes encapsulating ascorbic acid have been used in a competitive assay format for the pesticide atrazine using both lateral and horizontal flow formats and amperometric detection following Triton X-100–induced lysis [56]. The signal response was 1 to 3 min, and the sensitivity of the measurements in tap water was below 1 μg L−1 of atrazine and terbutylazine. Lee et al. reported a disposable liposome immunosensor for theophylline, combining an immunochromatographic membrane and a thick-film electrode [57]. An anti-theophylline antibody was immobilized in an antibody competition zone, and hexacyanoferrate(II)-loaded liposomes were immobilized in a signal generation zone. When a theophylline sample solution was applied to the immunosensor pre-loaded with theophylline–melittin conjugate in a sample loading zone, the theophylline and theophylline–melittin conjugate migrated through the anti-theophylline antibody zone where competitive binding occurs. Unbound theophylline–melittin conjugate further migrated into the signal generation zone where it disrupted the liposomes to release the electroactive hexacyanoferrate(II), which was then detected amperometrically. The detection limit of 5 μgmL−1 enabled the immunosensor to be used to monitor theophylline over the clinically relevant ranges (10 to 20 μg mL−1) with a one-step assay within 20 min.
Liposome encapsulating enzymes, such as HRP, have been used as labels to develop sensitive electrochemical immunosensors [58]. For example, Haga reported a liposome immunosensor for the detection of theophylline. The immunosensor is composed of a Clark-type oxygen electrode and HRP encapsulated liposomes. Sample theophylline and theophylline-tagged liposomes competed for antibody sites. Binding to the antibody initiated the activation of the complement, which lysed the liposomes and resulted in the release of the entrapped horseradish peroxidase. The released HRP catalyzed the conversion of NADH to NAD+, and the corresponding depletion of oxygen was monitored by an oxygen electrode. The authors reported a detection limit for theophylline of 0.72 ng/mL.
Recently, Chen et al. developed a novel piezoelectric immunoagglutination assay (PEIA) technique with antibody-conjugated liposome for direct quantitative detection of human immunoglobulin G (hIgG) [59]. This technique is based on specific agglutination of antibody-coated liposome particles in the presence of the corresponding antigen, which can be monitored by the frequency shift of a piezoelectric device. The frequency responses of the liposome-based PEIA are linearly correlated to hIgG concentration in the range of 0.05 to 6 μgmL−1 with a detection limit of 50 ng mL−1.
2.6 Silica nanoparticles
Silica nanoparticles have been successfully used in bio-imaging, optical bioassays, drug delivery, diagnosis, and therapeutic applications [60–64]. Recently, we developed an “electroactive” silica nanoparticle in which poly (guanine) (poly [G]) was used to functionalize silica nanoparticles, and then we demonstrated that the poly [G]-functionalized silica NPs could serve as a biological label for a sensitive electrochemical immunoassay [65,66]. The “electroactive” silica nanoparticle label was prepared by covalently binding poly [G] and avidin to the silica NP surface by using the conventional coupling reagent 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) and N-hydroxysuccinimide. It was found that there are ~60 strands of poly [G]20 per silica nanoparticle. Accordingly, the average surface coverage of poly [G]20 on a silica surface was determined to be ~ 8.5 ×1012 molecules cm−2. The functionalized silica NPs could be used as a label for an amplified electrochemical immunoassay. The principle of an electrochemical immunoassay based on a poly [G]-functionalized silica NP label is shown in Figure 8. After a complete sandwich immunoassay, a solution of Ru(bpy)3 2+ is added, and the catalytic current resulting from guanine oxidation is measured. This current is proportional to the amount of guanine in the vicinity of the electrode, which in turn depends on the concentration of target analyte in the original sample.
Figure 8.
