

Abstract
The interaction of insulin with its receptor is complex. Kinetic and equilibrium binding studies suggest co-existence of high and low affinity binding sites and/or negative cooperativity. These phenomena and high affinity interactions are dependent on the dimeric structure of the receptor. Structure-function studies of insulin analogs suggest insulin has two receptor binding sites, implying a bivalent interaction with the receptor. Alanine scanning studies of the secreted recombinant receptor implicate the L1 domain and a C-terminal peptide of the receptor α subunit as components of one ligand binding site. Functional studies suggest that the first and second Type III fibronectin repeats of the receptor contain a second ligand binding site. We have used structure directed alanine scanning mutagenesis to identify determinants in these domains involved in ligand interactions. cDNAs encoding alanine mutants of the holo-receptor were transiently expressed in 293 cells and the binding properties of the expressed receptor determined. Alanine mutations of Lys484, Leu552, Asp591, Ile602, Lys616, Asp620 and Pro621 compromised affinities for insulin 2- to 5- fold. With the exception of Asp620, none of these mutations compromised the affinity of the recombinant secreted receptor for insulin, indicating that the perturbation of the interaction is at the site of mutation and not an indirect effect on the interaction with the binding site of the secreted receptor. These residues thus form part of a novel ligand binding site of the insulin receptor. Complementation experiments demonstrate that insulin interacts in trans- with both receptor binding sites to generate high affinity interactions.
Insulin has pleiotropic physiological effects in mammals (see ref. 1 for review). It has long been known that it plays key roles in the maintenance of blood glucose by stimulating glucose uptake and inhibiting hepatic glucose release. It also plays roles in the regulation of protein synthesis and cell growth and survival. Furthermore, it has been implicated in organismal energy homeostasis, in the regulation of longevity and reproductive competence, and in male sex determination. All its biological actions are consequences of its interaction with its plasma membrane receptor (for review see ref.2). Binding of insulin to the receptor extra-cellular α subunits leads to activation of the receptor’s intra-cellular tyrosine kinase catalytic activity and the initiation of the canonical insulin signal transduction cascade (3;4). It is unclear how activation of signal transduction relates to specific insulin mediated biological events but there is an emerging body of evidence that the nature of the interaction with the receptor may influence biological outcomes. Insulin and insulin-like growth factor-II (IGF-II)1, a homologous peptide growth factor, both bind to the A isoform insulin receptor with high affinity (5), but generate very different biological effects; insulin produces predominantly metabolic effects and IGF-II predominantly mitogenic effects. Exposure of cells to these insulin receptor ligands produces selective patterns of recruitment of signaling molecules to the receptor and distinctive changes in gene expression (6). Denley et al. have demonstrated differences in the temporal patterns of changes in signaling molecules, mediated through the insulin receptor, in cells exposed to insulin and insulin-like growth factors (IGFs) (7). More recently, an insulin-mimetic receptor binding peptide, generated through phage display technology, has been shown to have metabolic but not mitogenic effects (8). These findings indicate that a more comprehensive understanding of the molecular and structural basis the interaction of insulin with its receptor will be essential for the for a complete understanding of insulin signal transduction.
The interaction of insulin with the insulin receptor, has been the subject of extensive investigation. Studies reveal complex interactions (for review see ref.2), which cannot be reconciled with a simple bi-molecular interaction. Analyses of equilibrium binding data suggest the co-existence of high and low affinity binding sites and/or negative co-operativity. Furthermore the dissociation of pre-bound labeled insulin from the receptor is accelerated by the presence of unlabeled insulin in the dissociation medium in a concentration dependent manner, suggesting possible negative co-operativity. This binding behavior, together with high affinity interactions with insulin and insulin regulated tyrosine kinase activity are dependent on the dimeric structure of the receptor (9–11). The secreted recombinant insulin receptor has very similar if not identical insulin binding properties to the receptor monomer (2).
The molecular mechanisms underlying this behavior are poorly understood. The structure and function of insulin and numerous naturally occurring and recombinant analogues have been characterized in extensive detail (12). The hormone is a small globular protein composed of two disulfide-linked peptide chains and is synthesized in the pancreatic β cell as a single chain precursor (13). This is post-translationally processed to the mature A chain, 21 amino acids, and B chain, 30 amino acids. The A chain is formed from two short α helices connected by a loop and the B chain from an N-terminal extended segment, a central α helix followed by a β turn leading to a C-terminal β strand. In contrast, our understanding of the structure and structure-function relationships of the insulin receptor is less well developed (for review see ref. 2). The receptor consists of two disulfide linked monomers and each monomer, in turn, consists of disulfide linked α and β subunits. The α subunits are wholly extra-cellular and contain the ligand binding site(s) and the β subunits have an extra-cellular component, a single trans-membrane helix and an intracellular component with tyrosine kinase catalytic activity. Determination of the structure of the receptor ectodomain dimer in complex with anti-receptor Fab fragments and an insulin –mimetic peptide (14) has provided more detailed insights into the structure of the extra-cellular domain (see Figure 1). The N-terminus is composed of two homologous globular L domains (L1 and L2) separated by a cysteine rich (CR) domain. The C-terminus is formed from three type 3 fibronectin (FnIII) repeats – FnIII-1, FnIII-2 and FnIII-3. Only FnIII-3 is typical; FnIII-1 and FnIII-2 are larger due to the presence of inserts between the C and C′ beta strands. The FnIII-1 insert contains Cys524 which contributes to the α–α cystine and in FnIII-2 there is a 120 amino acid insert domain (ID) which is not clearly visualized in the crystal structure. The ectodomain dimer has the form of an inverted V (Figure 1B). Each monomer has a folded over conformation in the form of a partial inverted V (Figure 1A). The N-terminal partial limb of the V is formed from the L1, CR and L2 domains and the C-terminal limb is formed from the three FnIII domains (Figure 1A).
Figure 1. Structure of the Insulin Receptor Extra-cellular Domain.

(A) The insulin receptor monomer: C-alpha backbone is shown in tube representation (coordinates obtained from PDB entry 2DTG, ref. (14)). Individual domains are designated according to ref.(14) as follows: L1, first globular L domain, amino acids 4-158; CR, cysteine rich domain, amino-acids 159-309; L2, second globular L domain, amino acids 310-470; FnIII-1, first Type 3 Fibronectin repeat, amino acids 471-595; FnIII-2, second Type 3 Fibronectin repeat, amino acids 596-820 (655 and 755 are the aminoa acids at the N- and C-termini of the insert domain which is absent from the crystallographic model) ; and FnIII-3, third Type 3 Fibronectin repeat, amino acids 821-909. (B) The insulin receptor dimer: Monomer 1 is shown in tube representation and monomer 2 in atomic sphere representation. (C) Insulin receptor ligand binding site. The L1, CR and L2 domains from monomer 1 and the FnIII domains of monomer 2 are shown in tube representation. The view is rotated 90° clockwise around the two-fold axis from those in (A) and (B). Alanine scanning mutagenesis studies has shown that determinants of insulin binding are located in the top surface of L1 (17). (D) Candidate residues for mutagenesis. The L1 domain of monomer 1 and the FnIII domains of monomer 2 are shown. Residues mutated to alanine are shown in atomic sphere representation. All structural Figures were generated using Pymol (Warren DeLano, Delano Scientific Inc., www.pymol.org)
A consensus has emerged from structure function studies of the insulin molecule that residues at the N-terminus of the A chain and in the B-chain α helix, β turn and β strand form a receptor binding site, frequently referred to as the “classical” receptor binding site (2;12). The insulin binding site of the secreted recombinant receptor has been shown to be composed of elements of the L1 domain and the amino acids 704–719 (15–17) in the ID at the C-terminus of the α subunit (CTα). Affinity labeling studies have shown that the “classical” receptor binding site of insulin interacts with the L1 domain and the CTα peptide of the secreted receptor ligand binding site (18–21). The affinity of this insulin binding site has been shown to be a major determinant of the affinity of the native receptor, although it is considerably lower than that of the native receptor (17). The lower affinity of the secreted receptor is, in part, due to the absence of membrane insertion (22). A large body of experimental evidence (23–28) suggests that there is a requirement for determinants in the L2, FnIII-1 or FnIII-2 domains of the receptor in addition to those in L1 and CTα for full affinity insulin binding of the native receptor. Recent studies from this laboratory suggest the possible existence of an insulin binding site in the L2 and FnIII-1 domains (26), suggesting high affinity insulin binding may arise from bivalent ligand-receptor interactions.
