

Abstract
Although notable progress has been made in the therapeutic management of patients with chronic kidney disease in both conservative and renal replacement treatments (dialysis and transplantation), the occurrence of medication-related problems (lack of efficacy, adverse drug reactions) still represents a key clinical issue. Recent evidence suggests that adverse drug reactions are major causes of death and hospital admission in Europe and the United States. The reasons for these conditions are represented by environmental/non-genetic and genetic factors responsible for the great inter-patient variability in drugs metabolism, disposition and therapeutic targets. Over the years several genetic settings have been linked, using pharmacogenetic approaches, to the effects and toxicity of many agents used in clinical nephrology. However, these strategies, analysing single gene or candidate pathways, do not represent the gold standard, being the overall pharmacological effects of medications and not typically monogenic traits. Therefore, to identify multi-genetic influence on drug response, researchers and clinicians from different fields of medicine and pharmacology have started to perform pharmacogenomic studies employing innovative whole genomic high-throughput technologies. However, to date, only few pharmacogenomics reports have been published in nephrology underlying the need to enhance the number of projects and to increase the research budget for this important research field. In the future we would expect that, applying the knowledge about an individual's inherited response to drugs, nephrologists will be able to prescribe medications based on each person's genetic make-up, to monitor carefully the efficacy/toxicity of a given drug and to modify the dosage or number of medications to obtain predefined clinical outcomes.
Keywords: chronic kidney disease, drugs, nephrology, pharmacogenetics, pharmacogenomics
Introduction
During the last 30 years, new medications (e.g. more selectively targeted immunosuppressants, angiotensin-converting enzyme inhibitors) have been introduced to treat major renal pathologies (e.g. acute and chronic glomerulonephritides) to slow down the progression of chronic kidney diseases (CKD) and to reduce the development of clinical complications associated to dialysis (peritoneal and haemodialysis) and renal transplantation [1–4]. However, the worldwide extensive use of these agents has been followed by several medication-related problems [e.g. overdose, subtherapeutic dosage, severe adverse drug reactions (ADRs)] with a large clinical impact and a consequent enormous cost for the health system. ADRs have been recognized as one of the most common causes of death and hospital admissions in the United States and Europe [5–7].
Recent evidence suggests that the latest methodologies used to adjust drug dosages (e.g. therapeutic drug monitoring) result most of the time in inadequate, non-reproducible and poor predictive efficacy/toxicity before drug administration [8,9]. Because of these limitations, researchers and clinicians are searching for new techniques to improve tailoring of drug therapy and to predict adverse events before drug administration.
Additionally, it has been well recognized that, despite the potential importance of non-genetic (e.g. age, gender, body mass index) and environmental factors (e.g. hepatic or renal function, hormonal levels and potential pharmacokinetic interactions with other co-administered drugs), inherited differences in drug metabolism and disposition and genetic variability in therapeutic targets (e.g. receptors) may have a predominant role in modulating drug effects [10–12]. Indeed, it has been estimated that genetics may account for 20–95% of variability in drug disposition and effect [13].
Despite the large amount of literature reports [10–12] suggesting a close link between genetic fingerprints and abnormal response to medications, to date a systematic approach to define the genetic contribution to different patterns of drug response is still lacking. Even if, for some drugs, it could be possible to predict their therapeutic effectiveness and dosage, established on the basis of characterization of polymorphic drug-related genes, this approach is not applicable for the majority of drugs. To this purpose, innovative automated genome-based research technologies derived from recent knowledge of the human genome project may represent a valuable tool to weight the genetic/genomic influence on pharmacological outcomes, to assist clinicians to optimize daily therapeutic strategies (Fig. 1) and to identify more selective and more appropriate targets for pharmacological interventions.
Fig. 1.
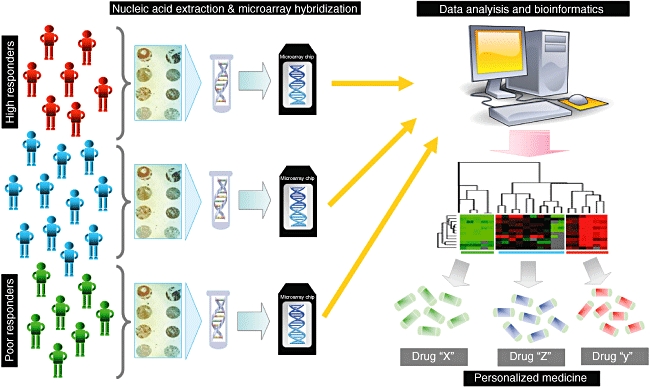
Integrated genomic methodology to personalize treatment. This schematic representation shows the main processes, from DNA or RNA harvesting to computational and statistical analysis, involved in the choice of the best treatment for different patient typologies (poor, normal or high responders to conventional medications).
Pharmacogenetics: the first step behind personalized medicine
Historical overview
For many years, several studies have emerged indicating that a substantial portion of variability in drug response is determined genetically. Approximately 40 years ago, Kalow and Gunn [14] described, for the first time, that subjects homozygous for a gene encoding for an atypical form of the enzyme butyrylcholinesterase (pseudocholinesterase) were predisposed to develop a delayed recovery from muscular paralysis and prolonged apnoea after administration of the neuromuscular blocker succinylcholine. At almost the same time, it was observed that a common genetic variation in a phase II pathway of drug metabolism (N-acetylation) could result in striking differences in the half-life and plasma concentrations of drugs metabolized by N-acetyltransferase. Such drugs included the anti-tuberculosis agent isionazid [15], the anti-hypertensive agent hydralazine [16] and the anti-arrhythmic drug procainamide [17]. In all cases these variations had clinical consequences [18]. These early examples of potential influence of inheritance on drug effects, followed by subsequent studies, gave rise to the field of ‘pharmacogenetics’.
However, the molecular genetic basis for such inherited traits began to be elucidated only in the late 1980s, with the initial cloning and characterization of polymorphic human genes encoding for drug-metabolizing enzymes [19,20].
Pharmacogenetics of drugs used commonly in clinical nephrology and transplantation
The use of different combinations of powerful drugs [e.g. calcineurin inhibitors, mammalian target of rapamycin (mTOR) inhibitors, corticosteroids] leads to a significant improvement in the treatment of several renal disorders and in the short- and long-term pharmacological management of renal transplantation recipients [1,21]. However, these drugs are hampered frequently by a narrow therapeutic index. Moreover, these agents are characterized by a high variability in pharmacokinetic behaviour and by a poor correlation between drug concentrations and pharmocodynamic effects [22–24]. ‘Tailoring’ the dose of such drugs to the specific requirements of the individual patient to minimize toxicity while maintaining efficacy is therefore a challenging goal in clinical nephrology. To achieve this objective, several research programmes have been undertaken analysing the genetic influence on the patient's response to these conventional treatments.
Azathioprine
Considerable evidence in the literature has reported that genetic polymorphisms have a major impact on the metabolism of azathioprine (AZA), a purine anti-metabolite used widely in nephrology [25–27]. This drug is catabolized mainly by the cytosolic enzyme thiopurine S-methyltransferase. This enzyme is encoded by the thiopurine S-methyltransferase coding gene (TPMT gene). The TPMT locus is subjected to several polymorphisms, with heterozygous individuals (6%–11% of Caucasian individuals) having intermediate TPMT activity and homozygous mutant individuals (0·2%–0·6% of Caucasian individuals) having very low TPMT activity. To date, 20 variant alleles (TPMT*2-*18) have been identified, which are associated with decreased activity compared with the TPMT*1 wild-type allele [28]. More than 95% of defective TPMT activity can be explained by the most frequent mutant alleles TPMT*2 and TPMT*3. The impaired or absent ability to metabolize AZA leads to high blood levels and an increased risk of developing severe and potentially life-threatening myelotoxicity when no dose reductions are performed [29,30]. Dervieux et al.[31], measuring the TPMT activity in red blood cells of paediatric patients after renal transplantation, demonstrated that elevated TPMT activity was associated with an increased risk of acute rejection. Genotyping for TPMT polymorphisms, before initiation of AZA therapy, may be a useful future tool to reduce clinical complications in patients undergoing this treatment.
Calcineurin inhibitors (CNIs)
For cyclosporine (CsA) and tacrolimus (TAC), potent agents used widely to treat a variety of autoimmune renal disorders and to prevent acute rejection after renal transplantation, the impact of genetic variability has not yet been defined completely. As shown in Table 1, for phase I metabolism it has already been demonstrated that expression of the multi-drug resistance 1 (MDR-1) gene that encodes for an efflux pomp which removes lipophilic drugs may influence significantly the pharmacokinetics and pharmacodynamics of both CsA and TAC. Regarding phase II metabolism, the relationship between polymorphisms in the P450 cytochrome system, an intracellular transporter system that is capable of carrying a variety of endogenous and exogenous compounds out of the cell, and pharmacological and clinical outcomes associated with CNI administration has been evaluated in several reports (Table 1). In particular, it has been reported that patients carrying CYP3A5*3, an allelic variant of the CYP3A5 gene that results in lack of enzyme expression, reaching high dose-adjusted levels, need lower dosages of CNIs compared to those with the wild-type genotype (CYP3A5*1/*1) [32,35,37,41–43,46,47,49,54,55]. However, the contribution of additional polymorphisms needs to be evaluated more clearly and addressed in future pharmacogenetic studies.
