

Abstract
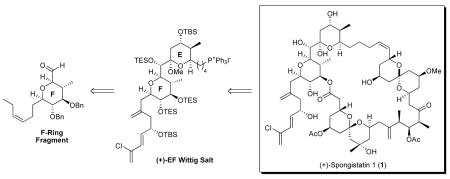
In a quest to develop an effective, scalable synthesis of (+)-spongistatin 1 (1), we devised a concise, third-generation scalable synthesis of (+)-7, the requisite F-ring tetrahydropyran aldehyde, employing a proline-catalyzed cross-aldol reaction. Subsequent elaboration to (+)-EF Wittig salt (+)-3, followed by union with advanced ABCD aldehyde (−)-4, macrolactonization and global deprotection permitted access to >1.0 gram of totally synthetic (+)-spongistatin 1 (1).
The spongipyrans comprise an architecturally unique family of macrolides that display extraordinary cytotoxicity against several highly chemo-resistant tumor cell lines.1 Among the natural congeners, (+)-spongistatin 1(1) is one of the most potent tumor cell growth inhibitors reported to date (average GI50 values of 25–35 pM against the NCI panel of 60 human cancer cell lines). Since their independent isolation by the research groups of Pettit,2 Kitagawa,3 and Fusetani,4 the spongistatins have been the focus of considerable attention in both the chemical and biological communities.5
The spongistatins possess a striking array of structural features, including a 42-membered macrolactone incorporating two spiroketals, in conjunction with a hemiketal, a fully-substituted tetrahydropyran unit, and a highly unsaturated side chain. The relative and absolute stereochemistries, first deduced by Kitagawa,3 were confirmed by the Evans total synthesis of spongistatin 2 (2)6 and the Kishi total synthesis of spongistatin 1 (1).7 More recently, successful total syntheses have been also achieved by the Smith,8 Paterson,9 Crimmins,10 Heathcock11 and the Ley12 laboratories. In addition, several synthetic approaches to the spongistatins have been disclosed.13
However, even with these seminal synthetic achievements, the scarcity of the spongistatins has prohibited further biological testing. Indeed, a reisolation by the Pettit group afforded only 35 mg of the natural product from 13 tons! of wet sponge.14 Given the limited supply from nature, our group initiated an ambitious program to develop a scalable approach to (+)-spongistatin 1 (1), capable of delivering ca. one gram, not only for further biological development, but also as an integral part of a program to design simpler congeners possessing potent tumor cell growth inhibitory activity.
To this end, we recently reported an effective synthesis of the EF fragment of (+)-spongistatin 1 (1) via (+)-7 (Scheme 2) exploiting the Petasis-Ferrier union/rearrangement,15 a synthetic tactic employed extensively in our laboratories to access cis-2,6-disubstituted tetrahydropyrans.16 This approach successfully provided more than 700 mg of the EF Wittig salt, which eventually led to 80 mg of spongistatin 1 (from 450 mg of the EF Wittig salt).17 Shortly thereafter, the MacMillan group reported an elegant two-step synthesis of carbohydrates,18 combining a highly enantioselective proline-catalyzed cross-aldol reaction of oxyaldehydes19 with a Mukaiyama aldol reaction. This approach to carbohydrates via the intermediacy of a lactone held the promise of an even more concise approach to (+)-7 possessing the requisite cis-tetrahydropyran F-ring of (+)-spongistatin 1.20 The potential benefits of this strategy would include: (1) a shorter reaction sequence, (2) excellent cost of goods, (3) stable intermediates, (4) simple operations, including purifications, (5) high reproducibility and efficiency, and most important, (6) scalability.
Scheme 2.

At the outset of what now constitutes a third-generation synthesis in the spongistatin area, we envisioned a five-step approach to (+)-7 from known21 aldehyde 6 taking advantage of the MacMillan protocol (Scheme 3). However, initial efforts indicated that the Mukaiyama aldol was not applicable for large-scale production of 9 (>100 g scale). The issue was attributed to the instability of the β-hydroxyaldehyde (+)-8.22 As a result, an approach that permitted production of (+)-7 on large scale had to be developed.
Scheme 3.