In the present study, we have used structure directed alanine scanning mutagenesis, to localize and further characterize a second insulin binding site of the insulin receptor. Candidate residues for mutagenesis, in the FnIII-1 and FnIII-2 domains, were identified by inspection of the receptor extra-cellular domain structure. The insulin binding properties of receptors with alanine mutations of these residues were evaluated. Alanine mutations of two residues in the Fn III-1 domain, Lys484 and Leu552, a residue in the inter-domain linker Asp591 and three residues in the FnIII-2 domain, Ile602, Lys616 and Pro621, produced 2- to 5- fold reductions in affinity of the holo-receptor for insulin without affecting the affinity of the secreted recombinant receptor. Complementation experiments with mutant hybrid receptors containing different combinations of inactivating mutations demonstrated that insulin interacts in trans- with the two binding sites to generate a high affinity interaction.
EXPERIMENTAL PROCEDURES
General procedures and Materials
All molecular biological procedures including agarose gel electrophoresis, restriction enzyme digestion, ligation, bacterial transformation and DNA sequencing were performed by standard methods (29). All oligonucleotides were purchased from Integrated DNA Technologies. Restriction and modifying enzymes were from New England Biolabs. Recombinant human insulin was from NovoNordisk A/S. HPLC-purified mono-iodinated 125I-[TyrA14] insulin was from GE Healthcare. Protease inhibitors were from Roche Molecular Biochemicals. PEAK Rapid cells (293 cells constitutively expressing SV40 large T antigen) were purchased from Edge Biosystems. Medium and serum for tissue culture were from Cellgro. The mammalian expression vector pcDNA3.1Zeo+ was from InVitrogen and was modified for C-terminal epitope tagging by subcloning an in-frame oligonucleotide cassette encoding in-frame triple repeats of the FLAG M2 epitope (Asp-Tyr-Lys-Asp-Asp-Asp-Asp-Lys) or of a triple repeat of the Glu-Glu epitope (Glu-Tyr-Met-Pro-Met-Glu) between the BamHI and XbaI restriction sites. The cDNA encoding the B isoform of the insulin receptor was as previously described (17). It was modified for subcloning into the modified expression vector by introduction of a BamHI site encoding an in-frame C-terminal Gly-Ser linker at its 3′ end just prior to the stop codon by site-directed mutagenesis (30). The cDNA for the secreted recombinant receptor encoded amino acids 1 to 929 of the extra-cellular domain. Monoclonal anti-FLAG M2 IgG was purchased from Sigma-Aldrich and monoclonal anti-Glu-Glu IgG was from Covance. Anti-insulin receptor monoclonal antibody IgG was kindly provided by Dr. K. Siddle (University of Cambridge).
Site Directed Mutagenesis
Alanine mutants of the insulin receptor cDNA were created by the method of Kirsch and Joly (30). In order to minimize the possibility of nucleotide mis-incorporation, Pfu Ultra II (Stratagene) was used for amplification and the number of PCR cycles restricted to a maximum of 15. Alanine mutations were confirmed by DNA sequencing.
Expression of Receptor cDNAs
The receptor cDNAs were expressed transiently in PEAK Rapid cells using polyethyleneimine as previously described (17). Cells were harvested by lysis in 0.15M NaCl, 0.1M Tris, pH 8, containing 1% (v/v) Triton X-100 and protease inhibitor cocktail (Roche) three days post-transfection when receptor expression was maximal. Lysates were stored at −80°C until assay.
Isolation of hybrid receptors
293Peak cells were co-transfected with equal amounts of cDNAs encoding mutant receptors tagged with either FLAG or Glu-Glu epitopes (see above). Detergent lysates of cells were prepared 72h post-transfection and enriched glycoprotein fractions were prepared by Wheat Germ Agglutinin chromatography (31). FLAG tagged receptors were purified from the glycoprotein fractions by immuno-affinity chromatography on anti-FLAG agarose with elution by 3XFLAG peptide (Sigma-Aldrich), 1 mg/ml, in 0.15M NaCl, 20mM HEPES, pH 7.5, according to the manufacturers’ protocol. Hybrid receptors were isolated from the FLAG eluates by capture in a microtiter plate coated with anti-Glu-Glu IgG.
Receptor Binding Assays
Insulin receptor binding assays were performed by a modification of the microtiter plate antibody capture assay that we have described previously (17). Microtiter strip plates (Nunc Maxisorb) were incubated overnight at 4°C with anti-FLAG or anti Glu-Glu IgG (100μl/well of a 40μg/ml solution in PBS) for soluble holo-receptor mutants and anti-insulin receptor antibody 18–44 IgG (32) for secreted receptor mutants. Washing, blocking, receptor binding and competitive binding assays with labeled and unlabeled peptides were performed as described (17). In all assays, the amount of added receptor was such that binding of 125I-[TyrA14] insulin in the absence of unlabeled insulin was less than 15%, in order to avoid ligand depletion artifacts. Binding data were analyzed by non-linear regression analysis using a two site sequential model to obtain IC50s and dissociation constants (33). This is a two site Adair model (34–36) which postulates insulin binds in an ordered sequential reaction to two binding sites, with apparent dissociation constants, Kd1 and Kd2, per receptor,. In the curve fitting algorithm, the concentrations of each binding site are constrained to be equal. Competitive binding assays with secreted recombinant receptor were performed as previously described (17) and data fitted to a single site model to obtain dissociation constants.
RESULTS
Selection of Candidate Residues for Mutagenesis
Inspection of the crystal structure extra-cellular domain dimer (14) reveals that the bottom of the first and the top of the second FnIII domains are adjacent to the top surface of L1 domain, which we have previously determined to contain determinants of a major ligand binding site(Figure 1C; ref.(17)). The insulin binding surface of the L1 domain (its top surface), the CR domain, and FnIII-1 and FnIII-2 domains form the walls of a cavity in the interface between the receptor monomers making up each limb of the inverted V (Figure 1C). The dimensions of these cavities are such (diameter 28–30Å) that they can accommodate an insulin molecule and that an insulin molecule bound to L1 could also contact FnIII-1 and FnIII-2 with relatively small conformational changes in the receptor. Inspection of these regions and determination of solvent accessibilities (GETAREA 1.1; http://pauli.utmb.edu/cgi-bin/get_a_form.tcl) suggested candidate residues in the α subunit which could potentially interact with insulin (see Figure 1D). In the FnIII-1 domain, these residues include Ser481 at the bottom of the A strand; Phe482 and Asp483 in the AB loop; Lys484 and Leu486 at the bottom of the B strand; Ser526, Asn527, Trp529, and Thr530 in the CC′ loop; Leu552 and Arg554 at the bottom of the E strand; Lys557 and Leu558 in the EF loop; Asp591 in the G strand; they are Thr593, Asn594, Pro595, Ser596, Val597, Leu599, Asp600, Pro601, and Ile602 in the intra-domain linker; and Lys616, Pro617, Pro618, Ser619, Asp620 and Pro621 in FnIII-2. In order to evaluate their possible role in insulin binding we individually mutated these residues to alanine and evaluated the insulin binding properties of the resultant mutant holo- receptors in transient expression experiments.