Table 1.
Relevant pharmacogenetic studies in nephrology
| Drug | Gene | Variant | Patients | Results | Ref. |
|---|---|---|---|---|---|
| AZA | TPMT, ITPA | TPMT*2, *3A,*3B, and *3C; ITPA (94C>A and IVS2 + 21A>C) | 157 renal TX | Mean white blood cell and platelet count were lower in carriers of variant TPMT alleles compared to patients with TPMT wild-type genotype. ITPA genotype did not influence AZA dose, haematological parameters or leucopenia risk | [25] |
| AZA | TPMT | TPMT*2, *3A, and *3C | 112 renal TX | Frequency of leucopenia episodes were significantly higher in heterozygous TPMT patients compared with those with TPMT wild-type genotype | [26] |
| AZA + CsA | TPMT | TPMT*2, *3A, *3B and *3C | 122 renal TX | TPMT*3C heterozygosity was associated with significant reductions in haematological indices and a significant decrease in CsA plasma concentrations in the first year after renal transplantation | [27] |
| CsA | CYP3A4, CYP3A5, MDR-1 | CYP3A4*18B; CYP3A5*3; MDR1 (C1236T, G2677T/A and C3435T) | 103 renal TX | Patients with a CYP3A4*1/*1 genotype had higher CsA dose-adjusted concentration compared with those with CYP3A4*18B/*18B. The dose-adjusted trough levels in patients with the CYP3A5*3/*3 genotype was higher than those with the wild-type genotype. The dose-adjusted trough levels were higher in patients with MDR-1 1236CC compared with those with 1236TT | [32] |
| CsA | CYP3A4, CYP3A5, MDR-1 | CYP3A4*1B and *3; CYP3A5*3 and *6; MDR-1 C3435T | 151 renal and heart TX | Patients carrying a CYP3A4*1B variant allele have a significantly higher oral CsA clearance compared with patients homozygous for CYP3A4*1. None of the other SNPs studied influenced any of the pharmacokinetic parameters significantly | [33] |
| CsA | CYP3A4, MDR-1 | CYP3A4-V and MDR-1 C3435T | 124 renal TX | CYP3A4-V and MDR-1 C3435T allele had no effect on CsA blood levels and dosage | [34] |
| CsA | CYP3A4, CYP3A5, MDR-1 | CYP3A4*18A, CYP3A5*3 and MDR-1 C3435T | 106 renal TX | CsA dose-adjusted trough levels were higher in CYP3A5*3/*3 than CYP3A5*1/*3 and CYP3A5*1/*1 genotype. Wild-type homozygotes for MDR-1 C3435T had a lower dose-adjusted trough levels compared with heterozygotes | [35] |
| CsA | CYP3A5 | CYP3A5*1 | 399 renal TX | CYP3A5*1 allele had no effect on CsA blood levels and dosage | [36] |
| CsA | CYP3A5, CYP3AP1 | CYP3A5*3 and CYP3AP1*3 | 67 renal TX | Patients with *1*1*1*1 CYP3A5- and CYP3AP1-linked genotypes needed higher dose of CsA compared to patients with *1*3*1*3 and *3*3*3*3 linked genotypes | [37] |
| CsA | CYP3A5, MDR-1 | CYP3A5*1/*3, MDR-1 (T-129C, C1236T, G2677 (T, A), C3435T) | 106 renal TX | None of the CsA pharmacokinetic parameters were associated with the CYP3A5 genetic polymorphism. Patients with the wild-type genotype in MDR-1 C1236T had significantly lower dose-adjusted peak drug concentrations and dose-adjusted AUC values over the first 4 h compared with mutated allele carriers. Haplotype analysis including MDR-1 C1236T, G2677(T,A) and C3435T SNPs showed no significant association between haplotypes and CsA pharmacokinetics or systemic exposure | [38] |
| CsA | MDR-1 | MDR-1 (C3435T, G2677T) | 69 renal TX | AUC (0–4)/mg dose CsA/kg was significantly higher in (C/C)3435 individuals than in C/T and T/T patients on postoperative day 3. MDR-1 G2677T variants were not correlated significantly with CsA absorption | [39] |
| CsA | MDR-1 | MDR-1 (C3435T) | 10 renal TX | Oral CsA clearance was significantly higher in subjects who carried at least one MDR-1 3435T allele compared to homozygous wild-type individuals. MDR-1 3435T allele carriers have enhanced oral clearance compared to individuals with the CC genotype | [40] |
| TAC | CYP3A4, CYP3A5 | CYP3A4*1B and CYP3A5*3 | 95 renal TX | Dose-corrected TAC levels were higher in patients with the CYP3A4*1/CYP3A5*3 genotype compared to wild-type | [41] |
| TAC | CYP3A4, CYP3A5, MDR-1 | CYP3A4*1B, CYP3A5*3, MDR-1 (C1236T, G2677T, A, C3435CT) | 832 renal TX | Homozygotes for the CYP3A4*1B, CYP3A5*3 haplotype achieved earlier therapeutic concentrations of TAC and a higher concentration to dose ratio at week 1 post-transplantation. MDR-1 haplotypes did not influence pharmacokinetic parameters | [42] |
| TAC | CYP3A5 | CYP3A5*3 | 136 renal TX | Post-transplantation TAC daily dose was lower in patients with CYP3A5*3/*3 compared to CYP3A5*1/*1 genotype | [43] |
| TAC | CYP3A5 | CYP3A5*3 | 154 renal TX | TAC dose/trough concentration ratios were lower in patients with CYP3A5*3/*3 compared to both CYP3A5*1/*3 and CYP3A5*1/*1 genotype | [44] |
| TAC | CYP3A5 | CYP3A5*3 | 118 renal TX | TAC dose-adjusted concentration was significantly higher in CYP3A5*3/*3 compared to CYP3A5*1/*3 and CYP3A5*1/*1 patients | [45] |
| TAC | CYP3A5 | CYP3A5*3 | 167 renal TX | TAC dose-adjusted trough levels were higher in the group of CYP3A5*3/*3 than in the group of CYP3A5*1 allele carriers. | [46] |
| TAC | CYP3A5 | CYP3A5*3 | 180 renal TX | Patients with CYP3A5*3/*3 homozygotes achieved TAC dose-adjusted levels compared to those with at least one CYP3A5*1 allele. | [47] |
| TAC | CYP3A5 | CYP3A5*3 | 73 renal TX | There was no difference in TAC dose-adjusted trough levels for individuals with the CYP3AP1*1/*3 genotype compared to CYP3A5*3/*3. | [48] |
| TAC | CYP3A5 | CYP3A5*3 | 80 renal TX | The mean TAC doses required to obtain the targeted concentration-to-dose ratio were significantly lower in patients with the CYP3A5*1/*1 genotype | [49] |
| TAC | MDR-1 | MDR-1 C3435T | 66 renal TX | The CC patients displayed a lower TAC level per dose than CT/TT patients | [50] |
| TAC | MDR-1 | MDR-1 (C1236T, G2677T/A, C3435T) | 206 renal TX | Lower dose-normalized blood TAC levels were achieved for MDR-1 2677-GG genotype patients, compared to 2677-TT, and for 3435-CC patients as compared to 3435-TT patients. There was a small, but significant, difference in dose requirements between haplotypes C-G-C and T-T T patients, which was not significant when patients were subclassified as producers and non-producers of cytochrome P450 3A5 (CYP3A5) | [51] |
| TAC | MDR-1 | MDR-1 (G2677T,A, T-129C, C1236T, C3435T) | 81 renal TX | The mean TAC dose required to obtain the target trough concentration was significantly higher in patients with the wild-type genotype than in those with one or two mutant alleles for the SNP G2677T,A. TAC dose requirements were not significantly different for the other SNPs | [52] |
| TAC | CYP3AP1 | CYP3AP1*3 | 178 renal TX | Patients with CYP3AP1*1/*3 or *1/*1 had lower mean TAC concentrations, with significant delay in achieving target concentrations | [53] |
| CsA and TAC | CYP3A4, CYP3A5 | CYP3A4*1B and CYP3A5*3 | 297 renal TX | For both CsA and TAC, dose-corrected trough levels were higher in CYP3A5*3/*3 than in CYP3A5*1/*3 and CYP3A5*1/*1 patients. No influence of CYP3A4*1B on CsA/TAC pharmacokinetics was observed | [54] |
| CsA and TAC | CYP3A5, MDR-1 | CYP3A5*3 and *6, MDR-1 (C1236T, G2677T/A, C3435T) | 100 renal TX | CsA and TAC dose-adjusted trough concentrations were higher in CYP3A5*3/*3 patients than in CYP3A5*1/*3 and CYP3A5*1/*1 patients. For both drugs, no association was found between trough blood concentrations or dose requirement and MDR-1 genotype | [55] |
| MPA | IMPDH II | IMPDH II 3757T>C | 101 renal TX | the IMPDH type II 3757T>C polymorphism was associated with an increased IMPDH activity in MMF-treated renal transplant patients | [56] |
| MPA and TAC | UGT1A7, UGT1A9 | UGT1A7*3, UGT1A9 1399C/C | 80 renal TX | UGT1A7 and UGT1A9 1399 polymorphisms do not contribute to interindividual differences in MPA pharmacokinetics | [57] |
| MPA | UGT1A8 | UGT1A8-999C>T; 255A>G; UGT1A8-277G>A | 74 renal TX | Adverse events were observed more frequently among individuals receiving MMF who carryied the variant UGT1A8 codon 277A, the haplotype UGT1A8H5 (−999C/codon 55A/codon 277A) and the diplotype UGT1A8H2/H5 (−999CC/codon 255AA/codon 277GA) | [58] |
