

Abstract
Activation of complement occurs during autoimmune retinal and intraocular inflammatory disease as well as neuroretinal degenerative disorders. The cleavage of C5 into fragments C5a and C5b is a critical event during the complement cascade. C5a is a potent proinflammatory anaphylatoxin capable of inducing cell migration, adhesion and cytokine release, while membrane attack complex C5b-9 causes cell lysis. Therapeutic approaches to prevent complement-induced inflammation include the use of blocking monoclonal antibodies (mAb) to prevent C5 cleavage. In these current experiments, the rat anti-mouse C5 mAb (BB5·1) was utilized to investigate the effects of inhibition of C5 cleavage on disease progression and severity in experimental autoimmune uveoretinitis (EAU), a model of organ-specific autoimmunity in the eye characterized by structural retinal damage mediated by infiltrating macrophages. Systemic treatment with BB5·1 results in significantly reduced disease scores compared with control groups, while local administration results in an earlier resolution of disease. In vitro, contemporaneous C5a and interferon-γ signalling enhanced nitric oxide production, accompanied by down-regulation of the inhibitory myeloid CD200 receptor, contributing to cell activation. These experiments demonstrate that C5 cleavage contributes to the full expression of EAU, and that selective C5 blockade via systemic and local routes of administration can suppress disease. This presents great therapeutic potential to protect against tissue damage during autoimmune responses in the retina or inflammation-induced degenerative disease.
Keywords: autoimmunity, complement C5, experimental autoimmune uveoretinitis, macrophage activation, therapy
Introduction
Experimental autoimmune uveoretinitis (EAU) is an animal model that correlates with the spectrum of clinico-pathological features of human uveitides and, as a result, is a successful preclinical model for translation of immunotherapies [1–3]. Furthermore, the model serves to dissect immunopathogenic mechanisms relating to immune-mediated tissue damage, which in turn highlight avenues for future treatment [4,5]. Murine EAU arises following immunization with specific ocular antigens and the local activation of ocular-specific CD4+ T cells, within or around photoreceptor segments [6–10]. Disease occurs subsequent to activated T cell infiltration into the target organ with consequent macrophage recruitment into the eye and activates them, generating structural damage via mechanisms that include the interferon (IFN)-γ-induced secretion of nitric oxide (NO) [11].
The versatility of macrophages is due largely to their ability to respond to both endogenous and exogenous signals via a multitude of cell surface receptors [12,13]. They facilitate the amplification of local inflammatory signals and the production of a cascade of damaging mediators. Targeting macrophages is an effective approach that can restrict tissue damage in EAU [14]. One key activating cytokine is tumour necrosis factor (TNF)-α and neutralization of it results in reduced tissue damage, despite leucocytic infiltration, indicating that macrophage activation is critical in determining disease severity [15,16]. Furthermore, the enhancement of normal homeostatic down-regulation via monoclonal antibody-mediated CD200R signalling can also suppress macrophage activation and protect against tissue damage [17]. Another less well-explored approach is to inhibit macrophage activation through the complement C5a receptor.
The complement system is a central component of innate immunity, consisting of a large family of plasma and membrane-bound proteins that are critical for protection against bacterial infection and immune complex deposition [18]. More recently, it has also been found to be functional in adaptive immune responses, with roles identified in antigen processing and presentation, T cell proliferation and differentiation and systemic tolerance [19,20]. In organ-specific autoimmune disease, inappropriate activation of the complement system amplifies the pathology of human and/or experimental diseases, including rheumatoid arthritis, multiple sclerosis and macular degeneration [21–25]. Complement activation exerts harmful effects through the generation of complement protein cleavage products, which promote and perpetuate inflammatory reactions.
In ocular immunity, these complement fragments are implicated in the development and progression of several immune-mediated ocular conditions, including age-related macular degeneration (AMD) and uveitis [26–29]. The presence and activation of complement has also been shown recently to be central to the development of experimental autoimmune anterior uveitis (EAAU), an animal model of idiopathic human anterior uveitis [28]. Furthermore, blocking the interaction of the complement fragment receptor, C3aR, has a protective effect on the initiation and progression of EAAU. Previous studies in EAU show that the depletion of complement system components by cobra venom factor, or the use of C3−/− mice, causes reduced severity and incidence of disease [30]. Also, in a rat uveitis model, complement deposition in the eye correlated with disease severity and ‘knock-down’ of endogenous complement regulatory proteins exacerbated disease [31,32]. Alternatively, approaches including the overexpression of complement regulatory proteins such as CD55 have been demonstrated recently to prevent T cell autoreactivity and generation of pathogenic effector T cells in EAU [33]. This highlights the role that specific complement pathways play in the pathogenesis of EAU and suggests that complement may be a target for therapy in uveitis.
A critical event in the cascade is the cleavage of C5 into fragments C5a and C5b, and the subsequent formation of the membrane attack complex (C5b-9) which is involved with cytolysis, cell activation and production of proinflammatory mediators [34]. The C5a fragment is a potent inflammatory mediator that induces cellular migration, cell adhesion and cytokine release [35], and its receptor is expressed in a variety of cell types, in particular immune cells including macrophages, neutrophils and T cells [36]. Therefore prevention of C5 cleavage would have anti-inflammatory consequences, and previous studies utilizing C5-specific monoclonal antibodies have demonstrated therapeutic efficacy in the experimental model of rheumatoid arthritis [37]. The primary objective of this study was to test whether prevention of cleavage of the C5 complement protein by systemic or local administration of a blocking anti-C5 monoclonal antibody (mAb) could suppress myeloid activation and thus tissue damage in EAU.
Materials and methods
Mice
B10.RIII and C57BL/6 mice were obtained originally from Harlan UK Limited (Oxford, UK) and breeding colonies established within the Animal Services Unit at Bristol University, Bristol, UK for further experimentation. All mice were housed under specific pathogen-free conditions with continuously available food and water. Female mice immunized for disease induction were aged between 6 and 8 weeks. Treatment of animals conformed to the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research.
Reagents
The peptide RBP-3 161–180 (SGIPYIISYLHPGNTILHVD) was obtained from Sigma-Genosys Limited (Poole, UK). Peptide purity was >95% as determined by high-performance liquid chromatography (HPLC). The anti-mouse C5 hybridoma BB5·1 was provided originally by Dr Brigita Stockinger (National Institute for Medical Research, London, UK) [38]. Antibody was purified from sterile culture supernatant by protein G affinity chromatography. BB5·1 mAb preparations used contained <0·1 ng of endotoxin/mg protein, as determined by the Limulus Amebocyte Lysate Pyrogen Testing kit, QCl-1000 (Lonza Biologics, Slough, Berkshire, UK).
Complete medium consisted of Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat inactivated fetal calf serum (TCS Cellworks, Buckingham, UK), 100 U/ml penicillin–streptomycin, 2 mmol/l l-glutamine, 1 mmol/l sodium pyruvate and 5 × 10−5 mol/l 2-mercaptoethanol (all from Invitrogen, Paisley, UK).