Insulin Binding to Alanine Mutant Holo-receptors
cDNAs encoding alanine mutant holo-receptors with C-terminal FLAG tags were created by site directed mutagenesis and transiently expressed in 293PEAK cells. The expressed receptors were harvested by detergent lysis of transfected cells and immobilized in microtiter plates coated with anti-FLAG antibody. Determination of tracer 125I-[TyrA14] insulin (insulin mono-iodinated at Tyr14 of the A chain) binding indicated that all mutant receptors were expressed (data not shown). Competitive equilibrium binding assays with 125I-[TyrA14] insulin and unlabeled insulin were performed to determine the effects of the alanine mutations on the affinity of the receptor. The IC50 of each mutant for insulin was obtained by fitting the binding data to a two site sequential model (33). Previous studies from this laboratory have shown that, under our assay conditions, the comparison of IC50s, determined in this manner, provides a reliable comparison of dissociation constants of mutant insulin receptors (17). For wild type receptor, the IC50 for insulin was 88 ± 3 pM (mean ± S.E.M., n=12). We considered any mutant with a twofold- or greater increase in IC50, i.e. IC50 of 176pM or greater, to exhibit a biologically significant perturbation of its affinity for insulin (37).
The results for the IC50s of the alanine mutant receptors are shown in Table 1. Seven alanine mutations resulted in significant increases in IC50, ranging from 2- to 5- fold. The largest increase (5- fold) was produced by alanine mutation of Leu552, the other six alanine mutations (Lys484, Asp591, Ileu602, Lys616, Asp620 and Pro621) produced increases in IC50 from 2- to 3- fold (Table 1 and see Fig. 2 for a representative experiment).
Table 1.
Alanine Scanning Mutagenesis of the FnIII-1 and FnIII-2 Domains of the Insulin Receptor
| Mutanta | IC50b (pM) | IC50Mut/IC50WT | Mutanta | IC50b (pM) | IC50Mut/IC50WT |
|---|---|---|---|---|---|
| WT | 88 ± 3 | 1.0 | D591A | 27 ± 9 | 3.1 |
| S481A | 85 ± 5 | 1.0 | T593A | 125 ± 5 | 1.4 |
| F482A | 81 ± 1 | 0.9 | N594A | 65 ± 5 | 0.7 |
| D483A | 82 ± 2 | 0.9 | S596A | 65 ± 5 | 0.7 |
| K484A | 187 ± 3 | 2.1 | V597A | 125 ± 5 | 1.4 |
| L486A | 89 ± 1 | 1.0 | L599A | 97 ± 1 | 1.1 |
| S526A | 90 ± 4 | 1.0 | D600A | 70 ± 2 | 0.8 |
| N527A | 120 ± 10 | 1.4 | P601A | 105 ± 5 | 1.2 |
| S528A | 89 ± 1 | 1.1 | I602A | 225 ± 9 | 2.6 |
| W529A | 105 ± 15 | 1.2 | K616A | 260 ± 15 | 2.9 |
| T530A | 127 ± 9 | 1.4 | P617A | 120 ± 10 | 1.4 |
| L552A | 430 ± 47 | 4.9 | S619A | 90 ± 1 | 1.o |
| R554A | 95 ± 15 | 1.1 | D620A | 208 ± 11 | 2.4 |
| K557A | 95 ± 5 | 1.1 | P621A | 233 ± 13 | 2.6 |
| L558A | 80 ± 2 | 0.9 |
The mutants are designated by the amino acid being mutated, in single letter code, followed by its position in the receptor sequence and then alanine.
cDNAs encoding alanine mutants of the insulin holo-receptor were transiently expressed in 293PEAK cells. Equilibrium binding assays with insulin and 125I-(TyrA14) insulin were performed. IC50’s were obtained from binding isotherms fitted to a sequential two-site model (33). The reported results represent the mean and S.E.M. of two to four independent determinations for each mutant. Twelve independent determinations were made for wild type receptor (WT) and four for each mutant with an IC50 twofold or greater than that of wild type.
Figure 2. Competitive Binding Assays of Disruptive Alanine Mutants.

cDNAs encoding alanine mutants of the insulin holo-receptor, which produced a twofold or greater increase in IC50 for insulin, were transiently expressed in 293PEAK cells. Equilibrium binding assays with insulin and 125I-(Tyr A14) insulin were performed. The mutants are designated by the amino acid being mutated, in single letter code, followed by its position in the receptor sequence and then alanine. Results are expressed as B/B0 where B is 125I-(Tyr A14) insulin bound by receptor at the designated insulin concentration and B0 is 125I-(Tyr A14) insulin bound by receptor in the absence of unlabeled insulin.
In order to analyze the perturbations in affinity in greater detail, the equilibrium binding data were fitted to a two site sequential model (33). This has been shown to reliably fit the complex binding isotherms obtained in insulin and IGF-I receptor competitive binding assays. The results are summarized in Table 2. Wild type receptor had a high affinity component of binding with a Kd of 42pM and a low affinity component with a Kd of 520pM, which is comparable with our previously published findings (Table 2; ref. 17). Changes in both high and low affinity components of binding produced by the alanine mutations correlated well with the respective changes in IC50.
Table 2.
Dissociation Constants of Disruptive Insulin Receptor Alanine Mutants
| Mutanta | IC50 (pM) | Dissociation Constants b | Hill Coefficientc | |
|---|---|---|---|---|
| Kd1 (pM) | Kd2 (nM) | |||
| WT | 88 ± 3 | 42 ± 2 | 0.52 ± 0.04 | 0.46 ± 0.006 |
| K484A | 187 ± 3 | 82 ± 5 | 0.84 ± 0.08 | 0.48 ± 0.007 |
| L552A | 430 ± 47 | 180 ± 6 | 2.00 ± 0.06 | 0.46 ± 0.001 |
| D591A | 270 ± 9 | 163 ± 18 | 1.47 ± 0.14 | 0.50 ± 0.01 |
| I602A | 225 ± 9 | 81 ± 5 | 0.92 ± 0.06 | 0.46 ± 0.008 |
| K616A | 260 ± 15 | 152 ± 10 | 1.70 ± 0.06 | 0.46 ± 0.006 |
| D620A | 208 ± 11 | 110 ± 4 | 1.09 ± 0.04 | 0.48 ± 0.005 |
| P621A | 233 ± 13 | 115 ± 5 | 1.15 ± 0.05 | 0.48 ± 0.007 |
The mutants apart from are designated by the amino acid being mutated, in single letter code, followed by its position in the receptor sequence and then alanine
The dissociation constants for insulin were obtained from a two site sequential fit (33) of the equilibrium binding data for the alanine mutants with IC50s greater than 2- fold that of wild type insulin receptor. Results are the mean ± SEM of at least 4 independent determinations.
The Hill coefficient, nH, at 50% receptor occupancy was calculated from the equation (35)
In order to evaluate the effects of the mutations on the cooperativity of the insulin-receptor interaction, the Hill coefficient at 50% receptor occupancy was calculated from the Kds (Table 2; ref. 35). This was less than 1 for the wild type and all mutants, consistent with the existence of negatively cooperative interactions. Only the alanine mutant of Asp591 produced a small but statistically significant increase in this parameter (p< 0.05; Table 2). This indicates that these mutations are producing changes in affinity of the receptor binding sites rather than allosteric changes in the cooperativity of the interaction.