| MPA and TAC | UGT1A9 | UGT1A9-275T>A; UGT1A9-2152C>T | 338 renal TX | TAC-treated patients who were UGT1A9 −275T>A and/or −2152C>T carriers displayed a 20% lower MPA AUC (0–12). UGT1A9*3 carriers displayed a 49% higher MPA AUC (0–12) when treated with TAC | [59] |
| MPA, CsA and TAC | UGT1A8, UGT1A9 | UGT1A8*2, UGT1A9-275T>A; UGT1A9-2152C>T | 93 renal TX, 11 pancreas TX, 13 simultaneous kidney and pancreas | MPA dose-corrected trough concentrations were 60% higher in subjects heterozygous or homozygous for UGT1A8*2 than in those with the wild-type. When subjects were stratified by calcineurin inhibitor status, the UGT1A8*2 effect was apparent only in the TAC group. MPA dose-corrected trough concentrations were 70% lower in carriers of the UGT1A9 −275T>A/−2152C>T polymorphism who received CsA. There was no effect of the UGT1A9 −275T>A/−2152C>T polymorphism in the TAC group | [60] |
| MPA and TAC | UGT1A8, UGT2B7 | UGT1A8*2, UGT2B7*2 | 72 renal TX | UGT1A8 and UGT2B7 allelic variants did not affected MPA concentrations | [61] |
| MPA | UGT1A9 | UGT1A9-275T>A; UGT1A9-2152C>T | 95 Renal TX | The T-275A and C-2152T SNPs of the UGT1A9 gene promoter were associated with significantly lower MPA exposure in renal recipients treated with 2 g MMF daily, and part of this effect is caused by interruption of enterohepatic recirculation of MPA | [62] |
| MPA | UGT1A8, UGT1A9, ABCC2, UGT2B7 | UGT1A8*3, UGT1A9-118(dT)(9)/(10), UGT1A9 T-440C/C-331T, UGT2B7 G211T, UGT2B7 C802T, ABCC2 C-24T, ABCC2 G1249A | 46 renal TX | The enterohepatic recirculation of MPA in the patients was more extensive in UGT1A9-118(dT)(10) allele carriers, and the exposure of acyl glucuronide (AcMPAG) was higher in patients carrying the ABCC2 G1249A genotype than those with the wild-type genotype. The other SNPs that were genotyped did not cause any significant variation in MPA and MPAG pharmacokinetic parameters | [63] |
| MPA and TAC | MRP2 | MRP2-C-24T; MRP-C-3972T | 95 renal TX | The MRP2 C-24T and C-3972T polymorphisms protect renal transplant recipients from a decrease in MPA exposure associated with mild liver dysfunction. MRP2 C-24T SNP was associated with a lower oral clearance of MPA in steady-state conditions | [64] |
| SRL | CYP3A5 | CYP3A5*3 | 50 renal TX | The SRL concentration/dose ratio (C/D) in patients with CYP3A5*3/*3 were significantly higher than that of those with *1 allele | [65] |
| SRL | CYP3A5 | CYP3A5*3 | 47 renal TX | Significantly lower AUC/dose, C(max)/dose, and C(0)/dose values were found at steady state in individuals carrying at least 1 CYP3A5*1 allele than in *3/*3 patients. Patients with the CYP3A5*1/*1 and *1/*3 genotypes required a significantly higher sirolimus daily dose to achieve the same blood concentration at steady state as *3/*3 patients | [66] |
| SRL | CYP3A5 | CYP3A5*3 | 22 renal TX | Patients with the CYP3A5*1/*1 and *1/*3 genotypes had higher oral clearance compared to CYP3A5*3 genotype | [67] |
| SRL | CYP3A4, CYP3A5, MDR-1 | CYP3A4*1B, CYP3A5*3, MDR-1 (T-129C; Cl236T; C3435T) | 149 renal TX | Patients carrying the CYP3A4*1B or the CYP3A5*1 alleles required significantly more SRL to achieve adequate blood trough concentrations. None of the MDR1 SNPs was associated with the SRL concentration/dose ratio | [68] |
AZA, azathioprine; AUC, area under the curve; CsA, cyclosporine; IMPDH, inosine monophosphate deydrogenase; ITPA, inosine triphosphatase; TAC, tacrolimus; MDR-1, multi-drug resistance 1; MPA, mycophenolic acid; MMF, mycophenolate mofetil; SRL, sirolimus; SNP, single nucleotide polymorphism; TMPT, thiopurine S-methyltransferase coding gene; TX, transplantation; Ref, reference.
Mycophenolic acid (MPA)
The metabolism of MPA, a selective inhibitor of the de novo purine synthesis via inosine monophosphate deydrogenase (IMPDH) enzyme inhibition, is also influenced largely by several genetic polymorphisms (Table 1). Uridine diphosphate-glucuronosyltransferases (UGTs) 1A8, 1A9 and 1A10 are the main enzymes responsible for the glucuronidation of MPA to its inactive metabolite 7-O-glucuronide (MPAG). UGT1A9 is the primary enzyme and is expressed predominantly in liver and kidneys and, to a lesser extent, in the gastrointestinal tract. UGT1A8 and 1A10 are expressed throughout the gastrointestinal tract [69]. In vitro studies have shown that polymorphisms in the UGT1A9 gene result in significant alteration of the UGT enzymatic activity. Two polymorphisms, both in the promoter region of the UGT1A9 gene (C-275T>A) and (C-2152C>T), result in higher MPA glucuronidation rates [70], whereas the UGT1A9*3 (P 33M>T) polymorphism results in decreased enzyme activity and lower glucuronidation rate of MPA compared to the wild-type [71]. Clinical investigation of the effects of the UGT1A9-275T>A and -2152C>T polymorphisms in kidney transplant recipients has demonstrated that carriers of either or both polymorphisms had lower MPA area under the curve (AUC) and trough concentrations [59,60,62]. Polymorphisms have been also identified in the UGT1A8 gene. It has been reported that UGT1A8*3 (P 277C>Y) polymorphism results in an approximately 30-fold reduction in MPAG formation. This reduction has been attributed to the mutation effects on substrate affinity and the rate of MPAG formation [72]. Additionally, in a prospective study Sombogaard et al.[56] have recently measured the target enzyme IMPDH activity, MPA plasma concentrations and eight polymorphisms of inosine IMPDH type II in de novo kidney transplant recipients 6 days post-transplantation while on mycophenolate mofetil (MMF) treatment. Ten of 101 patients (10%) were heterozygous and two of 101 patients (2%) homozygous for IMPDH type II 3757T>C. The allele frequency was 6·9%. The IMPDH activity over 12 h AUC was 49% higher for patients with an IMPDH type II 3757C variant. The IMPDH activity measured before transplantation was not significantly different between IMPDH type II 3757TT wild-type and variant carrier patients. However, additional in vivo studies need to be performed to assess the potential clinical utility of these findings.
mTOR inhibitors
Recent data indicate that genetic mutations may influence the sensitivity of mTOR inhibitors (Table 1). This represents a relatively new family of anti-cancer and immunosuppressive agents, currently including sirolimus and its derivates, CCI-779 and RAD001 (everolimus). These drugs form complexes with an intracellular immunophillin (FKBP) which bind to the kinase mTOR. This kinase controls the phosphorylation of proteins that regulate the translation of mRNAs encoding regulators of the cell cycle, such as p70S6 kinase. Thus, inhibition of this pathway arrests the cellular cycle to G1 phase [2]. Huang and Houghton [73] have recently reviewed the current knowledge of intrinsic mTOR resistance mechanism. This phenomenon could involve mutations of FKBP12 or mTOR as well as mutations or defects of mTOR-regulated proteins, including S6K1-, 4E-BP1- and PP2A-related phosphatases and p27 that can render mTOR inhibitors insensitive [74]. Furthermore, these drugs are metabolized by CYP3A4/CYP3A5 and transported by the MDR-1 P-gp system. Le Muer et al.[66] and Anglicheau et al.[68], in two different studies, reported an association between CYP3A and MDR1 genetic polymorphisms and sirolimus pharmacokinetics, demonstrating that patients expressing CYP3A5 (CYP3A5*1 carriers) required higher dosages of this drug to reach target through levels compared to CYP3A5*3/*3 carriers. Therefore, although clinical data are lacking, the possibility that pharmacogenetic considerations presented for calcineurin inhibitors may be applied to mTOR inhibitors exists.