EAU induction and scoring
B10.RIII mice were immunized subcutaneously in one flank with 50 µg/mouse RBP-3 161–180 peptide in phosphate-buffered saline (PBS) [2% dimethylsulphoxide (DMSO)] in complete Freund's adjuvant (CFA) (1 mg/ml; 1:1 v/v) supplemented with 1·5 mg/ml Mycobacterium tuberculosis complete H37 Ra (BD Biosciences, Oxford, UK) and 1 µg Bordetella pertussis toxin (Sigma-Aldrich) intraperitoneally (i.p.). At various time-points post-immunization (pi), mice were killed and eyes were enucleated and snap-frozen carefully, orientated in optimal cutting temperature (OCT) compound (ThermoFisher, Loughborough, UK). After they were made and stored at −80°C, serial 12-µm sections were thawed at room temperature and fixed in acetone for 10 min. Sections were stained with rat anti-mouse monoclonal anti-CD45 antibody (Serotec, Oxford, UK) and counterstained with haematoxylin (ThermoShandon, Pittsburgh, PA, USA). Sections were scored for inflammatory infiltrate (presence of CD45-positive cells) and structural disease (disruption of morphology). Cellular infiltrate is scored within the ciliary body, vitreous, vessels, rod outer segments and choroid, while structural disease is scored within the rod outer segments, neuronal layers and retinal morphology [39]. For C5, C5aR (which is known as C5R or CD88) and nitrotyrosine immunofluorescence staining, sections were fixed in 2% paraformaldehyde for 10 min, washed and incubated in blocking buffer (6% bovine serum albumin). They were stained with an anti-C5 rabbit polyclonal (Abcam, Cambridge, UK) and biotinylated rat anti-mouse monoclonal anti-C5R1 (Abcam), rabbit anti-mouse polyclonal anti-C5aR or rabbit anti-mouse nitrotyrosine (Sigma) for 1 h at room temperature. Sections were then washed in PBS, incubated with fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit immunoglobulin (Ig) with either phycoerythrin (PE)–streptavidin (for C5 and C5R1), propidium iodide (for C5aR) or 4,6-diamidino-2-phenylindole (DAPI) (for nitrotyrosine) for 1 h. Sections were then washed in PBS and mounted with Vectashield (Vector Laboratories, Peterborough, UK) before confocal microscopy. For control staining, rabbit serum was used instead of primary antibodies.
Therapeutic intervention
Systemic administration of the BB5·1 anti-C5 mAb was performed by i.p. injection in B10.RIII mice immunized as described above. Injections of 250 µg of the mAb or PBS control were performed on days 5 and/or 10. Local administration of mAb was performed by intravitreal injection in anaesthetized mice. Mice were anaesthetized by i.p. injection of 150 µl of Vetelar (ketamine hydrochloride 100 mg/ml; Pfizer, Sandwich, UK) and Rompun (xylazine hydrochloride 20 mg/ml; Bayer, Newbury, UK) mixed with sterile water in the ratio 0·6:1:84. The pupils of all animals were dilated using topical 1% tropicamide and 2·5% phenylepherine (Chauvin Pharmaceuticals, Kingston-Upon-Thames, Surrey, UK). Intravitreal injections of 25 µg of BB5·1 mAb or control IgG in 5 µl of PBS were performed under the direct control of a surgical microscope with the tip of a 12 mm 33-gauge hypodermic needle mounted on a 5-µl syringe (Hamilton AG, Bonaduz, Switzerland). The injection site was treated with chloramphenicol ointment. Injections were performed on days 10 or 14 after immunization.
Clinical assessment
Using a method adapted from Paques et al.[40,41] an endoscope with a 5-cm-long teleotoscope with a 3-mm outer diameter (1218AA; Karl Storz, Tuttlingen, Germany) was connected to a Nikon D80 digital camera with a 10-million pixel charge-coupled device (CCD) image sensor and Nikkor AF 85/F1·8 D objective (Nikon, Tokyo, Japan), with an additional + 4·00 dioptre magnifying lens. Through pupils dilated with topical tropicamide 1% and phenylepherine 2·5% (Minims, from Chauvin Pharmaceuticals, Kingston-Upon-Thames, Surrey UK), topical oxybropucaine 0·4% (Minims) and Viscotears (Novartis Pharmaceuticals, Camberley, UK) for corneal anaesthesia, images were obtained with direct corneal contact with the endoscope. Images were transferred to computer for processing using Adobe Photoshop (Adobe Corporation, Mountain View, CA, USA).
Total complement haemolytic assay
Female B10.RIII mice were treated with the BB5·1 anti-C5 mAb (250 µg or 1mg) on day 0 before killing on days 1 and 5 (n = 3). Blood was collected by cardiac puncture, serum prepared and total complement activity determined. Control serum was obtained from normal untreated mice on day 0. Briefly, sheep erythrocytes (E) (TCS Microbiology, Claydon, UK) were sensitized by incubating 1 volume of 4% E (v/v) with 1 volume of 1/250 rabbit anti-sheep E (Amboceptor, Behring Diagnostics, Marburg, Germany) for 30 min at 37°C. Sensitized cells (EA) were washed three times in complement fixation diluent (CFD; Oxoid Ltd, Basingstoke, UK) and resuspended to 2% (v/v). EA were then incubated for 30 min at 37°C with different dilutions of pretreatment, control or anti-C5 mAb-treated mouse serum. Cells were centrifuged to pellet and supernatant was removed for measurement of absorbance at 415 nm (haemoglobin release). Control incubations include cells with buffer only (0%) or with dH2O (100%). Percentage of lysis was calculated as described previously [42]. Percentage residual complement activity in mice that received anti-C5 mAb was calculated by taking complement activity in mice that did not receive anti-C5 mAb as 100%.
Splenocyte proliferation
Spleens were collected from immunized mice at various time-points and disrupted mechanically through a 40-µm cell strainer and red blood cells lysed. Cells were then seeded at 1 × 105 cells/well in 96-well round-bottomed plates, and stimulated with different concentrations of the immunizing peptide, RBP-3 161–180. Ovalbumin 323–339 peptide (OVA) was used as negative control for proliferation. To measure proliferation, cultures were pulsed with 18·5 kBq tritiated thymidine (GE Healthcare, Chalfont St.Giles, Bucks, UK) per well for the final 8 h of a 72-h incubation. Cells were harvested with a 96-well harvester, and thymidine uptake [measured in counts per minute (cpm)] was determined by liquid scintillation with a microbeta liquid scintillation counter (Microbeta 1450; Wallac Oy, Turku, Finland).
Generation of bone marrow-derived macrophages
Bone marrow-derived macrophages (BM-MΦ) were generated from C57BL/6 mice as described previously [43]. In brief, bone marrow cells were washed in DMEM and resuspended at 1 × 105 cells/ml in complete medium supplemented with 5% horse serum (PAA, Yeovil, UK) and 50 pg/ml macrophage–colony-stimulating factor. Cell suspension (50 ml) was transferred to Teflon-coated tissue culture bags (supplied by Dr M. Munder, University of Heidelberg, Germany) and incubated for 8 days at 37°C in 5% CO2. Cells were removed from the Teflon bags and plated at a concentration of 1 × 105 cells/well in 96-well, flat-bottomed plates (Corning-Costar, Corning, NY, USA) in complete media. BM-MΦ were stimulated with recombinant cytokines IFN-γ (100 U/ml) or IL-17 (200 ng/ml) (Peprotech, London, UK), Lipopolysaccharide (LPS) (0·1 µg/ml; Sigma-Aldrich), recombinant C5a (2·5 µg/ml to 0·01 µg/ml; R&D Systems, Abingdon, UK) or left unstimulated.