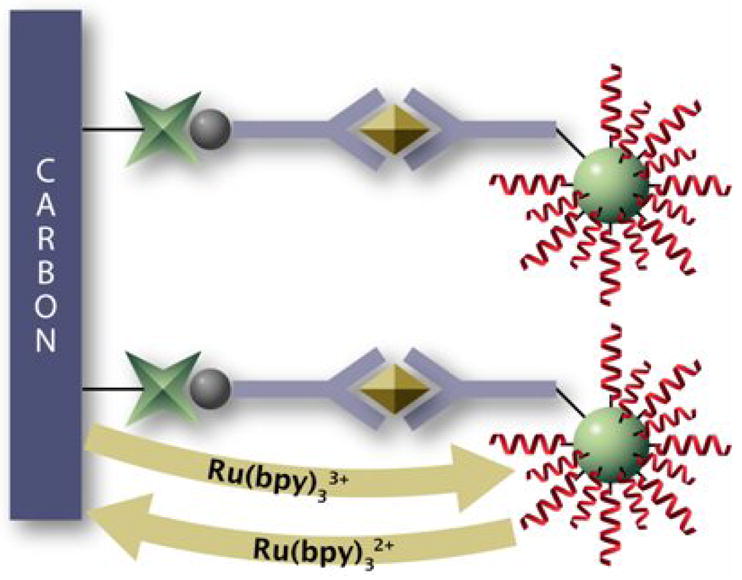
Electrochemical immunosensor based on the poly[guanine]-coated silica nanoparticle. (Reproduced from Ref. 65 with permission)
The application was first demonstrated by using IgG as model protein [65]. It was found that the current was proportional to the logarithm of IgG concentration. The limit of detection for this immunosensor (based on a signal-to-noise ratio S/N=3) is estimated to be about 0.2 ng mL−1 (about 1.3 pM), which corresponds to about 39 aM mouse IgG in a 30-μL sample solution. The developed electroactive silica nanoparticle label was also used to detect protein biomarker, TNF-α [66]. After optimizing the experimental parameters (e.g., concentration of Ru (bpy)3 2+, incubation time of TNF-α, etc.), the detection limit for TNF-α was found to be 5.0 ×10−11 g mL−1 (2.0 pM), which corresponds to 60 attomol of TNF-α in 30 μL of sample.
2.6 Other labels
Except for the nanomaterial labels described above, micrometer size materials, such as polystyrene beads and microcrystals, have been used as labels for highly sensitive electrochemical immunoassays. For example, Wang and co-workers reported on nucleic acid-coated polystyrene bead labels for an amplified electrochemical sandwich immunoassay of proteins [67]. This involves a sandwich immunoassay based on two antibodies linked to magnetic beads and DNA-functionalized polystyrene (PS) spheres, followed by the alkaline release of the oligonucleotide strands from the beads, the acidic dipurinization of the released DNA, and adsorptive chronopotentiometric stripping measurements of the free nucleobases at a pyrolytic graphite electrode transducer (Figure 9). The coupling of carrier-sphere amplifiers (7.5×104 oligonucleotides [containing 25 guanines] per sphere) with the preconcentration feature of electrochemical stripping detection leads to an extremely low detection limit of 2 pg mL−1 (13 fM). A similar use of different oligonucleotide tracers—based on different guanine-to-adenine ratios—should lead to an attractive multiplex protein assay. A ferrocene microcrystal was also used as a biolabel for the amplified electrochemical immunoassay [68]. In this case, LBL technology was employed to encapsulate electrochemical signal-generating microcrystals (ferrocene microcrystals). The encapsulated microcrystals were conjugated with antibody molecules through the adsorption process for sandwich immunoassays. The capture of the antibody-conjugated ferrocene microcrystal labels was followed by the release of a large amount of the redox marker through the capsule wall (by a releasing agent) and led to highly sensitive amperometric biodetection with a detection limit of 2.82 μg L−1.
Figure 9.
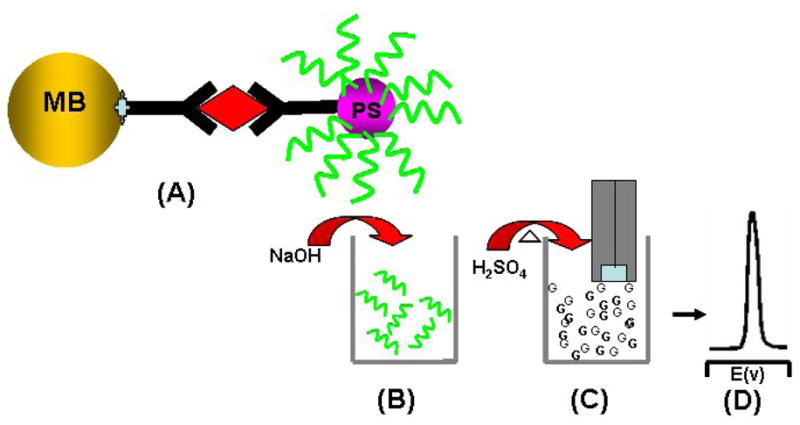
Use of oligonucleotide-loaded polystyrene sphere labels for the amplified electrochemical immunoassay of proteins: (A) immunocapturing oligonucleotide-loaded polystyrene spheres to a magnetic bead; (B) release of the oligonucleotides from the sphere using 0.05 M NaOH; (C) aid dipurinization; (D) detection of the releaded purine based at a graphite electrode (Based on ref. 67 with permission).