Insulin Binding to Alanine Mutant Secreted Receptors
In a bivalent interaction between insulin and the receptor, in which insulin interacts with the L1 and CTα binding site (Site1) and the possible FnIII domain site (Site 2), perturbations of the interactions of insulin with either site would be predicted to affect the affinity of the receptor for insulin. We have confirmed this prediction by demonstrating that Site 1 mutations, compromising the affinity of the secreted receptor for insulin, also produce compromise of the affinity of the native receptor for insulin (17). Thus, to exclude the possibility that the alanine mutations of Lys482, Leu552, Asp591, Ile602, Lys616, Asp620 and Pro621 impair affinity for insulin by compromising affinity of Site 1 by an indirect mechanism, we examined their impact on the insulin binding properties of the secreted insulin receptor extra-cellular domain; insulin binds only to Site 1 in this form of the receptor (2). cDNAs encoding mutant secreted receptors were transiently expressed in PEAK cells and conditioned medium was harvested 72h post-transfection. We have previously shown that the secretion of recombinant insulin receptor mutants is exquisitely sensitive to the integrity of the structure of the receptor protein (38). Therefore, in order to compare expression of the secreted mutants, we measured tracer insulin binding of conditioned medium of transfected cells in a capture assay utilizing anti-insulin receptor antibody 83-7(32). The epitope of this antibody is conformation sensitive and is located in the cysteine rich domain (32). The results of these determinations (Table 3) indicated that, with the possible exception of the Asp620 to Ala mutant, all mutant receptors bound comparably to 83-7 and that their expression was similar to or greater than that of wild type receptor. These findings, therefore, indicate that the structural integrity of the alanine mutants of Lys484, Leu552, Asp591, Ileu602, Lys616, and Pro621 is largely intact.
Table 3.
Dissociation Constants of Alanine Mutations of Secreted Recombinant Insulin Receptor Extra-cellular Domain
| Mutanta | B/T (%)b | Dissociation Constant (nM)c |
|---|---|---|
| WT | 36.0 ± 2.4 | 2.63 ± 0.01 |
| K484A | 43.1 ± 2.5 | 3.55 ± 0.05 |
| L552A | 34.0 ± 3.1 | 2.96 ± 0.11 |
| D591A | 30.9 ± 2.6 | 2.05 ± 0.15 |
| I602A | 41.1 ± 3.1 | 1.56 ± 0.10 |
| K616A | 37.6 ± 3.2 | 1.25 ± 0.30 |
| D620A | 26.6 ± 5.2 | 8.40 ± 0.56 |
| P621A | 41.6 ± 4.7 | 1.97 ± 0.04 |
The mutants are designated by the amino acid being mutated, in single letter code, followed by its position in the receptor sequence and then alanine.
cDNAs encoding alanine mutants of the recombinant secreted insulin receptor extra-cellular domain were transiently expressed in 293PEAK cells. To compare relative expression of receptor mutants, tracer 125I-(Tyr A14) insulin binding, B/T, of receptor, captured from a 1:20 dilution of conditioned medium by immobilized anti-receptor antibody 83-7 (32), was determined; under these conditions tracer binding of wild type secreted insulin receptor is 30–60% that obtained with undiluted conditioned medium (Whittaker, unpublished observations).
Equilibrium binding assays with insulin and 125I-(Tyr A14) insulin were performed. Dissociation constants were obtained from binding isotherms fitted to a single site model.. Results are the mean ± SEM of 3 independent determinations.
Competitive binding assays with 125I-[TyrA14] insulin and unlabeled insulin were then performed to further characterize their binding properties. Data from these experiments were fitted to a single site binding model to obtain the dissociation constant for insulin. These results are summarized in Table 3 and a representative assay is shown in Figure 3. The dissociation constant of the wild type secreted receptor was 2.6nM, which is comparable to our previously published values for this form of the receptor (17). With the exception of the alanine mutant of Asp620, which produced a 3- to 4- fold increase, the dissociation constants of the mutant secreted recombinant receptors for insulin were less than 2- fold greater than that of the wild type receptor. Thus these mutations had no biologically significant effect on the interaction between insulin and the secreted receptor and their effects on the affinity of the holo-receptor are due to perturbations of the interaction at the site of mutation. In contrast the alanine mutation of Asp620 must disrupt the affinity of the holo-receptor, in part, by perturbation of the interaction of Site 1 with insulin.
Figure 3. Insulin Binding to Alanine Mutant Secreted Receptors.

cDNAs encoding alanine mutants of the recombinant secreted insulin receptor extra-cellular domain were transiently expressed in 293PEAK cells. Equilibrium binding assays with insulin and 125I-(Tyr A14) insulin were performed. The mutants are designated by the amino acid being mutated, in single letter code, followed by its position in the receptor sequence and then alanine. Results are expressed as B/B0 where B is 125I-(Tyr A14) insulin bound by receptor at the designated insulin concentration and B0 is 125I-(Tyr A14) insulin bound by receptor in the absence of unlabeled insulin.
In view of the above and our previous finding that an insulin receptor L2, FnIII-1 domain – immunoglobulin Fc fusion protein contains determinants of insulin binding (26), we would conclude that the perturbations of insulin binding to the holo-receptor due to alanine mutations of Lys484, Leu552, Asp591, Ile602, Lys616 and Pro621 are the consequence of disruption of side chain interactions with insulin at the site of mutation and that they form the functional epitope of a second ligand binding site of the insulin receptor.
Complementation Studies
In order to further characterize the mechanism of high affinity interactions of insulin with the receptor and to determine the topology of the interaction, we performed complementation experiments. The published crystal structure of the secreted receptor extra-cellular domain (14) suggests that the bivalent binding of insulin by the receptor is a trans- interaction; the distance between the L1 domain of one monomer and the FnIII domains of the other monomer is too great for an insulin molecule to simultaneously bind to them. However as discussed above, this form of the receptor has a much lower affinity than the native receptor. Secreted extra-cellular domain fusion proteins, in which the receptor C-terminus is tethered by a coiled coil peptide (39), an immunoglobulin Fc fragment or lambda constant region (40) all have affinities for insulin which are indistinguishable from that of the native receptor. The structures of these fusion partners, particularly the immunoglobulin Fc fragment, suggest that the separation of the two limbs of the inverted V of the receptor in these proteins must be considerably closer than in the published ecto-domain structure and thus the possibility that insulin binds to its two cognate ligand binding sites in cis- must be considered.
Mutant IGF-I or insulin receptor monomers co-expressed in 293PEAK cells have a high propensity to form hybrid dimers (41). We, therefore, transiently co-transfected 293PEAK cells with combinations of cDNAs encoding mutant receptors with unique C-terminal epitope tags, and then purified the resulting hybrid receptors from lysates of transfected cells by sequential immuno-affinity chromatography. In order to exclude the possibility that non-hybrid receptors could co-purify in this procedure, we subjected a mixture of equal parts of detergent lysates from cells transfected with a cDNA encoding a FLAG-tagged wild type receptor and from cells transfected with a cDNA encoding Glu-Glu–tagged wild type receptor to the immuno-affinity chromatography protocol. Despite the presence of initial insulin binding activity equivalent to that present in lysates from cells co-transfected with the same cDNAs, no insulin binding activity was recovered at the completion of the procedure (data not shown), indicating that only covalently associated hybrid receptors are purified by this procedure.
We characterized the insulin binding properties of a variety of purified hybrid receptors formed from different combinations of an inactivating alanine mutation of Arg14 (17), alanine mutation of Leu552 and wild type receptors in competitive binding assays. The results of these experiments are summarized in Table 4 and a representative experiment is shown in Figure 4. Purified homomeric wild type hybrids (WT/WT) and homomeric hybrids with alanine mutations of Arg14 (R14A/R14A) or Leu552 (L552A/L552A) recapitulated the binding properties of mutant receptors with the corresponding mutations (37–39). Tracer binding of 125I-[TyrA14] insulin by R14A/R14A was too low to permit competitive binding assays for determination of affinity and the affinity of L552A/L552A was decreased 4- to 5- fold compared to that of WT/WT. However, it should be noted that the affinity of hybrid receptors for insulin (Table 4) was increased compared to that of the equivalent non-hybrid receptors (Table 2) but it is not clear whether this is an intrinsic property of the hybrids or a consequence of the purification procedures.