In keeping with the MacMillan aldol route, conversion of 6 to (+)-8 was easily performed on large scale (> 100 g). Exhaustive extraction with water eliminates > 90% of both the homoaldol byproduct and the reaction solvents (cf. DMF/dioxane) without compromise in yield23 or the need for chromatography. The resultant diastereomeric mixture of aldehydes (5:1) could then be treated without separation with (methoxycarbonyl-methylene)-triphenylphosphorane to furnish unsaturated esters 11 (still a 5:1 mixture) in 94% yield. Sharpless asymmetric dihydroxylation using AD-mix β,24 followed by cyclization with pyridinium p-toluenesulfonate successfully provided the corresponding lactone (−)-12 in 81% yield (based on the anti diastereomer). Pleasingly, the only triol diastereomer to undergo lactone formation was the anti isomer that places all substituents in pseudo-equitorial orientations; facile separation of the diastereomeric triols was thus possible. Treatment of the lactone diol (−)-12 with silver oxide, benzyl bromide and calcium sulfate in 1,2-dichloroethane then provided (−)-10 in 78% yield. In this transformation, monobenzylation occurs first at the α-position at 40 °C over a 24 hour time period; increasing the temperature to 60 °C, with additional silver oxide and benzyl bromide, leads to the second benzylation after 8 hours in 78% yield. Recovered monobenzylated lactone (ca. 17–20%) can be converted to the desired bis-benzylated product (−)-10 upon exposure to the original benzylation conditions. In this fashion, a two-step yield of 91% can be achieved. Continuing with the synthesis, addition of the Grignard reagent derived from cis-1-bromo-3-hexene to lactone (−)-10, followed by reduction of the derived lactol with Et3SiH and BF3·Et2O, employing the Kishi protocol, 25 cleanly produced the desired cis-tetrahydropyran (+)-13 in high yield (89%; two steps). Removal of the BPS protecting group also occurred during the reductive Et3SiH process.26 Final Parikh-Doering oxidation27 of the primary hydroxyl provided the F-ring aldehyde (+)-7 in 94% yield (50% overall yield from 6). To date, the third-generation route to the F-ring aldehyde (+)-7 has provided more than 68 grams.
Construction of Wittig salt (+)-3 from (+)-7 followed our previous reported15 sequence (Scheme 5). This sequence proved highly efficient and enabled the production of multigram quantities of (+)-3. Specifically, conversion of (+)-7 to EF Wittig salt (+)-3 begins by addition of dithiane (−)-1415 under chelation controlled conditions to construct the linear precursor of the E-ring of (+)-spongistatin 1, (+)-15. After a 9-step sequence that includes an acid-catalyzed spiroketalization, ozonolysis, and α-methylenation with Eschenmoser’s salt, allyl iodide (+)-16 is obtained in 43% overall yield.15 Alkylation of the latter with cyanohydrin 17 next successfully installs the chlorinated side-chain of (+)-spongisatin 1. Final conversion to the EF Wittig salt is then achieved in 4 steps and 68% yield. Employing the now scalable approach to (+)-7, we had in hand >5.8 g of EF Wittig salt (+)-3.
Scheme 5.

Completion of the third-generation synthesis of (+)-spongistatin 1 began with fragment union of EF Wittig salt (+)-3 with advanced aldehyde (−)-4.8d Wittig union employing the MeLi·LiBr conditions originally introduced by Crimmins10 in their synthesis of (+)-spongistatin 1 and 2, and subsequently utilized by Heathcock11 and Ley,12 provided alkene (+)-19 in 64% yield.
For final elaboration to (+)-spongistatin 1 (1), the two-step deprotection/reprotection sequence utilized in our second-generation synthesis to reveal seco-acid (+)-20 was not applicable, due to the failure of KF to remove cleanly the TES protecting groups (formally TMS groups).7c Use of TBAF, as employed by Heathcock to remove selectively the TES ethers at C(41) and C(42), and the TIPS ester,11 was therefore employed. Selective Yamaguchi macrolactonization then provided the desired 42-membered macrolide in 65–80% yield.15 Global deprotection employing 5 M HF in acetonitrile (1:1) completed the synthesis of (+)-spongistatin 1 (1) in 87% yield. Synthetic (+)-spongistatin 1 was identical in all respects with the natural product.
In summary, an effective, scalable synthesis of the F-ring subunit of (+)-spongistatin 1 (+)-7 exploiting an (L)-proline organocatalyzed cross-aldol reaction has been achieved; the sequence requires 8 steps compared to the 12 steps in our second-generation synthesis, and proceeds in 50% overall yield from 6. On ten to hundred gram scales, the average yield for each step was over 85%. With this achievement, the third-generation synthesis of Wittig-salt (+)-3 now proceeds with a longest linear sequence of 27 steps from commercially available cis-1,4-butenediol (9.5% overall), and as such represents a significant improvement, not only in yield, but also scalability. For (+)-spongistatin, the longest linear sequence is 31 steps (based on EF Wittig salt), and proceeds with an overall yield of 3.1%. To date, we have prepared 1.009 grams of totally synthetic (+)-spongistatin 1 (1).
Supplementary Material
Scheme 1.

Scheme 4.

Scheme 6.