Quantification of NO synthesis
Accumulated NO production was measured after 36 h in culture by assaying culture supernatants for the stable reaction product of NO (nitrite) against a sodium nitrite standard on the same plate. A 50-µl total volume of each cell-free supernatant was incubated with an equal volume of Griess reagent [0·5% sulphanilamide and 0·05% N-(1-napthyl) ethylenediamine dihydrochloride in 2·5% phosphoric acid] in 96-well flat-bottomed plates for 10 min at room temperature. The optical densities were measured at 540 nm, with a reference filter of 630 nm.
Flow cytometry
Following removal of cell culture supernatant for NO or enzyme-linked immunosorbent assay (ELISA) assays, stimulated BM-MΦ were incubated with 24G2 cell supernatant for 15 min on ice before incubation with allophycocyanin (APC)-Cy7-conjugated anti-mouse CD11b mAb, phycoerythrin (PE)-conjugated anti-mouse CD88 mAb (both BD Biosciences) and AF647-conjugated anti-mouse CD200R mAb (AbD Serotec, Oxford, UK) on ice. BM-MΦ were removed carefully from the bottom of the wells and cell suspensions acquired with a LSR-II flow cytometer (BD Cytometry Systems). Analysis was carried out using FlowJo software (TreeStar, San Carlos, CA, USA).
Statistical analyses
Using Prism4 software (GraphPad Software Inc., San Diego, CA, USA), statistical significance between treatment groups were assessed using the Mann–Whitney U-test.
Results
Systemic administration of anti-C5 mAb (BB5·1) suppresses EAU
As activated components of the complement system are potent mediators of inflammation, and targeted inhibition of the C5 component using monoclonal antibodies is considered a promising therapeutic option [37], we wished to test the efficacy of treatment with the murine BB5·1 anti-C5 mAb in EAU and test whether its effects were mediated via suppression of macrophage activation. To confirm that the C5-specific mAb was functional and could inhibit systemic late-stage complement activation, total complement haemolytic activity of serum from mice treated with the mAb was assessed. Female B10.RIII mice were treated with a single dose of 250 µg or 1 mg of BB5·1 anti-C5 mAb on day 0 and killed on days 1 and 5 to collect serum. Haemolytic activity was reduced significantly in mice that received anti-C5 mAb, compared to normal serum at both time-points (Fig. 1). At a dose of 250 µg, complement activity was reduced by between 40% and 60% (*P < 0·05), while at a dose of 1 mg complement activity was reduced by 75% to 80% (**P < 0·005), each through to day 5.
Fig. 1.
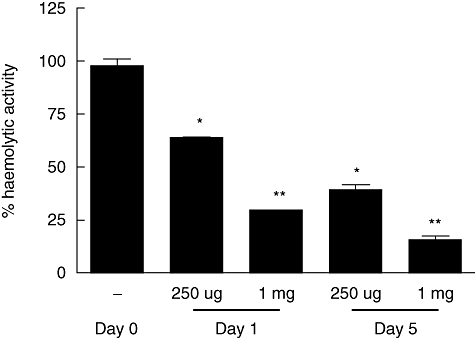
BB5·1 anti-C5 monoclonal antibody (mAb) treatment inhibits total serum complement haemolytic activity. The anti-C5 mAb was administered to mice intraperitoneally (250 µg or 1 mg per mouse) and serum was harvested at the time-points indicated. The total serum complement haemolytic activity was determined using a haemolysis assay. Results are expressed as percentage of activity compared to normal control serum levels, and ± standard deviation of haemolytic activity is shown (n = 3/time-point). Student's t-test was performed to determine P-values. *P < 0·05, **P < 0·005.
These data demonstrate that suppression of complement activation can be achieved when BB5·1 anti-C5 mAb is administered systemically in mice. To test the involvement and expression of complement component C5 and the C5a receptor (C5aR, also known as CD88) during progression of EAU, normal and inflamed retinal sections from peak disease animals were immunostained. Confocal microscopy demonstrates very low-level expression of C5 and CD88 in normal retinal sections (Fig. 2a,b). However, during the inflammatory response, C5 (green) and C5 receptor (red) co-localization and expression within inflammatory areas of the retina is clearly evident (Fig. 2c). Furthermore, complement receptor CD88 (green) expression was observed within cells located in subretinal folds, sites of maximal macrophage activation [11], as indicated by co-localization with nuclear PI staining (Fig. 2d).
Fig. 2.
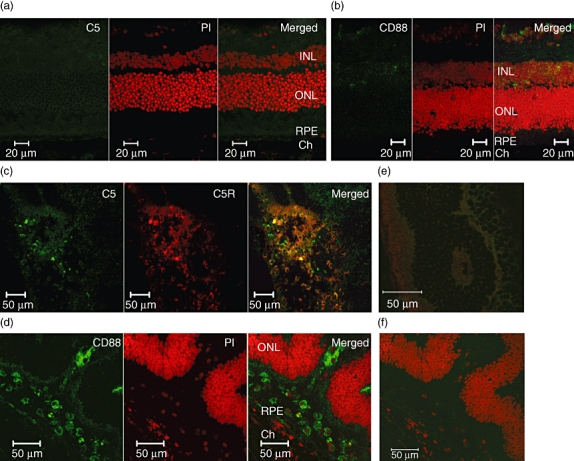
Expression of C5 and C5aR in normal and inflamed retinal tissue. Representative 12-µm sections from B10.RIII normal and day 14 post-immunization (PI) experimental autoimmune uveoretinitis (EAU). Dual stains for C5 (a) or the C5a receptor CD88 (b) in green and PI nuclear stain (red) in the normal retina. Dual stains for (c) C5 (green) and C5R (red) and (d) CD88 (green) and PI (red) expression in peak disease retina. Rabbit immunoglobulin (Ig)G control staining (e,f). Both anti-C5aR1 and anti-CD88 recognize the C5a receptor (CD88), but in different epitopes. Original magnification ×200.
To test the efficacy of the mAb in EAU we utilized the highly susceptible B10.RIII mouse strain, in which the immunizing regimen produces consistent moderate disease severity, ensuring that any changes would be clearly evident. Mice were immunized subcutaneously with 50 µg RBP-3161–180 emulsified in CFA and pertussis toxin was co-administered i.p. Complement activation may be important in both priming an immune response and amplifying inflammation within the target tissue. During the disease course of EAU in B10.RIII mice, antigen-specific cells are detectable in the retina at day 5 after immunization, several days prior to the development of clinical disease [44]. Thus, by this time-point, priming and dissemination of CD4+ antigen-specific cells has occurred and therefore this was when we chose to test the effects of systemic administration of the anti-C5 mAb. To this end we administered 250 µg anti-C5 mAb, or PBS control, by i.p. injection on days 5 and/or 10. On days 14 and 21, mice were killed and eyes were snap-frozen, sectioned and stained with anti-CD45 antibody. Three sections per treatment per time-point were scored for inflammatory infiltrate and structural damage.