Recent efforts focused on developing nanoparticle encoded spheres, submicometer metallic microrods, and metal or alloy nanowires [69–74]. Such materials have been used as tags for product identification [70], an electrochemical DNA hybridization assay [72], and an optical bioassay [69, 73, 74]. For example, indium/gold nanowire tags prepared by electrodepositing indium and gold into the pores of an alumina membrane offered a significantly low detection limit (250 zmol) for the electrochemical DNA hybridization assay [70]. Semiconductor nanoparticle-encoded polystyrene beads [69] and metallic striped nanowires have been used as tags for the multiplex optical immunoassay of proteins, DNA, and pathogens [73, 74]. Because of its big size (micrometer length), the content of metal in metallic nanowire is much more than that of the reported semiconductor nanoparticles. It will be possible to use such metallic nanowire tags for ultrasensitive electrochemical immunoassays.
3. Conclusion and Outlook
This review has summarized the recent advances of nanomaterial labels in EII. The unique and attractive properties of nanomaterials have paved the way for the development of highly sensitive electrochemical diagnosis devices. The studies described above demonstrate the broad potential of bioconjugated nanomaterials for the amplified electrochemical transduction of antibody-antigen recognition events. The remarkable sensitivity of the new nanomaterial-label-based sensing protocols opens the possibility for detecting disease markers, biothreat agents, or infectious agents that cannot be measured by conventional methods. Such highly sensitive biodetection schemes could provide for early detection of diseases or a warning of a terrorist attack. The use of super tags, such as electroactive-marker-loaded polymer beads and submicrometer metallic strips will further amplify the electrochemical signal of EII and thus bring about a lower detection limit. However, the challenges for current research are 1) “How will the nanomaterial-label-based EII technologies work in clinical samples, such as blood, plasma?” and 2) “Will it be possible to apply this technology to point-of-care and clinical diagnosis?” Future innovative research is expected to lead to advanced nanomaterial-based EII, which, coupled with other major technological advances, such as lateral-flow, test-strip technology and electronics technology, will result in powerful, easy-to-use, hand-held devices for fast, sensitive, and low-cost biomarker detection.
Acknowledgments
The work was done at Pacific Northwest National Laboratory (PNNL) supported partially by grant number NS058161-01 from the National Institutes of Health CounterACT Program through the National Institute of Neurological Disorders and Stroke, partially by CDC/NIOSH Grant R01 OH008173-01, and partially by grant number U54 ES16015 from the National Institute of Environmental Health Sciences (NIEHS), NIH. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the Federal Government. The research described in this paper was partly performed at the Environmental Molecular Sciences Laboratory, a national scientific user facility sponsored by the DOE’s Office of Biological and Environmental Research and located at PNNL. PNNL is operated by Battelle for DOE under Contract DE-AC05-76RL01830. G. Liu acknowledges the financial support from startup funds from NDSU.