Table 4.
Complementation studies; insulin binding properties of hybrid receptors
| Hybrida | IC50 (pM)b | Dissociation Constantsb | |
|---|---|---|---|
| Kd1 (pM) | Kd2 (nM) | ||
| WTFLAG/WTEE | 55 ± 3 | 21 ± 2 | 0.32 ± 0.04 |
| R14AFLAG/R14AEE | ND | ND | ND |
| L552AFLAG/L552AEE | 253 ± 20 | 153 ± 3 | 1.47 ± 0.23 |
| R14AFLAG/WTEE | 60 ± 1 | 25 ± 3 | 0.31 ± 0.05 |
| L552AFLAG/WTEE | 122 ± 8 | 64 ± 6 | 0.84 ± 0.12 |
| R14AFLAG/L552AEE | 71 ± 1 | 20 ± 1 | 0.25 ± 0.01 |
| R14A+L552AFLAG/WTEE | 275 ± 3 | 157 ± 3 | 1.65 ± 0.04 |
| R14A+L552AFLAG/R14AEE | ND | ND | ND |
| R14A+L552AFLAG/L552AEE | 362 ± 23 | 218 ± 12 | 2.00 ± 0.11 |
| R14A+L552AFLAG/R14A+L552AEE | ND | ND | ND |
The mutations in the hybrid receptor monomers are designated by the amino acid being mutated, in single letter code, followed by its position in the receptor sequence and then alanine. The epitope tag is shown in superscript. Thus R14A+L552AFLAG/L552AEE is a hybrid receptor, in which one monomer contains alanine mutations of Arg14 and Leu552 and a C-terminal FLAG-tag and the second monomer has an alanine mutation of Leu552 and a C-terminal Glu-Glu tag.
Hybrid insulin receptors were generated and purified as described in Experimental Procedures. Equilibrium binding assays with insulin and 125I-(Tyr A14) insulin were performed. Dissociation constants and IC50’s were obtained from binding isotherms fitted to a sequential two-site model as previously described (33). Results are the mean ± SEM of 3 independent determinations.
Figure 4. Competitve Binding Assays of Hybrid Receptors.

Hybrid insulin receptors were generated and purified as described in “Methods”. Equilibrium binding assays with insulin and 125I-(Tyr A14) insulin were performed. The mutations in the hybrid receptor monomers are designated by the amino acid being mutated, in single letter code, followed by its position in the receptor sequence and then alanine. The epitope tag is shown in superscript. Thus R14A+L552AFLAG/L552AEE is a hybrid receptor, in which one monomer contains alanine mutations of Arg14 and Leu552 and a C-terminal FLAG-tag and the second monomer has an alanine mutation of Leu552 and a C-terminal Glu-Glu tag. Results are expressed as B/B0 where B is 125I-(Tyr A14) insulin bound by receptor at the designated insulin concentration and B0 is 125I-(Tyr A14) insulin bound by receptor in the absence of unlabeled insulin.
To determine whether a wild type receptor monomer could rescue the insulin binding properties of an inactive receptor monomer in a hybrid receptor, we examined insulin binding to a hybrid receptor formed from a wild type receptor monomer and the inactive mutant receptor monomer with an alanine mutation of Arg14. This receptor hybrid had an IC50 of 0.072nM and Kds close to those of the wild type hybrid (Table 4), thus confirming that a wild type insulin receptor monomer can complement an inactive monomer and restore high affinity insulin binding. These findings are consistent with our previously published studies of the homologous IGF-I receptor (41).
In order to elucidate the topology of the interaction, we examined the insulin binding properties of hybrids with alanine mutations of Arg14 and of Leu552. Initial experiments were performed with one hybrid receptor with a single alanine mutation of Arg14 in one monomer and an alanine mutation of Leu552 in the other (R14A/L552A) and a second with alanine mutations of Arg14 and Leu552 in one monomer and a second wild type monomer (R14A +L552A/WT). If insulin bound to Sites 1 and 2 of the receptor in trans-, R14A/L552A would be predicted to have one intact high affinity binding site and one completely inactive high affinity site and should thus bind to this hybrid with a similar affinity to its affinity for wild type receptor. In contrast, if insulin bound to the two sites in cis-, it would be predicted that this hybrid would have one completely inactive high affinity site and one with an affinity similar to that of the Leu552 to alanine mutant. For the R14A + L552A/WT hybrid these predictions would be reversed. In equilibrium binding studies (Table 4 and Figure 4), R14A/L552A had an affinity for insulin near that of WT/WT, whereas R14A + L552A/WT had an affinity for insulin that was 4- to 5- fold lower than that of WT/WT and was similar to that L552A/L552A. Taken together these results indicate that the presence of one intact Site 1 on one receptor monomer and one intact Site 2 on the second monomer are sufficient for to confer full affinity insulin binding in trans-.
These findings were further substantiated by experiments with hybrids with other combinations of monomers with Site 1 and Site 2 mutations. When complemented with either wild type monomer or a monomer with the Leu552 to alanine mutation, double mutant monomers with both Arg14 and Leu552 to alanine mutations i.e. R14A+L552A/WT or R14A+L552A/L552A hybrids, bound insulin with a similar affinity to the L552A/L552A hybrid (Table 4 and Figure 4). Complementation with monomers with the Arg14 to alanine mutation or the Arg14 and Leu552 to alanine mutations (R14A+L552A/R14A and R14A+L552A/R14A+L552A respectively) resulted in tracer binding of 125I-[TyrA14] insulin that was too low to permit competitive binding assays.
Complementation of a monomer with the the Leu552 to alanine mutation with a wild type monomer resulted in a hybrid with an affinity for insulin that was only half that of the WT/WT hybrid (Table 4 and Figure 4). This would be consistent with the presence of two populations of functional binding sites. The first would be formed from trans- interactions between the intact Site 1 and Site 2 of each monomer and would thus have full affinity for insulin. The second would be formed from trans- interactions between intact Site 1 and the Leu552 to Ala mutant of Site 2 with compromised affinity for insulin. This results in the observed affinity for insulin intermediate between that of the homomeric Leu552 to Ala mutant receptor and that of the wild type receptor.
DISCUSSION
In the present study, we have used structure directed alanine mutagenesis to identify residues in the FnIII-1 and FnIII-2 domains of the insulin receptor whose alanine mutation perturbs insulin binding to the full length receptor but has minimal effect on the affinity of the ligand binding site of the secreted recombinant receptor (Site 1) for insulin. These findings provide evidence for the existence of a second ligand binding site (Site 2) of the insulin receptor located in these domains. The complementation experiments with hybrid receptors with inactivating mutations of each site described here also demonstrate that insulin interacts with and cross-links the two receptor binding sites in trans- to generate a high affinity interaction, i.e. the insulin molecule binds to Site 1 of one monomer and to Site 2 of the other monomer.
The identification of a second receptor ligand binding site, trans- interactions between insulin and the two ligand binding sites of the receptor and the anti-parallel organization of the receptor monomers in the extra-cellular domain dimer structure provide support for DeMeyts’ model of bivalent interaction of insulin with the receptor (2). This model and the similar model of Schaffer (42) were proposed to explain the complexities of insulin-receptor interactions, when it was found that substitutions of LeuA13 and LeuB17 located on the opposite side of the insulin molecule from the classical receptor binding site compromise affinity for the receptor (42). This model proposes that insulin has two distinct receptor binding sites and each receptor α subunit has two corresponding ligand binding sites organized in an anti-parallel topology in the receptor dimer. High affinity interactions are generated by trans- cross-linking of the receptor ligand binding sites and low affinity interactions arise from single site occupancy. The model further posits that two cross-links cannot occur simultaneously. Therefore, the binding and cross-linking of unoccupied ligand binding sites by a second insulin molecule would result in the disruption of the first cross-link and hence a decrease in affinity of the interaction, accompanied by an increase in the dissociation rate from this site. The model thus can account for both the existence of high and low affinity binding sites and negative cooperativity.