By the first time-point examined on day 14 (start of peak disease) mice treated at days 5 and 10 together, or on either day alone, with BB5·1 anti-C5 mAb, displayed reduced overall disease scores when compared to the controls, with the reduction in disease severity most significant (*P = 0·0062) by day 21 (Fig. 3a). The presence of CD45+ cellular infiltrate and accompanying structural damage, including retinal folding and vasculitis, were clearly visible in the PBS-treated mice at days 14 and 21 [Fig. 3c: panels (i) and (v)]. In contrast, the retinas from the anti-C5 mAb-treated groups appeared normal and healthy, with maintained photoreceptor layers and minimal infiltrate, when mAb was administered on days 5 and 10, or as a single administration on days 5 or 10 only [Fig. 3c: panels (ii) (iii) and (iv), respectively]. To determine whether C5 inhibition could influence the initiation of disease, in another group we also included an additional mAb injection 1 day before disease immunization. Histological assessment at day 14 demonstrated comparable suppression of disease (data not shown). Following previous reports that complement anaphylatoxin C5a regulated antigen presentation [45,46] we determined, using splenocytes from control and anti-C5-treated mice at day 14 (peak disease), peptide-specific T cell proliferation. The proliferative response of splenocytes to reactivation with RBP-3 161–180 from the anti-C5 mAb group was reduced compared to control mice (Fig. 3b).
Fig. 3.
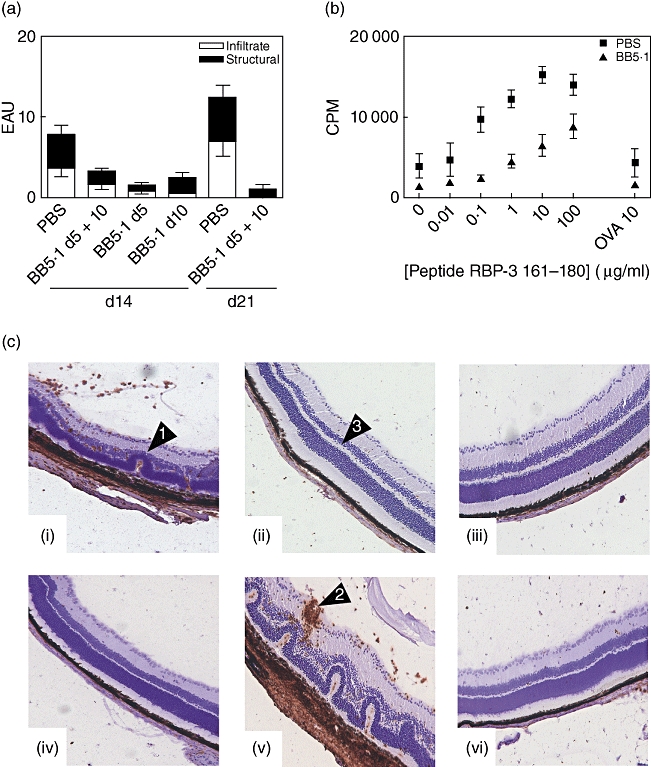
Systemic administration of BB5·1 anti-C5 monoclonal antibody (mAb) suppresses experimental autoimmune uveoretinitis (EAU). (a) Eyes were harvested at the days post-immunization indicated and average disease score ± standard deviation of inflammatory infiltrate and structural disease is shown (n = 6/time-point). Data are representative of two independent experiments. The Mann–Whitney U-test was performed on total disease score; *P = 0·0062. (b) Proliferation of splenocytes harvested on day 14 from B10.RIII mice treated with phosphate-buffered saline (PBS) or BB5·1 anti-C5 mAb on days 5 and 10. (c) Representative 12-µm sections on days 14 and 21 post-immunization. Photographs show sections stained with anti-CD45 antibody counterstained with haematoxylin from B10.RIII mice either treated with PBS control or BB5·1 anti-C5 mAb administered on days 5 and 10 (ii), or single administrations on days 5 (iii) or 10 (iv) only. In the control PBS-treated animals on days 14 (i) and 21 (v) there is marked retinal destruction, as indicated by arrows, retinal detachment, retinal folding (1) and vasculitis (2). In mice treated with anti-C5 (ii, iii, iv, vi), the retina appears normal with maintained photoreceptors (3). Original magnification ×100.
Activation of macrophages to produce NO following C5a stimulation requires IFN-γ
The systemic effect of anti-C5 mAb administration on the effector phase of EAU is clear, as demonstrated by the suppression of clinical disease development by treatment following priming. Complement activation generates the C3a and C5a fragments which promote and perpetuate inflammatory responses. The C5a fragment has a broad spectrum of functions [47], including being a chemoattractant for neutrophil recruitment, and having chemotactic activity for monocytes and macrophages [48]. C5a is reported as a potent inducer of proinflammatory activity, and C5a stimulation of human mononuclear phagocytes leads to secretion of IL-1β, IL-6, TNF-α and prostaglandin E2 (PGE2) [49–52]. Murine macrophage activation mediated by IFN-γ results in a similar pattern of proinflammatory cytokine expression, as well as the generation of NO, responsible for tissue damage observed during EAU. Therefore, we sought to determine whether C5a could similarly stimulate and classically activate murine macrophages.
Murine BM-Mφ were stimulated with recombinant C5a alone or in combination with IFN-γ or IL-17 and, after 36 h, culture supernatant analysed for NO production (Fig. 4a). Control stimulation of BM-Mφ with 100 U/ml recombinant IFN-γ or 0·1 µg/ml LPS demonstrate robust NO production, as reported previously [17], and stimulation with 200 ng/ml recombinant IL-17 resulted in only slightly elevated NO levels compared to unstimulated cells. Incubation of cells with C5a failed to induce any NO production at low concentrations (0·01–1 µg/ml), with only a small induction observed following the high 2·5 µg/ml stimulation. Addition of IFN-γ in combination with the lowest concentration of C5a resulted in NO levels similar to IFN-γ stimulation alone. However, increased concentrations of C5a mediated a dose-dependent and synergistic effect, with significantly elevated levels of NO, equivalent to high levels of NO production observed with LPS stimulation alone. Combined stimulation of cells with IL-17 with the range of C5a concentrations failed to elevate NO levels above that of unstimulated cells.
Fig. 4.
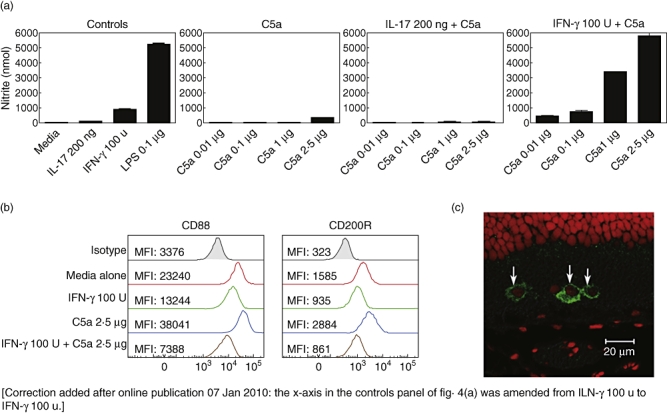
Activation of macrophages to produce nitric oxide (NO) following C5a stimulation requires interferon (IFN)-γ. C57BL/6 BM-MΦ were plated in 96-well plates in complete Dulbecco's modified Eagle's medium (DMEM) at a concentration of 1 × 105 cells/well, and stimulated with either recombinant interleukin (IL)-17 (200 ng/ml) or IFN-γ (100 U/ml) cytokines, lipopolysaccharide (LPS) (0·1 µg/ml), recombinant C5a (2·5 µg/ml to 0·01 µg/ml) or C5a in combination with IL-17 or IFN-γ, or left unstimulated. All treatments were performed in triplicate. After 36 h, NO production in culture supernatants was analysed using the Griess reaction (a), and surface expression of CD88 and CD200R examined by flow cytometry (b). Data are representative of three independent experiments (MFI: mean fluorescent intensity). A representative retinal section stained for CD88 (green) and PI nuclear stain (red) expression at peak experimental autoimmune uveoretinitis (EAU) disease, demonstrating the cytoplasmic expression of the receptor during inflammatory conditions (c).