Biographies
 From G. Liu
From G. Liu
I would like to congratulate Prof. Wang on his 60th birthday. HAPPY BIRTHDAY, Dr. Wang! Millions of thanks for your guidance, encouragement, and support during my career development! I am looking forward to reading your 1000th wonderful paper! Guodong
 From Y. Lin
From Y. Lin
I first met Dr. Wang when I joined his Biosensor group at Las Cruces in 1991 as a postdoctoral researcher working on organic-phase enzyme biosensors. The time I worked with Joe during the 1991–1992 is unforgettable to me and is very important to my career development. Since I joined Pacific Northwest National Laboratory in 1997, Joe has been a valuable long-term collaborator of mine in research of lab-on-a-chip and carbon nanotubes biosensors. Whether working with students or colleagues, Joe is an excellent role model: he is a creative, diligent, and productive scientist. Many thanks to you, Joe, for your inspiration and encouragement over the years, and HAPPY 60th Birthday! Yuehe
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Warsinke A, Benkert A, Scheller FW. Fresenius J Anal Chem. 2000;366:622. doi: 10.1007/s002160051557. [DOI] [PubMed] [Google Scholar]
- 2.Ghindilis AL, Atanasov P, Wilkinst M, Wilkins E. Biosens Bioelectron. 1998;13:113. doi: 10.1016/s0956-5663(97)00031-6. [DOI] [PubMed] [Google Scholar]
- 3.Lin J, Ju H. Biosensors & Bioelectronics. 2005;20:1461. doi: 10.1016/j.bios.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 4.Stradiotto NR, Yamanaka H, Zanoni MVB. J Braz Chem Soc. 2003;14:159. [Google Scholar]
- 5.Marquette CA, Blum LJ. Biosens Bioelectron. 2006;21:1424. doi: 10.1016/j.bios.2004.09.037. [DOI] [PubMed] [Google Scholar]
- 6.Bakker E, Qin Y. Anal Chem. 2006;78:3965. doi: 10.1021/ac060637m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang J. Electroanalysis. 2007;19:769. [Google Scholar]
- 8.Wang J. small. 2005;1:1036. doi: 10.1002/smll.200500214. [DOI] [PubMed] [Google Scholar]
- 9.Wang J. Biosens Bioelectron. 2006;21:1887. doi: 10.1016/j.bios.2005.10.027. [DOI] [PubMed] [Google Scholar]
- 10.Katz E, Willner I, Wang J. Electroanalysis. 2004;16:19. [Google Scholar]
- 11.Hernandez-Santos D, Gonzalez-Garcia MB, Garcia AC. Electroanalysis. 2002;14:1225. [Google Scholar]
- 12.Luo X, Morrin A, Killard AJ, Smyth MR. Electroanalysis. 2006;18:319. [Google Scholar]
- 13.Katz E, Willner I. Angew Chem Int Ed. 2004;43:6042. doi: 10.1002/anie.200400651. [DOI] [PubMed] [Google Scholar]
- 14.Liu G, Wang J, Wu H, Lin Y, Lin Y. Electroanalysis. 2007;19:777. [Google Scholar]
- 15.Hayatt MA. Colloidal gold-Principles, Methods and applications. Academic Press; San Diego: 1989. [Google Scholar]
- 16.Gonzalez-Garcia MB, Fernandez-Sanchez C. A Costa-Garcia Biosens. Bioelectron. 2000;15:315. doi: 10.1016/s0956-5663(00)00074-9. [DOI] [PubMed] [Google Scholar]
- 17.Dequaire M, Degrand C, Limoges B. Anal Chem. 2000;72:5521. doi: 10.1021/ac000781m. [DOI] [PubMed] [Google Scholar]
- 18.Liu G, Lin Y. J Nanosci Nanotech. 2005;5:1060. doi: 10.1166/jnn.2005.178. [DOI] [PubMed] [Google Scholar]
- 19.Ambrosi A, Castaneda MT, Killard AJ, Smyth MR, Alegret S, Merkoci A. Anal Chem. 2007;79:5232. doi: 10.1021/ac070357m. [DOI] [PubMed] [Google Scholar]