Glucocorticoids
Although few reports have indicated a genetic contribution on therapeutic efficacy/toxicity of glucocorticoids, powerful anti-inflammatory drugs used to treat glomerulonephritides and as primary agents for induction and maintenance immunonosupressive treatment, additional studies are needed [2]. They act by binding to a glucocorticoid receptor; the complex translocates to the nucleus and regulates gene expression decreasing transcription of various proinflammatory proteins and increasing transcription of anti-inflammatory genes. A subset of patients is resistant to glucocorticoids and they show overexpression of the glucocorticoid receptor [75] and changes in the activity of proinflammatory transcription factors AP-1 and nuclear factor kappa B (NF-κB) [76]. Recently, Miura et al.[77] have indicated that nuclear receptor subfamily 1, group I, member 2 (NR1I2, A7635G), rather than CYP3A5 or MRP1 allelic variants, affected patient variability of plasma prednisolone concentrations in renal transplant recipients on maintenance immunosuppressive treatment. Recipients carrying the NR1I27635G allele seemed to possess higher metabolic activity for prednisolone in the intestine, greatly reducing its maximal plasma concentration. Therefore, in the future glucocorticoid pharmocogenetics may represent an interesting field of nephrology research [78,79].
Renin–angiotensin–aldosterone system (RAAS) blockers
CKD constitutes a highly prevalent health problem worldwide [80,81] and is associated with a high risk of protein–energy malnutrition and adverse cardiovascular outcomes [82]. In the past two decades, considerable gains in retarding progression of CKD by enhancing clinical surveillance have been made, ameliorating patients' lifestyles (dietary intake, physical activity) and introducing, at an early stage, more effective drugs [83,84]. In particular, the effective blockade of the RAAS by angiotensin-converting enzyme inhibitors (ACE-I) and angiotensin II receptor blockers (ARB) has been recognized as one of the more effective targets for the treatment of CKD [85,86]. Although the use of these agents is generally safe and not followed by severe adverse events, their efficacy is largely variable and poorly predictive. The genetic contribution to such variability and the concordance between genotype/phenotype of the ACE, the key enzyme in the RAAS system, has been addressed in many studies [87,88]. ACE catalyzes the conversion of angiotensin I to angiotensin II and inactivation of the vasodilatory peptide bradykinin. The ACE gene has an insertion/deletion (I/D) polymorphism, which is due to the presence or absence of a 287 base pairs (bp) fragment inside intron 16. The D allele is associated with higher circulating and tissue ACE levels and low response to ACE-I and ARB medications [89,90]. These findings, however, appeared inconsistent, and the studies have been criticized because the effect on some outcomes has been modest in larger studies, suggesting a significant publication bias [91]. In addition, recent evidence suggests that the DD genotype is associated with a lower erythropoietin requirement in continuous ambulatory dialysis patients [92]. Thus, because the ACE I/D polymorphism may be a reliable and cost-effective tool to identify patients at risk and those who may benefit from these therapies, and to design clinical trials in progressive nephropathies, the necessity to design additional research projects to evaluate these important issues more effectively seems unquestionable [93,94].
Pharmacogenomics: a new strategy to study the polygenic influence of drug effects and toxicity
Although pharmacogenetic approaches, involving a single gene or a specific pathway, had reasonable success in identifying genetic variants linked to specific pharmacological phenotypes (e.g. drug metabolism, the mechanisms of action of drugs, adverse drug effects), they do not represent the gold standard, being the overall pharmacological effects of medications, and not typically monogenic traits [12]. Thus, instead of searching for a ‘dramatic genetic effect’ produced by one gene, it is more realistic to consider a group of genetic variants, each with a moderate effect, which together result in an overall genetic effect in drug efficacy or toxicity. Such polygenic traits are more difficult to elucidate in clinical studies, especially when a medication's metabolic fate and mechanisms of action are defined poorly. The completion of the Human Genome Project [95,96] and the development of innovative high-throughput screening technologies [including massive parallel gene analysis, DNA sequencing and synthesis and single nucleotide polymorphism (SNP) genotyping] have provided powerful tools to evaluate the multi-genetic influence to a specific drug therapy [21–23]. Several commercial techniques are currently available and researchers may choose the most appropriate platform to use in their projects. Among them, the DNA microarray (also referred to as gene or genome chip, DNA chip or biochip) represents the most utilized technique. This consists of an arrayed series of thousands of microscopic spots containing DNA oligonucleotide probes. The probes usually represent a short sequence of a gene specifically hybridizing a cDNA or cRNA sample (target) under high-stringency conditions. Probe–target hybridization is usually detected and quantified using fluorophore-, silver- or chemiluminescence-labelled targets to determine the relative abundance of specific nucleic acid sequences in the target. In standard microarrays, the probes are attached to a solid surface by a covalent bond to a chemical matrix (via epoxy-silane, amino-silane, lysine, polyacrylamide or others). DNA microarrays can be used to measure changes in gene expression levels to detect SNPs in genotyping or in resequencing mutant genomes [97]. The applicability of microarrays in genomics research has expanded with the evolution and maturation of the technology, but a major issue concerning these methods is still represented by complex data analysis and bioinformatics [98]. In fact, during the last few years, many bioinformatics approaches have been developed to identify more clearly the genetic/genomic bases of complex and polygenic diseases. Traditionally, this objective has been reached by measuring expression levels of thousands of genes simultaneously and identifying, through different statistical algorithms (e.g. t-tests, non-parametric tests, Bayesian models), those genes expressed differentially among two or more different phenotypic conditions. However, it now well known that results obtained by these methodologies are, most of the time, over-optimistic and poorly reproducible. In addition, it has been demonstrated extensively that pathway analysis rather than single gene evaluation has many advantages. In a recent paper, Abatangelo et al.[99] reviewed the main technical aspects of pathway analysis and provided practical advice to perform data analysis more efficiently. Therefore, it seems clear that, in future, researchers involved in pharmacogenomics studies should combine all available methods (associative, predictive) to obtain more reliable and reproducible results. However, considerable effort needs to be made to produce simple algorithms and statistical methods to identify easily genes expressed differentially or gene variants relevant to drug therapies.
Pharmacogenomics and immunosuppressive drugs used in clinical nephrology and renal transplantation
Nephrology researchers have begun to employ these innovative high-throughput procedures to identify the whole basal expression profile of normal or pathological human kidney [100], to select biomarkers predicting acute and chronic allograft outcomes [101,102] and to assess more clearly the intricate molecular pathways associated to the pathogenesis and onset of several immunological renal diseases [103,104]. On the contrary, only few reports to date have been published describing the multi-genetic influence on drug response in nephrology.
Recently, our group, applying a classical pharmacogenomic approach, has identified a new potential therapeutic target responsible for MPA anti-fibrotic and anti-proteinuric effects. Microarray analysis has revealed that neutral endopeptidase (NEP), a gene encoding for an enzyme involved primarily in the degradation of angiotensin-II, was the most significant up-regulated gene in a cohort of stable renal transplant recipients 3 months after conversion from AZA to MPA. Then, glomerular and tubular NEP protein levels, measured on graft biopsies from an independent group, were significantly higher in patients on CsA + MPA treatment compared to those on CsA + AZA and CsA alone. Glomerular NEP levels were correlated inversely with glomerulosclerosis and proteinuria measured at the time of biopsy. Tubular NEP levels were associated inversely with interstitial fibrosis. Incubation of proximal tubular cells with MPA led to a dose- and time-dependent increase of NEP gene expression. For the first time, these data suggested that MPA treatment may modulate this enzyme directly, contributing to the slow-down of the chronic glomerular progression and tubulointerstitial damage [105].
Additionally, a few other studies using in vitro and animal models have identified, using specifically designed microarray platforms, some genes with putative relevance to efficacy and toxicity of immunosuppressive drugs used in nephrology. However, none of the results obtained by these studies has been confirmed in a clinical setting.
Pharmacogenomics and immunological tolerance
Interestingly, high-throughput genomic screening technologies have been used to identify biomarkers associated with immunological tolerance in renal transplant patients. It is well known that long-term allograft survival requires lifelong immunosuppression, but recipients rarely display spontaneous ‘operational tolerance’ with stable graft function in the absence of immunosuppression [106]. The lack of biological markers of this phenomenon precludes identification of potentially tolerant patients in which immunosuppression could be tapered or interrupted early. Therefore, the objective of all these studies was to identify markers able to identify a tolerant population clearly. Several genes have been suggested as potentially useful predictors of tolerance, including genes involved in immune quiescence, apoptosis and memory T cell response [107]. However, further validation and prospective clinical trials using these selective biological elements are needed.
Pharmacogenomics and CKD
Microarray studies have been performed to identify specific genomic fingerprints modulated during acute [108] and chronic [109,110] dialysis therapy. Interestingly, several genes were de-regulated in CKD patients undergoing these renal replacement treatments. Among the genes selected were those encoding for several chemokines with proinflammatory and chemotactic activity [e.g. interleukin (IL)-8, chemokine (C-C motif) receptor 7 (CCR7), tumour necrosis factor (TNF)-α, chemokine (C-X-C motif) receptor 4 (CXCR4)], key regulators of oxidative stress [e.g. v-rel reticuloendotheliosis viral oncogene homologue A (RELA) and glutathione synthetase (GSS)] and those implicated in the mitochondrial oxidative phosphorylation system (e.g. ATP5O, COX6C, COX7C, NDUFS5, NDUFA6, UQCRH, NDUFA1, ATP5J, UQCRB, NDUFB1 and ATP5I). These results may be important in order to understand more clearly the complex alterations of the biochemical cell machinery induced by these treatments, but also to recognize new potential targets for future therapeutic approaches useful to reduce the development of clinical complications commonly present in this extensive patient population (e.g. atherosclerosis, cardiovascular diseases, malnutrition).