To determine if these treatments influenced the expression of activatory or inhibitory myeloid receptors, stimulated macrophages were analysed by flow cytometry for the surface expression of CD88 (C5a receptor) and the inhibitory macrophage receptor CD200R (Fig. 4b). Data are presented as histograms and demonstrate the mean receptor expression (geometric mean fluorescence intensity) from unstimulated macrophages (red) or cells treated with IFN-γ (green), C5a (blue) alone or in combination (brown). The pattern of receptor expression following C5a treatment demonstrated an up-regulation in the relative expression of both receptors, with a 1·5-fold increase in CD88 and CD200R expression, respectively. Combined stimulation of cells with C5a and IFN-γ resulted in the greatest decrease of CD88 and CD200R surface expression. Furthermore, increasing C5a concentration in the presence of IFN-γ caused reduced CD88 and CD200R expression in a dose-dependent manner (data not shown). Cellular responses to G-protein-coupled receptors including the C5a receptor are controlled tightly by receptor dimerization and internalization [53]. Therefore, the observed reduction in macrophage surface expression of CD88 following in vitro activation with C5a and IFN-γ and intracellular staining of C5aR in cells that infiltrate the retina at peak disease (Fig. 4c) are consistent with such internalization.
Local delivery of BB5·1 mAb reduces EAU disease severity
The in vitro results suggest that activation of macrophages within the target organ, via simultaneous C5a and IFN-γ signalling, leads to enhanced nitric oxide response and tissue damage. This effect is accompanied by down-regulation of the inhibitory macrophage receptor CD200R, which is permissive for a highly activated macrophage phenotype. This raises the question of whether inhibiting the activation of complement in the local environment of the target organ would be sufficient to impact the course of clinical disease. This is also relevant to further possible clinical application and future translation in the arena of locally delivered ocular therapeutics. One advantage of this approach would be the reduction of adverse effects of peripheral late stage complement or macrophage deactivation, particularly if escalating doses are required.
To investigate this groups of mice were immunized to induce EAU, and single intravitreal injections of BB5·1 anti-C5 mAb were performed in the left eyes on either days 10 or 14 following immunization; groups of control mice were immunized similarly, but injected with control IgG. The peak of disease in this model is on day 14, thus targeting at these times ensures BB5·1 anti-C5 mAb saturation of the target tissue during disease peak. On day 21 eyes were snap-frozen, sectioned and stained with anti-CD45 antibody, and scored as described previously. Histological assessment demonstrated that local administration of BB5·1 anti-C5 mAb on days 10 or 14 resulted in reduced infiltrate and structural disease scores on day 21, with a 50% reduction in total disease score compared to control IgG alone (Fig. 5a and b). Similar suppression of disease was also seen on day 25 (data not shown). Prior to killing on day 21, clinical assessment of injected eyes was performed using topical endoscopic fundal imaging, a non-invasive technique that can be used to monitor clinical disease changes of the retina in EAU [41,54]. Representative fundal images from mice that received a single injection of BB5·1 anti-C5 on day 10 demonstrated suppression of chorioretinal infiltrate, perivascular sheathing or optic nerve head swelling, compared to IgG control animals that also showed other features of severe disease, which include retinal folding and exudative retinal detachments (Fig. 5c). During peak EAU, infiltrating myeloid cells under the influence of the pronounced cytokine release from the T helper type 1 (Th1) T cell infiltrate, express NOS2 and nitrotyrosine, and express NO constitutively [55]. As the in vitro data demonstrate the synergistic effect of C5a and IFN-γ promoting NO generation, we wished to determine whether disease suppression achieved via local delivery of the BB5·1 anti-C5 mAb was therefore as a direct result of reduced macrophage activation. To this end, sections were determined with a surrogate of myeloid-derived NO production via staining for the presence of nitrotyrosine (Fig. 5d). Representative immunofluorescence images of sections from mice that received the BB5·1 anti-C5 mAb [Fig. 5d; (iii)] demonstrate significantly reduced nitrotyrosine staining compared to the IgG control group, in which positive cells are clearly evident within areas of retinal damage and disruption [Fig. 5d; (i)].
Fig. 5.
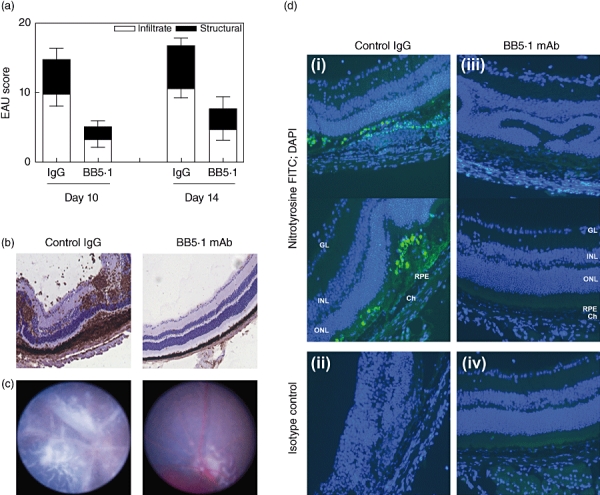
Local administration of BB5·1 anti-C5 monoclonal antibody (mAb) suppresses experimental autoimmune uveoretinitis (EAU). (a) Eyes from mice treated with the anti-C5 mAb on days 10 or 14 post-immunization were harvested on day 21 and average disease score ± standard deviation of inflammatory infiltrate and structural disease is shown (n = 3/time-point). (b) Representative 12-µm sections from day 21 post-immunization. Photographs show sections stained with anti-CD45 antibody counterstained with haematoxylin, from mice treated with either immunoglobulin (Ig)G control or BB5·1 anti-C5 mAbs administered locally on day 10. In the control animals treated with control IgG only on day 10 there is marked retinal destruction, retinal folding and vasculitis. In mice treated with the anti-C5 mAb at these time-points, the retina appears normal with maintained photoreceptors. Original magnification ×100. (c) Representative fundal images of the retina taken on day 21. The clinical images demonstrate that local administration of BB5·1 anti-C5 on day 10 results in marked suppression of chorioretinal infiltrate, perivascular sheathing or optic nerve head swelling, compared to IgG-treated control animals that demonstrate other features of severe disease, including retinal folds and exudative retinal detachments. (d) Representative photographs of retinal sections from day 21 post-immunization, dual-stained with anti-nitrotyrosine (green) conjugated antibody and 4,6-diamidino-2-phenylindole (DAPI) nuclear stain (blue) from mice treated with either IgG control or BB5·1 anti-C5 mAbs administered locally on day 10. In the control animals treated with control IgG there is increased staining of nitrotyrosine-positive cells within inflamed and damaged areas of the retina (i) compared to mice treated with the anti-C5 mAb (iii). Isotype rabbit Ig control staining (ii) and (iv). Original magnification ×200.