Acknowledgments
Financial support was provided by the NIH (NCI) through Grant No. CA-70329. CAR was supported by the American Cancer Society – Michael Schmidt Postdoctoral Fellowship. JBS was supported by a National Institutes of Health, Ruth L. Kirschstein National Research Service Award (FCA121716A) from the National Cancer Institute. We also like to thank Drs. George Furst (University of Pennsylvania) and Rakesh Kohli (University of Pennsylvania) for their assistance in obtaining NMR spectra and high-resolution mass spectra, respectively.
Footnotes
Supporting Information Available: Spectroscopic and analytical data and selected experimental procedures. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Pettit GA, Cichacz ZA, Gao F, Herald CL, Boyd MR, Schmidt JM, Hooper JN. J Org Chem. 1993;58:1302. [Google Scholar]; (b) Pettit GR. Pure Appl Chem. 1994;66:2271. [Google Scholar]
- 2.(a) Pettit GR, Cichacz ZA, Gao F, Herald CL, Boyd MR. J Chem Soc, Chem Commun. 1993:1166. [Google Scholar]; (b) Pettit GR, Cichacz ZA, Gao F, Herald CL, Boyd MR. J Chem Soc, Chem Commun. 1993:1805. [Google Scholar]
- 3.(a) Kobayashi M, Aoki S, Sakai H, Kawazoe K, Kihara N, Sasaki T, Kitagawa I. Tetrahedron Lett. 1993;34:2795. [Google Scholar]; (b) Kobayashi M, Aoki S, Sakai H, Kihara N, Sasaki T, Kitagawa I. Chem Pharm Bull. 1993;41:989. doi: 10.1248/cpb.41.989. [DOI] [PubMed] [Google Scholar]
- 4.Fusetani N, Shinoda K, Matsunaga S. J Am Chem Soc. 1993;115:3977. [Google Scholar]
- 5.(a) Catassi A, Cesario A, Arzani D, Menichini P, Alama A, Bruzzo C, Imperatori A, Rotolo N, Granone P, Russo P. Cell Mol Life Sci. 2006;63:2377. doi: 10.1007/s00018-006-6264-7. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Pettit R, Woyke T, Pon S, Cichacz Z, Pettit G, Herald C. Med Mycology. 2005;43:453. doi: 10.1080/13693780500050598. [DOI] [PubMed] [Google Scholar]; (c) Pettit RK, McAllister SC, Pettit GR, Herald CL, Johnson JM, Cichacz ZA. Int J Antimicrob Agents. 1998;9:147. doi: 10.1016/s0924-8579(97)00044-7. [DOI] [PubMed] [Google Scholar]; (d) Bai R, Taylor GF, Cichacz ZA, Herald CL, Kepler JA, Pettit GR, Hamel E. Biochemistry. 1995;34:9714. doi: 10.1021/bi00030a009. [DOI] [PubMed] [Google Scholar]
- 6.(a) Evans DA, Coleman PJ, Dias LC. Angew Chem, Int Ed Engl. 1997;36:2738. [Google Scholar]; (b) Evans DA, Trotter BW, Côté B, Coleman PJ. Angew Chem, Int Ed Engl. 1997;36:2741. [Google Scholar]; (c) Evans DA, Trotter BW, Côté B, Coleman PJ, Dias LC, Tyler AN. Angew Chem, Int Ed Engl. 1997;36:2744. [Google Scholar]; (d) Evans DA, Trotter BW, Coleman PJ, Côté B, Dias LC, Rajapakse HA, Tyler AN. Tetrahedron. 1999;55:8671. [Google Scholar]
- 7.(a) Guo J, Duffy KJ, Stevens KL, Dalko PI, Roth RM, Hayward MM, Kishi Y. Angew Chem, Int Ed. 1998;37:187. [Google Scholar]; (b) Hayward MM, Roth RM, Duffy KJ, Dalko PI, Stevens KL, Guo J, Kishi Y. Angew Chem, Int Ed. 1998;37:192. [Google Scholar]
- 8.(a) Smith AB, III, Doughty VA, Lin Q, Zhuang L, McBriar MD, Boldi AM, Moser WH, Murase N, Nakayama K, Sobukawa M. Angew Chem, Int Ed. 2001;40:191. [PubMed] [Google Scholar]; (b) Smith AB, III, Lin Q, Doughty VA, Zhuang L, McBriar MD, Kerns JK, Brook CS, Murase N, Nakayama K. Angew Chem, Int Ed. 2001;40:196. [PubMed] [Google Scholar]; (c) Smith AB, III, Zhu W, Shirakami S, Sfouggatakis C, Doughty VA, Bennett CS, Sakamoto Y. Org Lett. 2003;5:761. doi: 10.1021/ol034037a. [DOI] [PubMed] [Google Scholar]; (d) Smith AB, III, Doughty VA, Sfouggatakis C, Bennett CS, Koyanagi J, Takeuchi M. Org Lett. 2002;4:783. doi: 10.1021/ol017273z. [DOI] [PubMed] [Google Scholar]