It is unclear how the data obtained from the equilibrium binding studies on wild type and mutant holo-receptors in the present study relate to this model of the mechanism of insulin-receptor interactions. The two site sequential model used to obtain the dissociation constants for insulin by non-linear regression analysis utilizes an Adair equation (34) to describe the two step binding of insulin to high and low affinity sites. As such, this is a valid thermodynamic description of the interaction but does not provide any insights into the underlying molecular mechanisms (35). It is tempting to suggest that the high and low affinity dissociation constants of the holo-receptor obtained from these analyses are the dissociation constants of the respective bivalent and monovalent insulin-receptor interactions. If this were the case, this might suggest that these data are inconsistent with the De Meyts model (2), as in the wild type receptor the dissociation constant of the low affinity site (Site 1; Kd2 = 0.52nM, Table 2) is a significant underestimate of the dissociation constant of the monovalent interaction determined for the secreted receptor (2.6nM, Table 3) and, furthermore, the Site 2 alanine mutations that are disruptive of interactions of insulin with the holo-receptor but not the secreted receptor appear to affect both dissociation constants proportionately (Table 2). However given the potential complexities of the insulin-receptor interaction predicted by this model, a detailed mathematical description and more detailed kinetic and thermodynamic studies will be necessary to resolve this issue. Similarly such information will also be necessary to resolve other apparent inconsistencies with the De Meyts model, i.e. the apparent absence of the effects of Site 2 mutations on co-operativity of the insulin receptor interaction (Table 2) and the apparent complex binding behavior of the hybrid receptor formed from a wild type monomer and a monomer with an inactivating Site 1 mutation (R14AFLAG/WTEE, Table 4).
When viewed in the context of the structure of the receptor, the functional epitope of this second insulin binding site is formed from scattered side-chains rather than a contiguous patch (hot-spot), as is commonly observed in sites of protein-protein interaction (43). This may be a technical artifact. It is possible that some disruptive mutations have been overlooked because we have used a 2-fold increase in IC50 as the criterion for a biologically significant perturbation of side-chain interaction by alanine substitution, in accord with published studies of alanine scanning of protein–protein interfaces (37). This would be consistent with the relatively small decrease in affinity of the multiple alanine mutations. Also we have not directly examined the interaction of insulin with this epitope but have determined the effect of mutations on the affinity of the bivalently bound insulin. Thus it is possible that the energetic contributions of side chains of the other residues in the functional epitope have been obscured by compensatory changes in interactions elsewhere in the ligand–receptor interface. This notion is supported by our previous studies of the insulin receptor, which have demonstrated that the ligand receptor interface exhibits considerable plasticity (17). This has a precedent in one of the best studied protein-protein interactions, the interaction of growth hormone with its receptor (44). While the receptor is monomeric, growth hormone interacts with a non-covalent receptor dimer in an ordered sequential reaction which is analogous to that proposed for insulin and its receptor. Walsh et al. have demonstrated the two ligand-receptor interfaces of the non-covalent receptor dimer are allosterically coupled (45). Mutations compromising affinity of one interface produce conformational and energetic re-organization of the second interface leading to an absence of net change in overall affinity for growth hormone.
While it is possible that the apparent absence of a hot spot in this functional epitope is an artifact, there are also some precedents for the absence of hotspots in protein-protein interfaces. The binding sites of protein L for immunoglobulin-κ light chain lack hot spots (46). It was suggested that this might have been a consequence of the large number of main chain interactions between the two proteins. In the absence of a high resolution structure of the high affinity insulin-receptor complex, it is not possible determine whether this might apply to this insulin-receptor interface. It should also be noted that this study characterized a bivalent interaction of protein L with immunoglobulin-k light chain and that the monovalent interaction was low affinity (900nM). Roisman et al (47) also found that alanine mutations of residues in the interface between IFN-a2 and its receptor IFNA-R1, a low affinity interaction (dissociation constant, Kd = 1.5μM), produced reductions in affinity of fivefold or less without any hotspots. If these findings are characteristic of low affinity interactions, this may be the reason for the absence of a hotspot in this insulin-receptor interface. The apparent free energy contributions attributable to the mutated residues that perturb affinity for insulin are considerably smaller than those we have observed with mutations of residues forming the functional epitope of Site 1 (17), suggesting that the affinity of this site is considerably lower than that of Site 1, probably in the high nanomolar to low micromolar range. It should also be noted that they are consistent with the effects of alanine mutations of IGF-I on interactions at the equivalent interface of the homologous IGF-I-IGF-I receptor complex (48).
Our previous mutational analyses have demonstrated that the affinity of site 1 for insulin is the major determinant of the affinity of the receptor for insulin (17). Further we and other laboratories have demonstrated that it is also the major determinant of specificity for insulin (15;26). This dominant role for Site 1 is also supported by the results of our complementation studies, in particular the finding that complete inactivation of Site 1 is essential for full complementation. Taken together these findings suggest that, as proposed by McKern et al. (14), binding to Site 1 may be the initial step in the ordered sequential interaction of insulin with the receptor. This would also be consistent with the apparent low affinity of Site 2 for insulin. Binding to Site 1 of free insulin in solution would lead to a major increase in the effective concentration of insulin at the receptor; it would no longer be free to diffuse in three dimensions. This would thus facilitate the second step of binding of insulin to the low affinity Site 2.
CONCLUDING REMARKS
The simultaneous binding of insulin to Sites 1 and 2 of the receptor that we have described produces conformational changes in the receptor extra-cellular domain and generates a high affinity interaction that lead to the activation. It should also be noted that recent studies by Chan et al. (49) have shown that Site 1 binding involves trans- interactions between the L1 domain of one monomer and the CTα of the other and also probably produces conformational changes in the receptor. These combined conformational changes in the receptor extra-cellular domain lead to activation of the receptor’s intracellular tyrosine kinase activity and insulin signal transduction. The present structure provides little insight into the nature of these changes and further receptor structures, in the presence and absence of ligand, will be essential to fully elucidate the mechanisms of trans-membrane signaling in response to insulin binding
Figure 5. Second Insulin Binding Site of the Insulin Receptor.
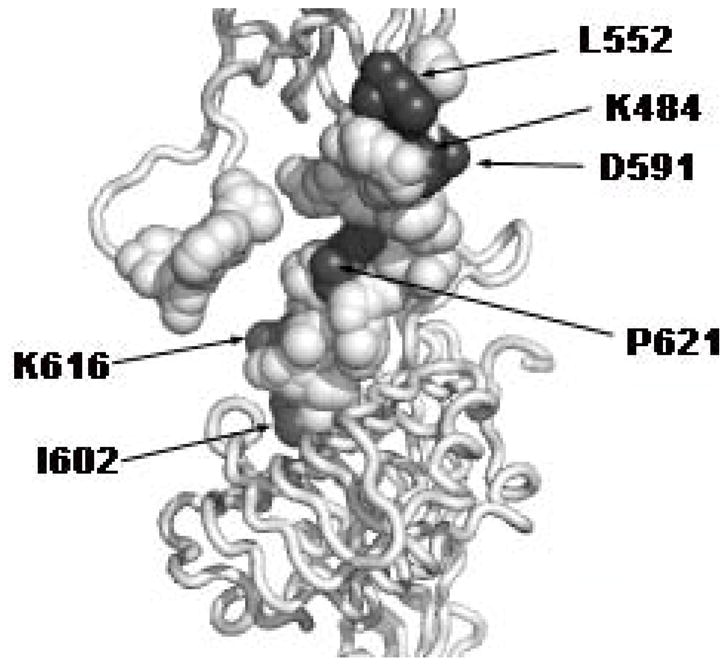
The L1 domain of monomer 1 and the FnIII domains of monomer 2 are shown (see legend to Figure 1). Residues mutated to alanine are shown in atomic sphere representation. Residues in dark grey are those, whose mutation to alanine produces a 2- to 5- fold decrease in affinity of the holo-receptor for insulin, without affecting the affinity of the secreted recombinant insulin receptor.