Pharmacogenomics and future perspectives
It is well recognized that, in the near future, nephrologists applying knowledge about an individual's inherited response to drugs and replacing the current methods of drug administration will be able to prescribe medications based on each person's genetic make-up [111]. This will maximize the therapy's value and decrease the likelihood of adverse drug effects. Knowing his own genetic background will allow a patient to make adequate lifestyle and environmental changes at an early age to avoid or lessen the severity of a genetic disease. Similarly, advanced knowledge of particular disease susceptibility will permit careful monitoring and the introduction of treatments at the most appropriate stage to maximize their effects.
Additionally, this will facilitate drug discovery by pharmaceutical companies and allow drug makers to produce a therapy more targeted to specific renal diseases (Fig. 2). This accuracy will not only maximize therapeutic effects, but also decrease damage to nearby healthy cells. Previously failed drug candidates may be revived as they are matched with the niche population they may serve. The drug approval process should be facilitated, as trials would be targeted for specific genetically defined population groups providing greater degrees of success. Targeting only those patients able to respond to a drug will reduce the cost and risk of clinical trials. Recently, to this purpose the Food and Drug Administration (FDA) released the ‘Guidance on pharmacogenomic data submissions on drug development’, a new industry guidance addressing the submission of pharmacogenomic data [112]. These guidelines are designed to assist drug companies to adopt pharmacogenomic technology in clinical development, and cover both targeted and exploratory aspects. While targeted pharmacogenomics must be included as part of any regulatory submission, exploratory approaches may be submitted voluntarily with assurances from the FDA that any such submissions will not be used to make regulatory decisions.
Fig. 2.
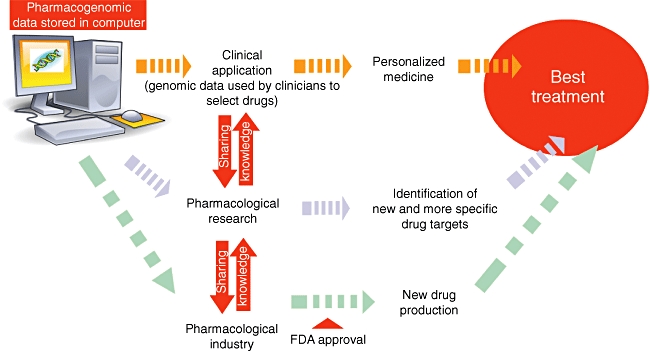
Future employment of pharmacogenomics. This figure describes potential practical applications of data and results obtained by pharmacogenomic studies. Data may be utilized easily by physicians to select appropriate treatments more effectively based on patient genomic make-up, to help researchers to identify more appropriate drug targets for future pharmacological interventions and to facilitate drug discovery by pharmaceutical companies.
In conclusion, the development of a co-operative framework among researchers, clinicians, industry and technology experts will be essential to fulfil the revolutionary promise that pharmacogeneomics hold for drug development, regulatory science, medical practice and public health (Fig. 2).
Disclosure
None.
References
- 1.Coppo R, Amore A. New perspectives in treatment of glomerulonephritis. Pediatr Nephrol. 2004;19:256–65. doi: 10.1007/s00467-003-1357-0. [DOI] [PubMed] [Google Scholar]
- 2.Halloran PF. Immunosuppressive drugs for kidney transplantation. N Engl J Med. 2004;351:2715–29. doi: 10.1056/NEJMra033540. [DOI] [PubMed] [Google Scholar]
- 3.Heung M, Mueller BA, Segal JH. Optimizing anemia management in hospitalized patients with end-stage renal disease. Ann Pharmacother. 2009;43:276–82. doi: 10.1345/aph.1L195. [DOI] [PubMed] [Google Scholar]
- 4.Lv J, Zhang H, Chen Y, et al. Combination therapy of prednisone and ACE inhibitor versus ACE-inhibitor therapy alone in patients with IgA nephropathy: a randomized controlled trial. Am J Kidney Dis. 2009;53:26–32. doi: 10.1053/j.ajkd.2008.07.029. [DOI] [PubMed] [Google Scholar]
- 5.Ernst FR, Grizzle AJ. Drug-related morbidity and mortality: updating the cost-of-illness model. J Am Pharm Assoc. 2001;41:192–9. doi: 10.1016/s1086-5802(16)31229-3. [DOI] [PubMed] [Google Scholar]
- 6.Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA. 1998;279:1200–5. doi: 10.1001/jama.279.15.1200. [DOI] [PubMed] [Google Scholar]
- 7.Patel H, Bell D, Molokhia M, et al. Trends in hospital admissions for adverse drug reactions in England: analysis of national hospital episode statistics 1998–2005. BMC Clin Pharmacol. 2007;7:9–14. doi: 10.1186/1472-6904-7-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Benet LZ. Relevance of pharmacokinetics in narrow therapeutic index drugs. Transplant Proc. 1999;31:1642–4. doi: 10.1016/s0041-1345(99)00083-4. [DOI] [PubMed] [Google Scholar]
- 9.Lindholm A, Sawe J. Pharmacokinetics and therapeutic drug monitoring of immunosuppressants. Ther Drug Monit. 1995;17:570–3. doi: 10.1097/00007691-199512000-00004. [DOI] [PubMed] [Google Scholar]
- 10.Weinshilboum R. Inheritance and drug response. N Engl J Med. 2003;348:529–37. doi: 10.1056/NEJMra020021. [DOI] [PubMed] [Google Scholar]
- 11.Evans WE, Relling MV. Moving towards individualized medicine with pharmacogenomics. Nature. 2004;429:464–8. doi: 10.1038/nature02626. [DOI] [PubMed] [Google Scholar]
- 12.Evans WE, Johnson JA. Pharmacogenomics: the inherited basis for interindividual differences in drug response. Annu Rev Genomics Hum Genet. 2001;2:9–39. doi: 10.1146/annurev.genom.2.1.9. [DOI] [PubMed] [Google Scholar]
- 13.Kalow W, Tang BK, Endrenyi I. Hypothesis: comparisons of inter- and intra-individual variations can substitute for twin studies in drug research. Pharmacogenetics. 1998;8:283–9. doi: 10.1097/00008571-199808000-00001. [DOI] [PubMed] [Google Scholar]
- 14.Kalow W, Gunn DR. The relation between dose of succinylcholine and duration of apnea in man. J Pharmacol Exp Ther. 1957;120:203–14. [PubMed] [Google Scholar]
- 15.Price Evans DAP, Manley KA, McKusick VA. Genetic control of isoniazid metabolism in man. BMJ. 1960;2:4484–91. doi: 10.1136/bmj.2.5197.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Timbrell JA, Harland SJ, Facchini V. Polymorphic acetylation of hydralazine. Clin Pharmacol Ther. 1980;28:350–5. doi: 10.1038/clpt.1980.173. [DOI] [PubMed] [Google Scholar]
- 17.Reidenberg MM, Drayer DE, Levy M, Warner H. Polymorphic acetylation of procainamide in man. Clin Pharmacol Ther. 1975;17:722–30. doi: 10.1002/cpt1975176722. [DOI] [PubMed] [Google Scholar]
- 18.Drayer DE, Reidenberg MM. Clinical consequences of polymorphic acetylation of basic drugs. Clin Pharmacol Ther. 1977;22:251–8. doi: 10.1002/cpt1977223251. [DOI] [PubMed] [Google Scholar]
- 19.Gonzalez FJ, Skoda RC, Kimura S, et al. Characterization of the common genetic defect in humans deficient in debrisoquine metabolism. Nature. 1988;331:442–6. doi: 10.1038/331442a0. [DOI] [PubMed] [Google Scholar]
- 20.Distlerath LM, Reilly PE, Martin MV, Davis GG, Wilkinson GR, Guengerich FP. Purification and characterization of the human liver cytochromes P-450 involved in debrisoquine 4-hydroxylation and phenacetin O-deethylation, two prototypes for genetic polymorphism in oxidative drug metabolism. J Biol Chem. 1985;260:9057–67. [PubMed] [Google Scholar]
- 21.Lechler RI, Sykes M, Thomson AW, Turka LA. Organ transplantation – how much of the promise has been realized? Nat Med. 2005;11:605–13. doi: 10.1038/nm1251. [DOI] [PubMed] [Google Scholar]
- 22.Plosker Gl, Foster RH. Tacrolimus: a further update of its pharmacology and therapeutic use in the management of organ transplantation. Drugs. 2000;59:323–89. doi: 10.2165/00003495-200059020-00021. [DOI] [PubMed] [Google Scholar]
- 23.Fulton B, Markham A. Mycophenolate mofetil: a review of its pharmacodynamic and pharmacokinetic properties and clinical efficacy in renal transplantation. Drugs. 1996;51:278–98. doi: 10.2165/00003495-199651020-00007. [DOI] [PubMed] [Google Scholar]
- 24.Jones TE. The use of other drugs to allow a lower dosage of cyclosporine to be used. Therapeutic and pharmacoeconomic considerations. Clin Pharmacokinet. 1997;32:357–67. doi: 10.2165/00003088-199732050-00002. [DOI] [PubMed] [Google Scholar]