- 9.Paterson I, Chen DYK, Coster MJ, Acenã JL, Bach J, Gibson KR, Keown LE, Oballa RM, Trieselmann T, Wallace DJ, Hodgson AP, Norcross RD. Angew Chem, Int Ed. 2001;40:4055. [PubMed] [Google Scholar]
- 10.Crimmins MT, Katz JD, Washburn DG, Allwein SP, McAtee LF. J Am Chem Soc. 2002;124:5661. doi: 10.1021/ja0262683. [DOI] [PubMed] [Google Scholar]
- 11.Heathcock CH, McLaughlin M, Medina J, Hubbs JL, Wallace GA, Scott R, Claffey MM, Hayes CJ, Ott GR. J Am Chem Soc. 2003;125:12844. doi: 10.1021/ja030317+. [DOI] [PubMed] [Google Scholar]
- 12.Ball M, Gaunt MJ, Hook DF, Jessiman AS, Kawahara S, Orsini P, Scolaro A, Talbot AC, Tanner HR, Yamanoi S, Ley SV. Angew Chem, Int Ed. 2005;44:5433. doi: 10.1002/anie.200502008. [DOI] [PubMed] [Google Scholar]
- 13.(a) Ciblat S, Kim J, Stewart CA, Wang J, Forgione P, Clyne D, Paquette LA. Org Lett. 2007;9:719. doi: 10.1021/ol063083i. [DOI] [PubMed] [Google Scholar]; (b) Allais F, Cossy J. Org Lett. 2006;8:3655. doi: 10.1021/ol060812l. [DOI] [PubMed] [Google Scholar]; (c) Holson EB, Roush WR. Org Lett. 2002;4:3723. doi: 10.1021/ol026688x. [DOI] [PubMed] [Google Scholar]; (d) Holson EB, Roush WR. Org Lett. 2002;4:3719. doi: 10.1021/ol0266875. [DOI] [PubMed] [Google Scholar]; (e) Lemaire-Audoire S, Vogel P. J Org Chem. 2000;65:3346. doi: 10.1021/jo991642b. [DOI] [PubMed] [Google Scholar]; (f) Zuev D, Paquette LA. Org Lett. 2000;2:679. doi: 10.1021/ol0000049. [DOI] [PubMed] [Google Scholar]
- 14.Pettit GR. Arizona State University; personal communication. [Google Scholar]
- 15.Smith AB, III, Sfouggatakis C, Gotchev DB, Shirakami S, Zhu W, Doughty VA. Org Lett. 2004;6:3637. doi: 10.1021/ol048418f. [DOI] [PubMed] [Google Scholar]
- 16.Smith AB, III, Fox RJ, Razler TM. Acc Chem Res. 2008;41:675. doi: 10.1021/ar700234r. [DOI] [PubMed] [Google Scholar]
- 17.Sfouggatakis C. PhD thesis. University of Pennsylvania; 2004. [Google Scholar]
- 18.Northrup AB, MacMillan DWC. Science. 2004;305:1753. doi: 10.1126/science.1101710. [DOI] [PubMed] [Google Scholar]
- 19.Northrup AB, Mangoin IK, Hettche F, MacMillan DWC. Angew Chem, Int Ed. 2004;43:2152. doi: 10.1002/anie.200453716. [DOI] [PubMed] [Google Scholar]
- 20.Lactones comprise well known precursors of cis-tetrahydropyrans. See: Lemaire-Audoire S, Vogel P. Tetrahedron Lett. 1998;39:1345.Zhao GL, Liao WW, Cordova A. Tetrahedron Lett. 2006;47:4929.
- 21.Choi WB, Wilson LJ, Yeola S, Liotta DC, Schinazi RF. J Am Chem Soc. 1991;113:9377. [Google Scholar]
- 22.Källström S, Erkkilä A, Pihko PM, Sjöholm R, Sillanpää R, Leino R. Synlett. 2005:751. [Google Scholar]
- 23.Twenty water extractions successfully removed most major impurities, unreacted propanal, homoaldol byproduct, and reaction solvents (cf. DMF/dioxane).
- 24.Hermitage SA, Murphy A, Nielsen P, Roberts SM. Tetrahedron. 1998;54:13185. [Google Scholar]
- 25.Lewis MD, Cha JK, Kishi Y. J Am Chem Soc. 1982;104:4976. [Google Scholar]
- 26.Yoshimitsu T, Song JJ, Wang GQ, Masamune S. J Org Chem. 1997;62:8978. [Google Scholar]
- 27.Parikh JP, Doering WE. J Am Chem Soc. 1967;89:5505. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.