Acknowledgments
We thank Michael Weiss (Case Western Reserve University, Cleveland) and Dr. Pierre De Meyts (Hagedorn Research Institut, Bagsvaerd, Denmark) for helpful discussion.
Footnotes
Abbreviations: IGF-II, insulin-like growth factor II; IGFs, insulin-like growth factors; CR, cysteine rich; FnIII, type 3 fibronectin repeat; ID, insert domain; CTα, C-terminal peptide (amino acids 704-719) of the insulin receptor α subunit; 125I-[TyrA14] insulin, insulin mono-iodinated on Tyrosine 14 of the A chain.
This work was supported by a Grant DK065890 (to J.W.) from the National Institute of Diabetes, Digestive and Kidney Diseases of the National Institutes of Health (NIH)
References
- 1.Kitamura T, Kahn CR, Accili D. Insulin receptor knockout mice. Annu Rev Physiol. 2003;65:313–332. doi: 10.1146/annurev.physiol.65.092101.142540. [DOI] [PubMed] [Google Scholar]
- 2.De Meyts P, Whittaker J. Structural biology of insulin and IGF1 receptors: implications for drug design. Nat Rev Drug Discov. 2002;1:769–783. doi: 10.1038/nrd917. [DOI] [PubMed] [Google Scholar]
- 3.Chang L, Chiang SH, Saltiel AR. Insulin signaling and the regulation of glucose transport. Mol Med. 2004;10:65–71. doi: 10.2119/2005-00029.Saltiel. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- 5.Frasca F, Pandini G, Scalia P, Sciacca L, Mineo R, Costantino A, Goldfine ID, Belfiore A, Vigneri R. Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol Cell Biol. 1999;19:3278–3288. doi: 10.1128/mcb.19.5.3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pandini G, Conte E, Medico E, Sciacca L, Vigneri R, Belfiore A. IGF-II binding to insulin receptor isoform A induces a partially different gene expression profile from insulin binding. Ann N Y Acad Sci. 2004;1028:450–456. doi: 10.1196/annals.1322.053. [DOI] [PubMed] [Google Scholar]
- 7.Denley A, Carroll JM, Brierley GV, Cosgrove L, Wallace J, Forbes B, Roberts CT., Jr Differential activation of insulin receptor substrates 1 and 2 by insulin-like growth factor-activated insulin receptors. Mol Cell Biol. 2007;27:3569–3577. doi: 10.1128/MCB.01447-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jensen M, Hansen B, De Meyts P, Schaffer L, Urso B. Activation of the insulin receptor by insulin and a synthetic peptide leads to divergent metabolic and mitogenic signaling and responses. 2007 doi: 10.1074/jbc.M704599200. [DOI] [PubMed] [Google Scholar]
- 9.Boni-Schnetzler M, Scott W, Waugh SM, DiBella E, Pilch PF. The insulin receptor. Structural basis for high affinity ligand binding. J Biol Chem. 1987;262:8395–8401. [PubMed] [Google Scholar]
- 10.Deger A, Kramer H, Rapp R, Koch R, Weber U. The nonclassical insulin binding of insulin receptors from rat liver is due to the presence of two interacting alpha-subunits in the receptor complex. BIOCHEM BIOPHYS RES COMMUN. 1986;135:458–464. doi: 10.1016/0006-291x(86)90016-1. [DOI] [PubMed] [Google Scholar]
- 11.Sweet LJ, Morrison BD, Pessin JE. Isolation of functional alpha beta heterodimers from the purified human placental alpha 2 beta 2 heterotetrameric insulin receptor complex. A structural basis for insulin binding heterogeneity. J Biol Chem. 1987;262:6939–6942. [PubMed] [Google Scholar]
- 12.Baker EN, Blundell TL, Cutfield JF, Cutfield SM, Dodson EJ, Dodson GG, Hodgkin DM, Hubbard RE, Isaacs NW, Reynolds CD. The structure of 2Zn pig insulin crystals at 1.5 A resolution. Philos Trans R Soc Lond B Biol Sci. 1988;319:369–456. doi: 10.1098/rstb.1988.0058. [DOI] [PubMed] [Google Scholar]
- 13.Zhou A, Webb G, Zhu X, Steiner DF. Proteolytic processing in the secretory pathway. J Biol Chem. 1999;274:20745–20748. doi: 10.1074/jbc.274.30.20745. [DOI] [PubMed] [Google Scholar]
- 14.McKern NM, Lawrence MC, Streltsov VA, Lou MZ, Adams TE, Lovrecz GO, Elleman TC, Richards KM, Bentley JD, Pilling PA, Hoyne PA, Cartledge KA, Pham TM, Lewis JL, Sankovich SE, Stoichevska V, Da SE, Robinson CP, Frenkel MJ, Sparrow LG, Fernley RT, Epa VC, Ward CW. Structure of the insulin receptor ectodomain reveals a folded-over conformation. Nature. 2006;443:218–221. doi: 10.1038/nature05106. [DOI] [PubMed] [Google Scholar]
- 15.Andersen AS, Kjeldsen T, Wiberg FC, Vissing H, Schaffer L, Rasmussen JS, De Meyts P, Moller NP. Identification of determinants that confer ligand specificity on the insulin receptor. J Biol Chem. 1992;267:13681–13686. [PubMed] [Google Scholar]
- 16.Kristensen C, Wiberg FC, Schaffer L, Andersen AS. Expression and characterization of a 70-kDa fragment of the insulin receptor that binds insulin. Minimizing ligand binding domain of the insulin receptor. J Biol Chem. 1998;273:17780–17786. doi: 10.1074/jbc.273.28.17780. [DOI] [PubMed] [Google Scholar]
- 17.Whittaker J, Whittaker L. Characterization of the functional insulin binding epitopes of the full-length insulin receptor. J Biol Chem. 2005;280:20932–20936. doi: 10.1074/jbc.M411320200. [DOI] [PubMed] [Google Scholar]
- 18.Huang K, Xu B, Hu SQ, Chu YC, Hua QX, Qu Y, Li B, Wang S, Wang RY, Nakagawa SH, Theede AM, Whittaker J, De Meyts P, Katsoyannis PG, Weiss MA. How insulin binds: the B-chain alpha-helix contacts the L1 beta-helix of the insulin receptor. J Mol Biol. 2004;341:529–550. doi: 10.1016/j.jmb.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 19.Huang K, Chan SJ, Hua QX, Chu YC, Wang RY, Klaproth B, Jia W, Whittaker J, De MP, Nakagawa SH, Steiner DF, Katsoyannis PG, Weiss MA. The A-chain of insulin contacts the insert domain of the insulin receptor. Photo-cross-linking and mutagenesis of a diabetes-related crevice. J Biol Chem. 2007 doi: 10.1074/jbc.M705996200. [DOI] [PubMed] [Google Scholar]
- 20.Kurose T, Pashmforoush M, Yoshimasa Y, Carroll R, Schwartz GP, Burke GT, Katsoyannis PG, Steiner DF. Cross-linking of a B25 azidophenylalanine insulin derivative to the carboxyl-terminal region of the alpha-subunit of the insulin receptor. Identification of a new insulin-binding domain in the insulin receptor. J Biol Chem. 1994;269:29190–29197. [PubMed] [Google Scholar]
- 21.Xu B, Hu SQ, Chu YC, Huang K, Nakagawa SH, Whittaker J, Katsoyannis PG, Weiss MA. Diabetes-associated mutations in insulin: consecutive residues in the B chain contact distinct domains of the insulin receptor. Biochemistry. 2004;43:8356–8372. doi: 10.1021/bi0497796. [DOI] [PubMed] [Google Scholar]