- 25.Kurzawski M, Dziewanowski K, Lener A, Drozdzik M. TPMT but not ITPA gene polymorphism influences the risk of azathioprine intolerance in renal transplant recipients. Eur J Clin Pharmacol. 2009;65:533–40. doi: 10.1007/s00228-009-0630-y. [DOI] [PubMed] [Google Scholar]
- 26.Kurzawski M, Dziewanowski K, Gawrońska-Szklarz B, Domański L, Drozdzik M. The impact of thiopurine s-methyltransferase polymorphism on azathioprine-induced myelotoxicity in renal transplant recipients. Ther Drug Monit. 2005;27:435–41. doi: 10.1097/01.ftd.0000164393.09402.c9. [DOI] [PubMed] [Google Scholar]
- 27.Song DK, Zhao J, Zhang LR. TPMT genotype and its clinical implication in renal transplant recipients with azathioprine treatment. J Clin Pharm Ther. 2006;31:627–35. doi: 10.1111/j.1365-2710.2006.00775.x. [DOI] [PubMed] [Google Scholar]
- 28.Yates CR, Krynetski EY, Loennechen T, et al. Molecular diagnosis of thiopurine S-methyltransferase deficiency: genetic basis for azathioprine and mercaptopurine intolerance. Ann Intern Med. 1997;126:608–14. doi: 10.7326/0003-4819-126-8-199704150-00003. [DOI] [PubMed] [Google Scholar]
- 29.Evans WE, Hon YY, Bomgaars L, et al. Preponderance of thiopurine S-methyltransferase deficiency and heterozygosity among patients intolerant to mercaptopurine or azathioprine. J Clin Oncol. 2001;19:2293–301. doi: 10.1200/JCO.2001.19.8.2293. [DOI] [PubMed] [Google Scholar]
- 30.Evans WE. Thiopurine S-methyltransferase: a genetic polymorphism that affects a small number of drugs in a big way. Pharmacogenetics. 2002;12:421–3. doi: 10.1097/00008571-200208000-00001. [DOI] [PubMed] [Google Scholar]
- 31.Dervieux T, Médard Y, Baudouin V, et al. Thiopurine methyltransferase activity and its relationship to the occurrence of rejection episodes in paediatric renal transplant recipients treated with azathioprine. Br J Clin Pharmacol. 1999;48:793–800. doi: 10.1046/j.1365-2125.1999.00087.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qiu XY, Jiao Z, Zhang M, et al. Association of MDR1, CYP3A4*18B, and CYP3A5*3 polymorphisms with cyclosporine pharmacokinetics in Chinese renal transplant recipients. Eur J Clin Pharmacol. 2008;64:1069–84. doi: 10.1007/s00228-008-0520-8. [DOI] [PubMed] [Google Scholar]
- 33.Hesselink DA, van Gelder T, van Schaik RH, et al. Population pharmacokinetics of cyclosporine in kidney and heart transplant recipients and the influence of ethnicity and genetic polymorphisms in the MDR-1, CYP3A4, and CYP3A5 genes. Clin Pharmacol Ther. 2004;76:545–56. doi: 10.1016/j.clpt.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 34.von Ahsen N, Richter M, Grupp C, Ringe B, Oellerich M, Armstrong VW. No influence of the MDR-1 C3435T polymorphism or a CYP3A4 promoter polymorphism (CYP3A4-V allele) on dose-adjusted cyclosporin A trough concentrations or rejection incidence in stable renal transplant recipients. Clin Chem. 2001;47:1048–52. [PubMed] [Google Scholar]
- 35.Hu YF, Qiu W, Liu ZQ, et al. Effects of genetic polymorphisms of CYP3A4, CYP3A5 and MDR1 on cyclosporine pharmacokinetics after renal transplantation. Clin Exp Pharmacol Physiol. 2006;33:1093–98. doi: 10.1111/j.1440-1681.2006.04492.x. [DOI] [PubMed] [Google Scholar]
- 36.Kreutz R, Zürcher H, Kain S, Martus P, Offermann G, Beige J. The effect of variable CYP3A5 expression on cyclosporine dosing, blood pressure and long-term graft survival in renal transplant patients. Pharmacogenetics. 2004;14:665–71. doi: 10.1097/00008571-200410000-00004. [DOI] [PubMed] [Google Scholar]
- 37.Eng HS, Mohamed Z, Calne R, et al. The influence of CYP3A gene polymorphisms on cyclosporine dose requirement in renal allograft recipients. Kidney Int. 2006;69:1858–64. doi: 10.1038/sj.ki.5000325. [DOI] [PubMed] [Google Scholar]
- 38.Anglicheau D, Thervet E, Etienne I, et al. CYP3A5 and MDR1 genetic polymorphisms and cyclosporine pharmacokinetics after renal transplantation. Clin Pharmacol Ther. 2004;75:422–33. doi: 10.1016/j.clpt.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 39.Foote CJ, Greer W, Kiberd B, et al. Polymorphisms of multidrug resistance gene (MDR1) and cyclosporine absorption in de novo renal transplant patients. Transplantation. 2007;83:1380–4. doi: 10.1097/01.tp.0000264197.88129.2e. [DOI] [PubMed] [Google Scholar]
- 40.Yates CR, Zhang W, Song P, et al. The effect of CYP3A5 and MDR1 polymorphic expression on cyclosporine oral disposition in renal transplant patients. J Clin Pharmacol. 2003;43:555–64. [PubMed] [Google Scholar]
- 41.Kuypers DR, de Jonge H, Naesens M, Lerut E, Verbeke K, Vanrenterghem Y. CYP3A5 and CYP3A4 but not MDR1 single-nucleotide polymorphisms determine long-term tacrolimus disposition and drug-related nephrotoxicity in renal recipients. Clin Pharmacol Ther. 2007;82:711–25. doi: 10.1038/sj.clpt.6100216. [DOI] [PubMed] [Google Scholar]
- 42.Bandur S, Petrasek J, Hribova P, Novotna E, Brabcova I, Viklicky O. Haplotypic structure of ABCB1/MDR1 gene modifies the risk of the acute allograft rejection in renal transplant recipients. Transplantation. 2008;86:1206–13. doi: 10.1097/TP.0b013e318187c4d1. [DOI] [PubMed] [Google Scholar]
- 43.Quteineh L, Verstuyft C, Furlan V, et al. Influence of CYP3A5 genetic polymorphism on tacrolimus daily dose requirements and acute rejection in renal graft recipients. Basic Clin Pharmacol Toxicol. 2008;103:546–52. doi: 10.1111/j.1742-7843.2008.00327.x. [DOI] [PubMed] [Google Scholar]
- 44.Renders L, Frisman M, Ufer M, et al. CYP3A5 genotype markedly influences the pharmacokinetics of tacrolimus and sirolimus in kidney transplant recipients. Clin Pharmacol Ther. 2007;81:228–34. doi: 10.1038/sj.clpt.6100039. [DOI] [PubMed] [Google Scholar]
- 45.Zhang X, Liu ZH, Zheng JM, et al. Influence of CYP3A5 and MDR1 polymorphisms on tacrolimus concentration in the early stage after renal transplantation. Clin Transplant. 2005;19:638–43. doi: 10.1111/j.1399-0012.2005.00370.x. [DOI] [PubMed] [Google Scholar]
- 46.Zhao Y, Song M, Guan D, et al. Genetic polymorphisms of CYP3A5 genes and concentration of the cyclosporine and tacrolimus. Transplant Proc. 2005;37:178–81. doi: 10.1016/j.transproceed.2005.01.077. [DOI] [PubMed] [Google Scholar]
- 47.Macphee IA, Fredericks S, Mohamed M, et al. Tacrolimus pharmacogenetics: the CYP3A5*1 allele predicts low dose-normalized tacrolimus blood concentrations in whites and South Asians. Transplantation. 2005;79:499–502. doi: 10.1097/01.tp.0000151766.73249.12. [DOI] [PubMed] [Google Scholar]
- 48.Mai I, Perloff ES, Bauer S, et al. MDR1 haplotypes derived from exons 21 and 26 do not affect the steady-state pharmacokinetics of tacrolimus in renal transplant patients. Br J Clin Pharmacol. 2004;58:548–53. doi: 10.1111/j.1365-2125.2004.02182.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thervet E, Anglicheau D, King B, et al. Impact of cytochrome p450 3A5 genetic polymorphism on tacrolimus doses and concentration-to-dose ratio in renal transplant recipients. Transplantation. 2003;76:1233–5. doi: 10.1097/01.TP.0000090753.99170.89. [DOI] [PubMed] [Google Scholar]
- 50.Li D, Gui R, Li J, Huang Z, Nie X. Tacrolimus dosing in Chinese renal transplant patients is related to MDR1 gene C3435T polymorphisms. Transplant Proc. 2006;38:2850–2. doi: 10.1016/j.transproceed.2006.08.089. [DOI] [PubMed] [Google Scholar]
- 51.Fredericks S, Moreton M, Reboux S, et al. Multidrug resistance gene-1 (MDR-1) haplotypes have a minor influence on tacrolimus dose requirements. Transplantation. 2006;82:705–8. doi: 10.1097/01.tp.0000234942.78716.c0. [DOI] [PubMed] [Google Scholar]
- 52.Anglicheau D, Verstuyft C, Laurent-Puig P, et al. Association of the multidrug resistance-1 gene single-nucleotide polymorphisms with the tacrolimus dose requirements in renal transplant recipients. J Am Soc Nephrol. 2003;14:1889–96. doi: 10.1097/01.asn.0000073901.94759.36. [DOI] [PubMed] [Google Scholar]