- 20.Chu X, Fu X, Chen K, Shen G, Yu R. Biosens Bioelectron. 2005;20:1805. doi: 10.1016/j.bios.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 21.Chu X, Xiang Z, Fu X, Wang S, Shen G, Yu R. J Immunological Methods. 2005;301:77. doi: 10.1016/j.jim.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 22.Chumbimuni-Torres Ky, Dai Z, Rubinova N, Xiang Y, Pretsch E, Wang J, Bakker E. J Am Chem Soc. 2006;128:13676. doi: 10.1021/ja065899k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Velev OD, Kaler EW. Langmuir. 1999;15:3693. [Google Scholar]
- 24.Chu X, Zhao Z, Shen G, Yu R. Sensors and Actuators B. 2006;114:696. [Google Scholar]
- 25.Mao X, Jiang JH, Chen JW, Huang Y, Shen GL, Yu RQ. Anal Chim Acta. 2006;557:159. [Google Scholar]
- 26.Zhou G, Li J, Jiang J, Shen G, Yu R. Acta Chimi Sinica. 2005;63:2093. [Google Scholar]
- 27.Liao K, Huang H. Anal Chim Acta. 2005;538:159. [Google Scholar]
- 28.Das J, Aziz MA, Yang H. 2006;128:16022. doi: 10.1021/ja0672167. [DOI] [PubMed] [Google Scholar]
- 29.Han M, Gao X, Su J, Nie S. Nature Biotech. 2001;19:631. doi: 10.1038/90228. [DOI] [PubMed] [Google Scholar]
- 30.Wang J, Liu G, Polsky R. Electrochem Commun. 2002;4:722. [Google Scholar]
- 31.Wang J, Liu G, Merkoçi A. J Am Chem Soc. 2003;125:3214. doi: 10.1021/ja029668z. [DOI] [PubMed] [Google Scholar]
- 32.Liu G, Wang J, Kim J, Jan MR, Collins GE. Anal Chem. 2004;76:7126. doi: 10.1021/ac049107l. [DOI] [PubMed] [Google Scholar]
- 33.Wu H, Liu G, Wang J, Lin Y. Electrochem Commun. 2007;9:1573. [Google Scholar]
- 34.Hansen JA, Wang J, Kawde A, Xiang Y, Gothelf KV, Collins G. J Am Chem Soc. 2006;126:2228. doi: 10.1021/ja060005h. [DOI] [PubMed] [Google Scholar]
- 35.Thurer R, Vigassy T, Hirayama M, Wang J, Bakker E, Pretsch E. Anal Chem. 2007;79:5107. doi: 10.1021/ac070932m. [DOI] [PubMed] [Google Scholar]
- 36.Wang J. Electroanalysis. 2005;17:7. [Google Scholar]
- 37.He P, Xu Y, Fang Y. Microchim Acta. 2006;152:175. [Google Scholar]
- 38.Gong K, Yan Y, Zhang M, Su L, Xiong S, Mao L. Analytical Sciences. 2005;21:1383. doi: 10.2116/analsci.21.1383. [DOI] [PubMed] [Google Scholar]
- 39.Trojanowicz M. Trends in Analytical Chemistry. 2006;25:480. [Google Scholar]
- 40.Lin Y, Yantasee W, Wang J. Front Biosci. 2005;10:492. doi: 10.2741/1545. [DOI] [PubMed] [Google Scholar]
- 41.Wang J, Liu G, Jan MR. J Am Chem Soc. 2004;126:3010. doi: 10.1021/ja031723w. [DOI] [PubMed] [Google Scholar]
- 42.Munge B, Liu G, Collins G, Wang J. Anal Chem. 2004;76:7126. [Google Scholar]
- 43.Yu X, Munge B, Patel V, Jensen G, Bhirde A, Gong JD, Kim SN, Gillespie J, Gutkind JS, Papadimitrakopoulos F, Rusling JF. J Am Chem Soc. 2006;128:11199. doi: 10.1021/ja062117e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu G, Wang J, Lin Y. ChemBioChem. 2006;7:1315. doi: 10.1002/cbic.200600225. [DOI] [PubMed] [Google Scholar]
- 45.Liu G, Wang J, Wu H, Lin Y. Analytical Chemistry. 2006;78:7417. doi: 10.1021/ac060653j. [DOI] [PubMed] [Google Scholar]
- 46.(a) Liu G, Wu H, Wang J, Lin Y. Small. 2006;2:1139. doi: 10.1002/smll.200600206. [DOI] [PubMed] [Google Scholar]; (b) Liu G, Lin Y. J Am Chem Soc. 2007;129:10394. doi: 10.1021/ja070429r. [DOI] [PubMed] [Google Scholar]
- 47.Liu G, Wu H, Dohnalkova A, Lin Y. Anal Chem. 2007;79:5614. doi: 10.1021/ac070086f. [DOI] [PubMed] [Google Scholar]
- 48.Ford GC, Harrison PM, Rice DW, Smith JMA, Treffry A, White YJ. Philos Trans R Soc London Ser B. 1984;304:551. doi: 10.1098/rstb.1984.0046. [DOI] [PubMed] [Google Scholar]