- 22.Whittaker J, Garcia P, Yu GQ, Mynarcik DC. Transmembrane domain interactions are necessary for negative cooperativity of the insulin receptor. Mol Endocrinol. 1994;8:1521–1527. doi: 10.1210/mend.8.11.7877620. [DOI] [PubMed] [Google Scholar]
- 23.Benyoucef S, Surinya KH, Hadaschik D, Siddle K. Characterization of insulin/IGF hybrid receptors: contributions of the insulin receptor L2 and Fn1 domains and the alternatively spliced exon 11 sequence to ligand binding and receptor activation. 403. 2007. pp. 603–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brandt J, Andersen AS, Kristensen C. Dimeric fragment of the insulin receptor alpha-subunit binds insulin with full holoreceptor affinity. J Biol Chem. 2001;276:12378–12384. doi: 10.1074/jbc.M009402200. [DOI] [PubMed] [Google Scholar]
- 25.Fabry M, Schaefer E, Ellis L, Kojro E, Fahrenholz F, Brandenburg D. Detection of a new hormone contact site within the insulin receptor ectodomain by the use of a novel photoreactive insulin. J Biol Chem. 1992;267:8950–8956. [PubMed] [Google Scholar]
- 26.Hao C, Whittaker L, Whittaker J. Characterization of a second ligand binding site of the insulin receptor. BIOCHEM BIOPHYS RES COMMUN. 2006;347:334–339. doi: 10.1016/j.bbrc.2006.06.089. [DOI] [PubMed] [Google Scholar]
- 27.Schumacher R, Soos MA, Schlessinger J, Brandenburg D, Siddle K, Ullrich A. Signaling-competent receptor chimeras allow mapping of major insulin receptor binding domain determinants. J Biol Chem. 1993;268:1087–1094. [PubMed] [Google Scholar]
- 28.Zhang B, Roth RA. A region of the insulin receptor important for ligand binding (residues 450–601) is recognized by patients’ autoimmune antibodies and inhibitory monoclonal antibodies. Proc Natl Acad Sci U S A. 1991;88:9858–9862. doi: 10.1073/pnas.88.21.9858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2001. [Google Scholar]
- 30.Kirsch RD, Joly E. An improved PCR-mutagenesis strategy for two-site mutagenesis or sequence swapping between related genes. Nucleic Acids Res. 1998;26:1848–1850. doi: 10.1093/nar/26.7.1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hedo JA, Harrison LC, Roth J. Binding of insulin receptors to lectins: evidence for common carbohydrate determinants on several membrane receptors. Biochemistry. 1981;20:3385–3393. doi: 10.1021/bi00515a013. [DOI] [PubMed] [Google Scholar]
- 32.Prigent SA, Stanley KK, Siddle K. Identification of epitopes on the human insulin receptor reacting with rabbit polyclonal antisera and mouse monoclonal antibodies. J Biol Chem. 1990;265:9970–9977. [PubMed] [Google Scholar]
- 33.Christoffersen CT, Bornfeldt KE, Rotella CM, Gonzales N, Vissing H, Shymko M, Ten Hoeve J, Groffen J, Heisterkamp N, De Meyts P, Wood S, Vogt PH. Negative cooperativity in the insulin-like growth factor-I receptor and a chimeric IGF-I/insulin receptor. Endocrinology. 1994;135:472–475. doi: 10.1210/endo.135.1.8013387. [DOI] [PubMed] [Google Scholar]
- 34.Adair GS. The Hemoglobin System, VI. The Oxygen Dissociation Curve of Hemoglobin. J Biol Chem. 1925;63:529–545. [Google Scholar]
- 35.Levitzki A. Quantitative Aspects of Allosteric Mechanisms. Springer Verlag; Berlin;Heidelberg;New York: 1978. [DOI] [PubMed] [Google Scholar]
- 36.Limbird LE. Receptors: A Short course on theory and methods. Springer; New York: 2005. [Google Scholar]
- 37.Wells JA. Systematic mutational analyses of protein-protein interfaces. Methods Enzymol. 1991;202:390–411. doi: 10.1016/0076-6879(91)02020-a. [DOI] [PubMed] [Google Scholar]
- 38.Williams PF, Mynarcik DC, Yu GQ, Whittaker J. Mapping of an NH2-terminal ligand binding site of the insulin receptor by alanine scanning mutagenesis. J Biol Chem. 1995;270:3012–3016. doi: 10.1074/jbc.270.7.3012. [DOI] [PubMed] [Google Scholar]
- 39.Hoyne PA, Cosgrove LJ, McKern NM, Bentley JD, Ivancic N, Elleman TC, Ward CW. High affinity insulin binding by soluble insulin receptor extracellular domain fused to a leucine zipper. FEBS Lett. 2000;479:15–18. doi: 10.1016/s0014-5793(00)01872-x. [DOI] [PubMed] [Google Scholar]
- 40.Bass J, Kurose T, Pashmforoush M, Steiner DF. Fusion of insulin receptor ectodomains to immunoglobulin constant domains reproduces high-affinity insulin binding in vitro. J Biol Chem. 1996;271:19367–19375. doi: 10.1074/jbc.271.32.19367. [DOI] [PubMed] [Google Scholar]
- 41.Chakravarty A, Hinrichsen J, Whittaker L, Whittaker J. Rescue of ligand binding of a mutant IGF-I receptor by complementation. BIOCHEM BIOPHYS RES COMMUN. 2005;331:74–77. doi: 10.1016/j.bbrc.2005.03.122. [DOI] [PubMed] [Google Scholar]
- 42.Schaffer L. A model for insulin binding to the insulin receptor. Eur J Biochem. 1994;221:1127–1132. doi: 10.1111/j.1432-1033.1994.tb18833.x. [DOI] [PubMed] [Google Scholar]
- 43.Bogan AA, Thorn KS. Anatomy of hot spots in protein interfaces. J Mol Biol. 1998;280:1–9. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- 44.Wells JA. Binding in the growth hormone receptor complex. Proc Natl Acad Sci U S A. 1996;93:1–6. doi: 10.1073/pnas.93.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Walsh ST, Sylvester JE, Kossiakoff AA. The high- and low-affinity receptor binding sites of growth hormone are allosterically coupled. Proc Natl Acad Sci U S A. 2004;101:17078–17083. doi: 10.1073/pnas.0403336101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Svensson HG, Wedemeyer WJ, Ekstrom JL, Callender DR, Kortemme T, Kim DE, Sjobring U, Baker D. Contributions of amino acid side chains to the kinetics and thermodynamics of the bivalent binding of protein L to Ig kappa light chain. Biochemistry. 2004;43:2445–2457. doi: 10.1021/bi034873s. [DOI] [PubMed] [Google Scholar]
- 47.Roisman LC, Jaitin DA, Baker DP, Schreiber G. Mutational analysis of the IFNAR1 binding site on IFNalpha2 reveals the architecture of a weak ligand-receptor binding-site. J Mol Biol. 2005;353:271–281. doi: 10.1016/j.jmb.2005.08.042. [DOI] [PubMed] [Google Scholar]
- 48.Gauguin L, Delaine C, Alvino CL, McNeil KA, Wallace JC, Forbes BE, De MP. Alanine scanning of a putative receptor binding surface of insulin-like growth factor-I. J Biol Chem. 2008;283:20821–20829. doi: 10.1074/jbc.M802620200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chan SJ, Nakagawa S, Steiner DF. Complementation analysis demonstrates that insulin cross-links both alpha subunits in a truncated insulin receptor dimer. J Biol Chem. 2007;282:13754–13758. doi: 10.1074/jbc.M700724200. [DOI] [PubMed] [Google Scholar]