- 53.MacPhee IA, Fredericks S, Tai T, et al. The influence of pharmacogenetics on the time to achieve target tacrolimus concentrations after kidney transplantation. Am J Transplant. 2004;4:914–19. doi: 10.1111/j.1600-6143.2004.00435.x. [DOI] [PubMed] [Google Scholar]
- 54.Singh R, Srivastava A, Kapoor R, Sharma K, Mittal D. Impact of CYP3A5 and CYP3A4 gene polymorphisms on dose requirement of calcineurin inhibitors, cyclosporine and tacrolimus, in renal allograft recipients of North India. Naunyn Schmiedebergs Arch Pharmacol. 2009;380:169–77. doi: 10.1007/s00210-009-0415-y. [DOI] [PubMed] [Google Scholar]
- 55.Haufroid V, Mourad M, Van Kerckhove V, et al. The effect of CYP3A5 and MDR1 (ABCB1) polymorphisms on cyclosporine and tacrolimus dose requirements and trough blood levels in stable renal transplant patients. Pharmacogenetics. 2004;14:147–54. doi: 10.1097/00008571-200403000-00002. [DOI] [PubMed] [Google Scholar]
- 56.Sombogaard F, van Schaik RH, Mathot RA, et al. Interpatient variability in IMPDH activity in MMF-treated renal transplant patients is correlated with IMPDH type II 3757T >C polymorphism. Pharmacogenet Genomics. 2009;19:626–34. doi: 10.1097/FPC.0b013e32832f5f1b. [DOI] [PubMed] [Google Scholar]
- 57.Inoue K, Miura M, Satoh S, et al. Influence of UGT1A7 and UGT1A9 intronic I399 genetic polymorphisms on mycophenolic acid pharmacokinetics in Japanese renal transplant recipients. Ther Drug Monit. 2007;29:299–304. doi: 10.1097/FTD.0b013e3180686146. [DOI] [PubMed] [Google Scholar]
- 58.Betônico GN, Abbud-Filho M, Goloni-Bertollo EM, et al. Influence of UDP-glucuronosyltransferase polymorphisms on mycophenolate mofetil-induced side effects in kidney transplant patients. Transplant Proc. 2008;40:708–10. doi: 10.1016/j.transproceed.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 59.Van Schaik R, van Agteren M, de Fijter J, et al. UGT1A9-275T>A/-2152C>T polymorphisms correlate with low MPA exposure and acute rejection in MMF/tacrolimus-treated kidney transplant patients. Clin Pharmacol Ther. 2009;86:319–27. doi: 10.1038/clpt.2009.83. [DOI] [PubMed] [Google Scholar]
- 60.Johnson LA, Oetting WS, Basu S, Prausa S, Matas A, Jacobson PA. Pharmacogenetic effect of the UGT polymorphisms on mycophenolate is modified by calcineurin inhibitors. Eur J Clin Pharmacol. 2008;64:1047–56. doi: 10.1007/s00228-008-0501-y. [DOI] [PubMed] [Google Scholar]
- 61.Kagaya H, Inoue K, Miura M, et al. Influence of UGT1A8 and UGT2B7 genetic polymorphisms on mycophenolic acid pharmacokinetics in Japanese renal transplant recipients. Eur J Clin Pharmacol. 2007;63:279–88. doi: 10.1007/s00228-006-0248-2. [DOI] [PubMed] [Google Scholar]
- 62.Kuypers DR, Naesens M, Vermeire S, Vanrenterghem Y. The impact of uridine diphosphate-glucuronosyltransferase 1A9 (UGT1A9) gene promoter region single-nucleotide polymorphisms T-275A and C-2152T on early mycophenolic acid dose-interval exposure in de novo renal allograft recipients. Clin Pharmacol Ther. 2005;78:351–61. doi: 10.1016/j.clpt.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 63.Zhang WX, Chen B, Jin Z, et al. Influence of uridine diphosphate (UDP)-glucuronosyltransferases and ABCC2 genetic polymorphisms on the pharmacokinetics of mycophenolic acid and its metabolites in Chinese renal transplant recipients. Xenobiotica. 2008;38:1422–36. doi: 10.1080/00498250802488585. [DOI] [PubMed] [Google Scholar]
- 64.Naesens M, Kuypers DR, Verbeke K, Vanrenterghem Y. Multidrug resistance protein 2 genetic polymorphisms influence mycophenolic acid exposure in renal allograft recipients. Transplantation. 2006;82:1074–84. doi: 10.1097/01.tp.0000235533.29300.e7. [DOI] [PubMed] [Google Scholar]
- 65.Miao LY, Huang CR, Hou JQ, Qian MY. Association study of ABCB1 and CYP3A5 gene polymorphisms with sirolimus trough concentration and dose requirements in Chinese renal transplant recipients. Biopharm Drug Dispos. 2008;29:1–5. doi: 10.1002/bdd.577. [DOI] [PubMed] [Google Scholar]
- 66.Le Meur Y, Djebli N, Szelag JC, et al. CYP3A5*3 influences sirolimus oral clearance in de novo and stable renal transplant recipients. Clin Pharmacol Ther. 2006;80:51–60. doi: 10.1016/j.clpt.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 67.Djebli N, Rousseau A, Hoizey G, et al. Sirolimus population pharmacokinetic/pharmacogenetic analysis and Bayesian modelling in kidney transplant recipients. Clin Pharmacokinet. 2006;45:1135–48. doi: 10.2165/00003088-200645110-00007. [DOI] [PubMed] [Google Scholar]
- 68.Anglicheau D, Le Corre D, Lechaton S, et al. Consequences of genetic polymorphisms for sirolimus requirements after renal transplant in patients on primary sirolimus therapy. Am J Transplant. 2005;5:595–603. doi: 10.1111/j.1600-6143.2005.00745.x. [DOI] [PubMed] [Google Scholar]
- 69.Li X, Bratton S, Radominska-Pandya A. Human UGT1A8 and UGT1A10 mRNA are expressed in primary human hepatocytes. Drug Metab Pharmacokinet. 2007;22:152–61. doi: 10.2133/dmpk.22.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Girard H, Court MH, Bernard O, et al. Identification of common polymorphisms in the promoter of the UGT1A9 gene: evidence that UGT1A9 protein and activity levels are strongly genetically controlled in the liver. Pharmacogenetics. 2004;14:501–15. doi: 10.1097/01.fpc.0000114754.08559.27. [DOI] [PubMed] [Google Scholar]
- 71.Bernard O, Guillemette C. The main role of UGT1A9 in the hepatic metabolism of mycophenolic acid and the effects of naturally occurring variants. Drug Metab Dispos. 2004;32:775–8. doi: 10.1124/dmd.32.8.775. [DOI] [PubMed] [Google Scholar]
- 72.Bernard O, Tojcic J, Journault K, Perusse L, Guillemette C. Influence of nonsynonymous polymorphisms of UGT1A8 and UGT2B7 metabolizing enzymes on the formation of phenolic and acyl glucuronides of mycophenolic acid. Drug Metab Dispos. 2006;34:1539–45. doi: 10.1124/dmd.106.010553. [DOI] [PubMed] [Google Scholar]
- 73.Huang S, Houghton PJ. Mechanisms of resistance to rapamycins. Drug Resist Updat. 2001;4:378–91. doi: 10.1054/drup.2002.0227. [DOI] [PubMed] [Google Scholar]
- 74.Bliskovsky V, Ramsay ES, Scott J, et al. Frap, FKBP12 rapamycin-associated protein, is a candidate gene for the plasmacytoma resistance locus Pctr2 and can act as a tumor suppressor gene. Proc Natl Acad Sci USA. 2003;100:14982–7. doi: 10.1073/pnas.2431627100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sousa AR, Lane SJ, Cidlowski JA, Staynov DZ, Lee TH. Glucocorticoid resistance in asthma is associated with elevated in vivo expression of the glucocorticoid receptor beta-isoform. J Allergy Clin Immunol. 2000;105:943–50. doi: 10.1067/mai.2000.106486. [DOI] [PubMed] [Google Scholar]
- 76.Fenech A, Hall IP. Pharmacogenetics of asthma. Br J Clin Pharmacol. 2002;53:3–15. doi: 10.1046/j.0306-5251.2001.01509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Miura M, Satoh S, Inoue K, et al. Influence of CYP3A5, ABCB1 and NR1I2 polymorphisms on prednisolone pharmacokinetics in renal transplant recipients. Steroids. 2008;73:1052–9. doi: 10.1016/j.steroids.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 78.Koper JW, Stolk RP, de Lange P, et al. Lack of association between five polymorphisms in the human glucocorticoid receptor gene and glucocorticoid resistance. Hum Genet. 1997;99:663–8. doi: 10.1007/s004390050425. [DOI] [PubMed] [Google Scholar]
- 79.Lane SJ, Arm JP, Staynov DZ, Lee TH. Chemical mutational analysis of the human glucocorticoid receptor cDNA in glucocorticoid-resistant bronchial asthma. Am J Respir Cell Mol Biol. 1994;11:42–8. doi: 10.1165/ajrcmb.11.1.8018337. [DOI] [PubMed] [Google Scholar]
- 80.Fernandez-Fresnedo G, Rodrigo E, de Francisco AL, de Castro SS, Castañeda O, Arias M. Role of pulse pressure on cardiovascular risk in chronic kidney disease patients. J Am Soc Nephrol. 2006;17:246–9. doi: 10.1681/ASN.2006080921. [DOI] [PubMed] [Google Scholar]