- 49.Rongen HAH, Bult A, Van Bennekom WP. J Immunological Methods. 1997;204:105. doi: 10.1016/s0022-1759(97)00041-0. [DOI] [PubMed] [Google Scholar]
- 50.Edwards KA, Baeumner AJ. Talanta. 2006;68:1432. doi: 10.1016/j.talanta.2005.08.031. [DOI] [PubMed] [Google Scholar]
- 51.Edwards KA, Baeumner AJ. Talanta. 2006;68:1421. doi: 10.1016/j.talanta.2005.08.044. [DOI] [PubMed] [Google Scholar]
- 52.D’Orazlo P, Rechnltz GA. Anal Chem. 1977;49:2083. doi: 10.1021/ac50021a047. [DOI] [PubMed] [Google Scholar]
- 53.D’Orazlo P, Rechnltz GA. Anal Chim Acta. 1979;109:25. [Google Scholar]
- 54.Shiba K, Wanstabe Y, Ogawa S, Fujiwara S. anal Chem. 1980;52:1610. doi: 10.1021/ac50061a018. [DOI] [PubMed] [Google Scholar]
- 55.Kannuck RM, Bellama JM. Anal Chem. 1988;60:142. doi: 10.1021/ac00153a009. [DOI] [PubMed] [Google Scholar]
- 56.Baumner AJ, Schmid RD. Biosens Bioelectron. 1998;13:519. doi: 10.1016/s0956-5663(97)00131-0. [DOI] [PubMed] [Google Scholar]
- 57.Lee KS, Kim T, Shin M, Lee W, Park J. Anal Chim Acta. 1999;380:17. [Google Scholar]
- 58.Haga M, Sugawara S, Itagaki H. Anal Biochem. 1981;118:286. doi: 10.1016/0003-2697(81)90584-4. [DOI] [PubMed] [Google Scholar]
- 59.Chen H, Jiang J, Li Y, Deng T, Shen G, Yu R. Biosens Bioelectron. 2007;22:993. doi: 10.1016/j.bios.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 60.Chen JF, Ding HM, Wang JX, Shao L. Biomaterials. 2004;25:723. doi: 10.1016/s0142-9612(03)00566-0. [DOI] [PubMed] [Google Scholar]
- 61.Lian W, Litherland SA, Badrane H, Tan WH, Wu DH, Baker HV, Gulig PA, Lim DV, Jin SG. Anal Biochem. 2004;334:135. doi: 10.1016/j.ab.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 62.Qhobosheane M, Santra S, Zhang P, Tan WH. Analyst. 2001;126:1274. doi: 10.1039/b101489g. [DOI] [PubMed] [Google Scholar]
- 63.Roy I, Ohulchanskyy TY, Bharali DJ, Pudavar HE, Mistretta RA, Kaur N, Prasad PN. Proc Natl Acad Sci USA. 2005;102:279. doi: 10.1073/pnas.0408039101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Santra S, Zhang P, Wang K, Tapec R, Tan W. Anal Chem. 2001;73:4988. doi: 10.1021/ac010406+. [DOI] [PubMed] [Google Scholar]
- 65.Wang J, Liu G, Lin Y. Small. 2006;2:1134. doi: 10.1002/smll.200600189. [DOI] [PubMed] [Google Scholar]
- 66.Wang J, Liu G, Engelhard MH, Lin Y. Anal Chem. 2006;78:6974. doi: 10.1021/ac060809f. [DOI] [PubMed] [Google Scholar]
- 67.Wang J, Liu G, Munge B, Lin L, Zhu Q. Angew Chemie Int Ed. 2004;43:2158. doi: 10.1002/anie.200453832. [DOI] [PubMed] [Google Scholar]
- 68.Mak WC, Cheung KY, Trau D, Warsinke A, Scheller F, Renneberg R. Anal Chem. 2005;77:2835. doi: 10.1021/ac048505l. [DOI] [PubMed] [Google Scholar]
- 69.Han M, Gao X, Su J, Nie Nature Biotech. 2001;19:631. doi: 10.1038/90228. [DOI] [PubMed] [Google Scholar]
- 70.Wang J, Liu G, Rivas G. Anal Chem. 2003;75:4661. doi: 10.1021/ac034411k. [DOI] [PubMed] [Google Scholar]
- 71.Wang J, Liu G. Anal Chem. 2006;78:2461. doi: 10.1021/ac051975a. [DOI] [PubMed] [Google Scholar]
- 72.Wang J, Liu G, Zhu Q. Anal Chem. 2003;75:6218. doi: 10.1021/ac034730b. [DOI] [PubMed] [Google Scholar]
- 73.Nicewarner-Pena SR, Freeman RG, Reiss BD, He L, Pena DJ, Walton ID, Cromer R, Keating CD, Natan MJ. 2001;294:137. doi: 10.1126/science.294.5540.137. [DOI] [PubMed] [Google Scholar]
- 74.Tok JBH, Chuang FYS, Kao MC, Rose KA, Pannu SS, Sha MY, Chakarova G, Penn SG, Dougherty GM. Angew Chem Int Ed. 2006;45:6900. doi: 10.1002/anie.200601104. [DOI] [PubMed] [Google Scholar]