Discussion
The work presented in this study shows for the first time that inhibition of C5 activation, both systemically and locally, suppresses the development and progression of an antigen-specific T cell-mediated model of intraocular inflammation, EAU. The EAU model used in this study parallels and reflects the disease chronicity observed in humans and moreover, although not tested directly in current experiments, this model shows maintained chronic inflammatory responses and macrophage infiltration associated with angiogenesis beyond 70 days post-immunization (Xu et al. unpublished observations).
Systemic administration of the BB5·1 anti-C5 mAb during the afferent phase of disease results in significantly reduced infiltrate and disease when compared to controls. This indicates that prevention of C5 cleavage interferes, either directly or indirectly, with the effector responses of propagation of activation and expansion of and/or migration of inflammatory cells in EAU. Local treatment demonstrates the importance of C5-mediated accentuation of inflammation within the target organ, as it confirmed the suppressive effects of direct administration of anti-C5 mAb to the eye. Intravitreal therapy reduces infiltration and structural damage by preventing myeloid activation, as demonstrated by the reduction in positive nitrotyrosine staining. Local administration to regulate complement is arguably a more therapeutically useful option for the treatment of idiopathic non-infectious intra-ocular inflammatory disease, because this approach will minimize adverse systemic effects of late-stage complement inhibition, such as suppression of innate immunity and macrophage activation.
For the first time we demonstrate the synergistic role that IFN-γ and C5a together play in the stimulation of murine macrophages to produce NO. This implies that the effects of C5a release on macrophage activation are much greater in cells which have been exposed to Th1 cytokines and, conversely, implies that C5a will have much less impact on the activation of macrophages when they do not encounter it in the context of an inflammatory microenvironment. Additionally, the effect of C5a release on macrophage activation in a correlate of Th17 inflammatory cytokine environment does not result in any amplification of the NO response (see Fig. 4a).
During EAU, retinal isolated macrophages are activated and produce NO in response to T cell-derived IFN-γ secretion [55,56], contributing to the local control of T cell proliferation as well as contributing directly to tissue damage, while in addition tissue resident microglia on IFN-γ stimulation generate IL-10 [57]. We have demonstrated that the combination of direct C5a stimulation and IFN-γ synergize in stimulating the production of NO when compared to the action of IFN-γ or C5a alone to levels observed with LPS stimulation. While such large amounts of NO would have a direct effect on tissue damage via peroxynitrite production and lipid peroxidation of cell membranes, the action of NO on T cells is also known to limit their proliferative capacity [58], and recent work from our laboratory demonstrates that IFN-γ-mediated myeloid derived-NO production inhibits T cell proliferation within the eye during disease [59]. Therefore, as there was markedly reduced tissue damage and nitrotyrosine expression (observed particularly in sites of infiltrate within the retina), the effect of anti-C5 therapy observed following local administration has targeted the myeloid cell compartment, and specifically the tissue infiltrating macrophages. Despite the lack of direct evidence, these results suggest strongly that preventing C5 cleavage reduces the levels of C5a within the tissue, as demonstrated by the surrogate of reduced nitrotyrosine expression. However, measurement of the C5a levels in diseased eyes is experimentally difficult but, considering the striking synergy observed between C5a and IFN-γin vitro, could provide avenues of future investigation to determine whether its action is exerted across the whole tissue or is limited to the inflammatory cell microenvironment.
Furthermore, our in vitro data show that increased production of the effector molecule NO is accompanied by down-regulation of the inhibitory CD200R macrophage receptor, which would also be expected to intensify the response. It has been demonstrated that CD200R-mediated signalling is a critical homeostatic control mechanism, which prevents expression of IFN-γ-mediated macrophage activation and protects against tissue damage during autoimmune responses [17]. Therefore, we postulate that preventing C5 cleavage, and subsequent C5a-mediated signalling, prevents activation of macrophages and consequent recruitment of cells to sites of inflammation.
Selective inhibition of C5 using monoclonal antibodies is considered a promising therapeutic option with current preclinical development and clinical testing for a range of immune disorders, including psoriasis, arthritis, systemic lupus erythematosus, asthma and AMD [60]. It has been shown recently that the anaphylatoxins C5a and C3a interact with their receptors C5aR and C3aR, expressed on antigen-presenting cells (APCs) and T cells, and are involved integrally in T cell proliferation and differentiation [45,61]. The activating signals from these complement fragments are produced in the local microenvironment by APCs and T cells during cognate interactions and participate in the activation and cytokine production from both cell types [46]. Our data support these findings, as preventing C5 cleavage and the generation of C5a systemically results in reduced proliferative response of splenic T cells, thus limiting the expansion and migration of these effector cells. This also influences the subsequent recruitment, infiltration and activation of macrophages to the eye which ultimately mediate tissue damage. Intravitreal administration experiments demonstrate that prevention of local complement activation in EAU acts predominantly towards the infiltrating macrophages through suppression of NO production. Furthermore, blockade of this critical step of the complement cascade also prevents formation of the membrane attack complex (C5b-9) in cellular membranes, and therefore cell lysis and consequent damage to retinal tissues in EAU.
The likelihood of therapeutic potential can be evaluated well in EAU, where discrimination between inflammatory infiltrate and tissue destruction can be made. Furthermore, this model offers the possibility of investigating further the impact of local administration and efficacy at the site of inflammation, as has been demonstrated both experimentally and clinically. Continuous improvement and clinical testing of C5-specific mAbs have resulted in the translation of Eculizumab™, which is currently the only complement-specific antibody approved for therapy of paroxysmal nocturnal haemoglobinuria, a rare immunological disorder characterized by the destruction of red blood cells. Further efficacy and preclinical testing is required, but preventing the cleavage of C5 and generation of the major proinflammatory factors C5a and C5b-9 has therapeutic potential to protect against tissue damage in organ-specific immune responses within the retina.
Acknowledgments
The authors wish to thank the National Eye Research Centre, UK for their generous funding of this research. Sivasankar Baalasubramanian is funded through a project grant funded by Wellcome Trust (ref. no: 079115). T. R. H. is funded by a Wellcome Trust programme grant awarded to B. P. M. (ref. no: 068590).
Disclosure
No conflict of interest to declare.