- 81.Goodkin DA, Bragg-Gresham JL, Koenig KG, et al. Association of co-morbid conditions and mortality in hemodialysis patients in Europe, Japan, and the United States: the Dialysis Outcomes and Practice Patterns Study (DOPPS) J Am Soc Nephrol. 2003;14:3270–7. doi: 10.1097/01.asn.0000100127.54107.57. [DOI] [PubMed] [Google Scholar]
- 82.Zimmermann J, Herrlinger S, Pruy A, Metzger T, Wanner C. Inflammation enhances cardiovascular risk and mortality in hemodialysis patients. Kidney Int. 1999;55:648–58. doi: 10.1046/j.1523-1755.1999.00273.x. [DOI] [PubMed] [Google Scholar]
- 83.Fink JC, Brown J, Hsu VD, Seliger SL, Walker L, Zhan M. CKD as an underrecognized threat to patient safety. Am J Kidney Dis. 2009;53:681–8. doi: 10.1053/j.ajkd.2008.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Brenner B. Retarding the progression of renal disease. Kidney Int. 2003;64:370–8. doi: 10.1046/j.1523-1755.2003.t01-2-00052.x. [DOI] [PubMed] [Google Scholar]
- 85.Laverman GD, de Zeeuw D, Navis G. Between patient differences in the renal response to rennin–angiotensin system intervention: clue to optimising renoprotective therapy? J Renin Angiotensin Aldosterone Syst. 2002;3:205–13. doi: 10.3317/jraas.2002.042. [DOI] [PubMed] [Google Scholar]
- 86.Jacobsen P, Andersen S, Jensen BR, Parving HH. Additive effect of ACE inhibition and angiotensin II receptor blockade in type 1 diabetic patients with diabetic nephropathy. J Am Soc Nephrol. 2003;14:992–9. doi: 10.1097/01.asn.0000054495.96193.bf. [DOI] [PubMed] [Google Scholar]
- 87.Bhatnagar V, O'Connor DT, Schork NJ, et al. Angiotensin-converting enzyme gene polymorphism predicts the time–course of blood pressure response to angiotensin converting enzyme inhibition in the AASK trial. J Hypertens. 2007;25:2082–92. doi: 10.1097/HJH.0b013e3282b9720e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Jacobsen PK, Tarnow L, Parving HH. Time to consider ACE insertion/deletion genotypes and individual renoprotective treatment in diabetic nephropathy? Kidney Int. 2006;69:1293–5. doi: 10.1038/sj.ki.5000283. [DOI] [PubMed] [Google Scholar]
- 89.Rigat B, Hubert C, Alhenc-Gelas F, Cambien F, Corvol P, Soubrier F. An insertion–deletion polymorphism in the angiotensin I converting enzyme gene accounting for half the variance of serum enzyme levels. J Clin Invest. 1990;86:1343–6. doi: 10.1172/JCI114844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Danser AH, Schalekamp MA, Bax WA, et al. Angiotensin-converting enzyme in the human heart: effect of the deletion/insertion polymorphism. Circulation. 1995;92:1387–8. doi: 10.1161/01.cir.92.6.1387. [DOI] [PubMed] [Google Scholar]
- 91.Morgan TM, Coffey CS, Krumholz HM. Overestimation of genetic risks owing to small sample sizes in cardiovascular studies. Clin Genet. 2003;64:7–17. doi: 10.1034/j.1399-0004.2003.00088.x. [DOI] [PubMed] [Google Scholar]
- 92.Ruggenenti P, Bettinaglio P, Pinares F, Remuzzi G. Angiotensin converting enzyme insertion/deletion polymorphism and renoprotection in diabetic and non-diabetic nephropathies. Clin J Am Soc Nephrol. 2008;3:1511–25. doi: 10.2215/CJN.04140907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Scharplatz M, Puhan MA, Steurer J, Perna A, Bachmann LM. Does the angiotensin-converting enzyme (ACE) gene insertion/deletion polymorphism modify the response to ACE inhibitor therapy? A systematic review. Curr Control Trials Cardiovasc Med. 2005;6:16–25. doi: 10.1186/1468-6708-6-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sharples EJ, Varagunam M, Sinnott PJ, McCloskey DJ, Raftery MJ, Yaqoob MM. The effect of proinflammatory cytokine gene and angiotensin-converting enzyme polymorphisms on erythropoietin requirements in patients on continuous ambulatory peritoneal dialysis. Perit Dial Int. 2006;26:64–8. [PubMed] [Google Scholar]
- 95.Lander ES, Linton LM, Birren B, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 96.Venter JC, Adams MD, Myers EW, et al. The sequence of the human genome. Science. 2001;291:1304–51. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 97.Möröy T. DNA microarrays in medicine: can the promises be kept? J Biomed Biotechnol. 2002;2:1–2. doi: 10.1155/S1110724302000360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hu J, Zou F, Wright FA. Practical FDR-based sample size calculations in microarray experiments. Bioinformatics. 2005;21:3264–72. doi: 10.1093/bioinformatics/bti519. [DOI] [PubMed] [Google Scholar]
- 99.Abatangelo L, Maglietta R, Distaso A, et al. Comparative study of gene set enrichment methods. BMC Bioinformatics. 2009;10:275. doi: 10.1186/1471-2105-10-275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yasuda Y, Cohen CD, Henger A, et al. Gene expression profiling analysis in nephrology: towards molecular definition of renal disease. Clin Exp Nephrol. 2006;10:91–8. doi: 10.1007/s10157-006-0421-z. [DOI] [PubMed] [Google Scholar]
- 101.Perco P, Kainz A, Wilflingseder J, Soleiman A, Mayer B, Oberbauer R. Histogenomics: association of gene expression patterns with histological parameters in kidney biopsies. Transplantation. 2009;87:290–5. doi: 10.1097/TP.0b013e318191b4c0. [DOI] [PubMed] [Google Scholar]
- 102.Saint-Mezard P, Berthier CC, Zhang H, et al. Analysis of independent microarray datasets of renal biopsies identifies a robust transcript signature of acute allograft rejection. Transpl Int. 2009;22:293–302. doi: 10.1111/j.1432-2277.2008.00790.x. [DOI] [PubMed] [Google Scholar]
- 103.Hauser PV, Perco P, Mühlberger I, et al. Microarray and bioinformatics analysis of gene expression in experimental membranous nephropathy. Nephron Exp Nephrol. 2009;112:43–58. doi: 10.1159/000213505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Izumi Y, Izumiya Y, Shiota M, et al. Gene expression profile in experimental mesangial proliferative glomerulonephritis. J Pharmacol Sci. 2004;96:91–4. doi: 10.1254/jphs.rc0040012. [DOI] [PubMed] [Google Scholar]
- 105.Zaza G, Dell'oglio MP, Rossini M, et al. Mycophenolic acid induces the expression of neutral endopeptidase (NEP): a new therapeutic target defined by a pharmacogenomic approach. J Am Soc Nephrol. 2008;19:604A. [Google Scholar]
- 106.Yabu JM, Vincenti F. Kidney transplantation: the ideal immunosuppression regimen. Adv Chronic Kidney Dis. 2009;16:226–33. doi: 10.1053/j.ackd.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 107.Brouard S, Mansfield E, Braud C, et al. Identification of a peripheral blood transcriptional biomarker panel associated with operational renal allograft tolerance. Proc Natl Acad Sci USA. 2007;104:15448–53. doi: 10.1073/pnas.0705834104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Friedrich B, Alexander D, Janessa A, Häring HU, Lang F, Risler T. Acute effects of hemodialysis on cytokine transcription profiles: evidence for C-reactive protein-dependency of mediator induction. Kidney Int. 2006;70:2124–30. doi: 10.1038/sj.ki.5001865. [DOI] [PubMed] [Google Scholar]
- 109.Zaza G, Pontrelli P, Pertosa G, et al. Dialysis-related systemic microinflammation is associated with specific genomic patterns. Nephrol Dial Transplant. 2008;23:1673–81. doi: 10.1093/ndt/gfm804. [DOI] [PubMed] [Google Scholar]
- 110.Granata S, Zaza G, Simone S, et al. Mitochondrial dysregulation and oxidative stress in patients with chronic kidney disease. BMC Genomics. 2009;10:388. doi: 10.1186/1471-2164-10-388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kapur G, Mattoo T, Aranda JV. Pharmacogenomics and renal drug disposition in the newborn. Semin Perinatol. 2004;28:132–40. doi: 10.1053/j.semperi.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 112.Savage DR US Food and Drug Administration (FDA) FDA guidance on pharmacogenomics data submission. Nat Rev Drug Discov. 2003;2:937–8. doi: 10.1038/nrd1274. [DOI] [PubMed] [Google Scholar]