References
- 1.Forrester JV, Liversidge J, Dua HS, Towler H, McMenamin PG. Comparison of clinical and experimental uveitis. Curr Eye Res. 1990;9(Suppl):75–84. doi: 10.3109/02713689008999424. [DOI] [PubMed] [Google Scholar]
- 2.Dick AD. Experimental approaches to specific immunotherapies in autoimmune disease: future treatment of endogenous posterior uveitis? Br J Ophthalmol. 1995;79:81–8. doi: 10.1136/bjo.79.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kerr EC, Copland DA, Dick AD, Nicholson LB. The dynamics of leukocyte infiltration in experimental autoimmune uveoretinitis. Prog Retin Eye Res. 2008;27:527–35. doi: 10.1016/j.preteyeres.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 4.Caspi RR. Regulation, counter-regulation, and immunotherapy of autoimmune responses to immunologically privileged retinal antigens. Immunol Res. 2003;27:149–60. doi: 10.1385/IR:27:2-3:149. [DOI] [PubMed] [Google Scholar]
- 5.de Smet MD, Chan CC. Regulation of ocular inflammation – what experimental and human studies have taught us. Prog Retin Eye Res. 2001;20:761–97. doi: 10.1016/s1350-9462(01)00011-8. [DOI] [PubMed] [Google Scholar]
- 6.Wacker WB. Retinal autoimmunity: two decades of research. Jpn J Ophthalmol. 1987;31:188–96. [PubMed] [Google Scholar]
- 7.Wacker WB. Proctor lecture. Experimental allergic uveitis. Investigations of retinal autoimmunity and the immunopathologic responses evoked. Invest Ophthalmol Vis Sci. 1991;32:3119–28. [PubMed] [Google Scholar]
- 8.Atalla L, Linker-Israeli M, Steinman L, Rao NA. Inhibition of autoimmune uveitis by anti-CD4 antibody. Invest Ophthalmol Vis Sci. 1990;31:1264–70. [PubMed] [Google Scholar]
- 9.Caspi RR. Immunogenetic aspects of clinical and experimental uveitis. Reg Immunol. 1992;4:321–30. [PubMed] [Google Scholar]
- 10.Thurau SR, Chan CC, Nussenblatt RB, Caspi RR. Oral tolerance in a murine model of relapsing experimental autoimmune uveoretinitis (EAU): induction of protective tolerance in primed animals. Clin Exp Immunol. 1997;109:370–6. doi: 10.1046/j.1365-2249.1997.4571356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoey S, Grabowski PS, Ralston SH, Forrester JV, Liversidge J. Nitric oxide accelerates the onset and increases the severity of experimental autoimmune uveoretinitis through an IFN-gamma-dependent mechanism. J Immunol. 1997;159:5132–42. [PubMed] [Google Scholar]
- 12.Gordon S. Pattern recognition receptors: doubling up for the innate immune response. Cell. 2002;111:927–30. doi: 10.1016/s0092-8674(02)01201-1. [DOI] [PubMed] [Google Scholar]
- 13.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 14.Forrester JV, Huitinga I, Lumsden L, Dijkstra CD. Marrow-derived activated macrophages are required during the effector phase of experimental autoimmune uveoretinitis in rats. Curr Eye Res. 1998;17:426–37. doi: 10.1080/02713689808951224. [DOI] [PubMed] [Google Scholar]
- 15.Dick AD, Duncan L, Hale G, Waldmann H, Isaacs J. Neutralizing TNF-alpha activity modulates T-cell phenotype and function in experimental autoimmune uveoretinitis. J Autoimmun. 1998;11:255–64. doi: 10.1006/jaut.1998.0197. [DOI] [PubMed] [Google Scholar]
- 16.Dick AD, Forrester JV, Liversidge J, Cope AP. The role of tumour necrosis factor (TNF-alpha) in experimental autoimmune uveoretinitis (EAU) Prog Retin Eye Res. 2004;23:617–37. doi: 10.1016/j.preteyeres.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 17.Copland DA, Calder CJ, Raveney BJ, et al. Monoclonal antibody-mediated CD200 receptor signaling suppresses macrophage activation and tissue damage in experimental autoimmune uveoretinitis. Am J Pathol. 2007;171:580–8. doi: 10.2353/ajpath.2007.070272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344:1058. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 19.Carroll MC. The complement system in regulation of adaptive immunity. Nat Immunol. 2004;5:981–6. doi: 10.1038/ni1113. [DOI] [PubMed] [Google Scholar]
- 20.Sohn JH, Bora PS, Suk HJ, Molina H, Kaplan HJ, Bora NS. Tolerance is dependent on complement C3 fragment iC3b binding to antigen-presenting cells. Nat Med. 2003;9:206–12. doi: 10.1038/nm814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Linton SM, Morgan BP. Complement activation and inhibition in experimental models of arthritis. Mol Immunol. 1999;36:905–14. doi: 10.1016/s0161-5890(99)00113-3. [DOI] [PubMed] [Google Scholar]
- 22.Woodruff TM, Strachan AJ, Dryburgh N, et al. Antiarthritic activity of an orally active C5a receptor antagonist against antigen-induced monarticular arthritis in the rat. Arthritis Rheum. 2002;46:2476–85. doi: 10.1002/art.10449. [DOI] [PubMed] [Google Scholar]
- 23.Prineas JW, Kwon EE, Cho ES, et al. Immunopathology of secondary-progressive multiple sclerosis. Ann Neurol. 2001;50:646–57. doi: 10.1002/ana.1255. [DOI] [PubMed] [Google Scholar]
- 24.Morgan BP, Griffiths M, Khanom H, Taylor SM, Neal JW. Blockade of the C5a receptor fails to protect against experimental autoimmune encephalomyelitis in rats. Clin Exp Immunol. 2004;138:430–8. doi: 10.1111/j.1365-2249.2004.02646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kijlstra A, La Heij E, Hendrikse F. Immunological factors in the pathogenesis and treatment of age-related macular degeneration. Ocul Immunol Inflamm. 2005;13:3–11. doi: 10.1080/09273940590909185. [DOI] [PubMed] [Google Scholar]
- 26.Mullins RF, Russell SR, Anderson DH, Hageman GS. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J. 2000;14:835–46. [PubMed] [Google Scholar]
- 27.Nozaki M, Raisler BJ, Sakurai E, et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc Natl Acad Sci USA. 2006;103:2328–33. doi: 10.1073/pnas.0408835103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jha P, Bora PS, Bora NS. The role of complement system in ocular diseases including uveitis and macular degeneration. Mol Immunol. 2007;44:3901–8. doi: 10.1016/j.molimm.2007.06.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Montalvo V, Campos MM, Chan CC, et al. Complement deposits on ocular tissues adjacent to sites of inflammation. Curr Eye Res. 2007;32:917–22. doi: 10.1080/02713680701656343. [DOI] [PubMed] [Google Scholar]
- 30.Read RW, Szalai AJ, Vogt SD, McGwin G, Barnum SR. Genetic deficiency of C3 as well as CNS-targeted expression of the complement inhibitor sCrry ameliorates experimental autoimmune uveoretinitis. Exp Eye Res. 2006;82:389–94. doi: 10.1016/j.exer.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 31.Jha P, Sohn JH, Xu Q, et al. The complement system plays a critical role in the development of experimental autoimmune anterior uveitis. Invest Ophthalmol Vis Sci. 2006;47:1030–8. doi: 10.1167/iovs.05-1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jha P, Sohn JH, Xu Q, et al. Suppression of complement regulatory proteins (CRPs) exacerbates experimental autoimmune anterior uveitis (EAAU) J Immunol. 2006;176:7221–31. doi: 10.4049/jimmunol.176.12.7221. [DOI] [PubMed] [Google Scholar]
- 33.An F, Li Q, Tu Z, et al. Role of DAF in protecting against T-cell autoreactivity that leads to experimental autoimmune uveitis. Invest Ophthalmol Vis Sci. 2009;50:3778–82. doi: 10.1167/iovs.08-3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Turnberg D, Botto M. The regulation of the complement system: insights from genetically-engineered mice. Mol Immunol. 2003;40:145–53. doi: 10.1016/s0161-5890(03)00110-x. [DOI] [PubMed] [Google Scholar]
- 35.Gerard NP, Gerard C. The chemotactic receptor for human C5a anaphylatoxin. Nature. 1991;349:614–7. doi: 10.1038/349614a0. [DOI] [PubMed] [Google Scholar]
- 36.Monk PN, Scola AM, Madala P, Fairlie DP. Function, structure and therapeutic potential of complement C5a receptors. Br J Pharmacol. 2007;152:429–48. doi: 10.1038/sj.bjp.0707332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y, Rollins SA, Madri JA, Matis LA. Anti-C5 monoclonal antibody therapy prevents collagen-induced arthritis and ameliorates established disease. Proc Natl Acad Sci USA. 1995;92:8955–9. doi: 10.1073/pnas.92.19.8955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frei Y, Lambris JD, Stockinger B. Generation of a monoclonal antibody to mouse C5 application in an ELISA assay for detection of anti-C5 antibodies. Mol Cell Probes. 1987;1:141–9. doi: 10.1016/0890-8508(87)90022-3. [DOI] [PubMed] [Google Scholar]
- 39.Dick AD, Cheng YF, Liversidge J, Forrester JV. Immunomodulation of experimental autoimmune uveoretinitis: a model of tolerance induction with retinal antigens. Eye. 1994;8:52–9. doi: 10.1038/eye.1994.10. [DOI] [PubMed] [Google Scholar]
- 40.Paques M, Guyomard JL, Simonutti M, et al. Panretinal, high-resolution color photography of the mouse fundus. Invest Ophthalmol Vis Sci. 2007;48:2769–74. doi: 10.1167/iovs.06-1099. [DOI] [PubMed] [Google Scholar]
- 41.Copland DA, Wertheim MS, Armitage J, Nicholson LB, Raveney BJ, Dick AD. The clinical time-course of experimental autoimmune uveoretinitis using topical endoscopic fundal imaging with histological and cellular infiltrate correlation. Invest Ophthalmol Vis Sci. 2008;49:5458–65. doi: 10.1167/iovs.08-2348. [DOI] [PubMed] [Google Scholar]
- 42.Holt DS, Botto M, Bygrave AE, Hanna SM, Walport MJ, Morgan BP. Targeted deletion of the CD59 gene causes spontaneous intravascular hemolysis and hemoglobinuria. Blood. 2001;98:442–9. doi: 10.1182/blood.v98.2.442. [DOI] [PubMed] [Google Scholar]
- 43.Munder M, Eichmann K, Moran JM, Centeno F, Soler G, Modolell M. Th1/Th2-regulated expression of arginase isoforms in murine macrophages and dendritic cells. J Immunol. 1999;163:3771–7. [PubMed] [Google Scholar]
- 44.Kerr EC, Raveney BJ, Copland DA, Dick AD, Nicholson LB. Analysis of retinal cellular infiltrate in experimental autoimmune uveoretinitis reveals multiple regulatory cell populations. J Autoimmun. 2008;31:354–61. doi: 10.1016/j.jaut.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 45.Liu J, Lin F, Strainic MG, et al. IFN-gamma and IL-17 production in experimental autoimmune encephalomyelitis depends on local APC-T cell complement production. J Immunol. 2008;180:5882–9. doi: 10.4049/jimmunol.180.9.5882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Strainic MG, Liu J, Huang D, et al. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity. 2008;28:425–35. doi: 10.1016/j.immuni.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Manthey HD, Woodruff TM, Taylor SM, Monk PN. Complement component 5a (C5a) Int J Biochem Cell Biol. 2009;41:2114–17. doi: 10.1016/j.biocel.2009.04.005. [DOI] [PubMed] [Google Scholar]
- 48.Guo RF, Ward PA. Role of C5a in inflammatory responses. Annu Rev Immunol. 2005;23:821–52. doi: 10.1146/annurev.immunol.23.021704.115835. [DOI] [PubMed] [Google Scholar]
- 49.Cavaillon JM, Fitting C, Haeffner-Cavaillon N. Recombinant C5a enhances interleukin 1 and tumor necrosis factor release by lipopolysaccharide-stimulated monocytes and macrophages. Eur J Immunol. 1990;20:253–7. doi: 10.1002/eji.1830200204. [DOI] [PubMed] [Google Scholar]
- 50.Scholz W, McClurg MR, Cardenas GJ, et al. C5a-mediated release of interleukin 6 by human monocytes. Clin Immunol Immunopathol. 1990;57:297–307. doi: 10.1016/0090-1229(90)90043-p. [DOI] [PubMed] [Google Scholar]
- 51.Ember JA, Sanderson SD, Hugli TE, Morgan EL. Induction of interleukin-8 synthesis from monocytes by human C5a anaphylatoxin. Am J Pathol. 1994;144:393–403. [PMC free article] [PubMed] [Google Scholar]
- 52.Kunkel SL, Kaercher K, Plewa M, Fantone JC, Ward PA. Production of cyclooxygenase products and superoxide anion by macrophages in response to chemotactic factors. Prostaglandins. 1982;24:789–99. doi: 10.1016/0090-6980(82)90059-4. [DOI] [PubMed] [Google Scholar]
- 53.Rabiet MJ, Huet E, Boulay F. Complement component 5a receptor oligomerization and homologous receptor down-regulation. J Biol Chem. 2008;283:31038–46. doi: 10.1074/jbc.M805260200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu H, Koch P, Chen M, Lau A, Reid DM, Forrester JV. A clinical grading system for retinal inflammation in the chronic model of experimental autoimmune uveoretinitis using digital fundus images. Exp Eye Res. 2008;87:319–26. doi: 10.1016/j.exer.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 55.Dick AD, Carter D, Robertson M, et al. Control of myeloid activity during retinal inflammation. J Leukoc Biol. 2003;74:161–6. doi: 10.1189/jlb.1102535. [DOI] [PubMed] [Google Scholar]
- 56.Robertson MJ, Erwig LP, Liversidge J, Forrester JV, Rees AJ, Dick AD. Retinal microenvironment controls resident and infiltrating macrophage function during uveoretinitis. Invest Ophthalmol Vis Sci. 2002;43:2250–7. [PubMed] [Google Scholar]
- 57.Broderick C, Duncan L, Taylor N, Dick AD. IFN-gamma and LPS-mediated IL-10-dependent suppression of retinal microglial activation. Invest Ophthalmol Vis Sci. 2000;41:2613–22. [PubMed] [Google Scholar]
- 58.van der Veen RC, Dietlin TA, Dixon Gray J, Gilmore W. Macrophage-derived nitric oxide inhibits the proliferation of activated T helper cells and is induced during antigenic stimulation of resting T cells. Cell Immunol. 2000;199:43–9. doi: 10.1006/cimm.1999.1597. [DOI] [PubMed] [Google Scholar]
- 59.Raveney BJ, Copland DA, Dick AD, Nicholson LB. TNFR1-dependent regulation of myeloid cell function in experimental autoimmune uveoretinitis. J Immunol. 2009;183:2321–9. doi: 10.4049/jimmunol.0901340. [DOI] [PubMed] [Google Scholar]
- 60.Ricklin D, Lambris JD. Complement-targeted therapeutics. Nat Biotechnol. 2007;25:1265–75. doi: 10.1038/nbt1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Peng Q, Li K, Patel H, Sacks SH, Zhou W. Dendritic cell synthesis of C3 is required for full T cell activation and development of a Th1 phenotype. J Immunol. 2006;176:3330–41. doi: 10.4049/jimmunol.176.6.3330. [DOI] [PubMed] [Google Scholar]