

Abstract
Type 1 diabetes results from a T cell-mediated destruction of insulin-producing pancreatic β cells. Little is known on local factors contributing to migration of T cells to pancreatic tissue. We recently demonstrated evidence of viral infection in β cells in several recent-onset type 1 diabetes patients. Islet inflammation was analysed in a series of new- or recent-onset type 1 diabetic patients and non-diabetic control subjects. Autoimmune T cell reactivity was studied in lymphocytes derived from pancreas-draining lymph nodes of one recent-onset type 1 diabetes patient in partial clinical remission. Insulitic lesions were characterized by presence of β cells, elevated levels of the chemokine CXCL10 and infiltration of lymphocytes expressing the corresponding chemokine receptor CXCR3 in all pancreatic lesions of type 1 diabetes patients, regardless of enterovirus infection of β cells. CXCR3 and CXCL10 were undetectable in pancreata of non-diabetic control subjects. T cells isolated from draining lymph nodes of a recent-onset patient with virally infected β cells and in clinical remission reacted with multiple islet autoantigens and displayed a mixed interferon (IFN)-γ/interleukin (IL)-10 cytokine pattern. Our data point to CXCL10 as an important cytokine in distressed islets that may contribute to inflammation leading to insulitis and β cell destruction, regardless of local viral infection. We demonstrate further pro- and anti-inflammatory islet autoreactivity, indicating that different adaptive and innate immune responses may contribute to insulitis and β cell destruction.
Keywords: autoreactive T cells, CXCL10, CXCR3, IP-10, type 1 diabetes
Introduction
Type 1 diabetes results from a T cell-mediated destruction of the insulin-producing pancreatic β cells [1]. T cell autoreactivity in peripheral blood of patients can serve as a surrogate marker of ongoing insulitis [2,3], but detection of circulating islet autoreactive T cells is hampered by low precursor frequencies and possibly regulatory T cells [4–8]. It is unclear to what extent peripheral T cell autoreactivity bears relevance to the pathogenesis of type 1 diabetes. Studies to identify diabetes-associated T cells in men have been hindered thus far by the inaccessibility of the insulitic lesions.
In both humans and the non-obese diabetic (NOD) mouse strain, that develops autoimmune diabetes spontaneously, β cell destruction is preceded by leucocyte infiltration of the pancreatic islets (insulitis). We have demonstrated recently that T cells isolated from peripheral blood of prediabetic subjects and reactive against the islet autoantigen glutamic acid decarboxylase 65 (GAD65) home to pancreatic tissue and pancreas-draining lymph nodes but not to other secondary lymphoid tissues when injected into NOD/severe combined immunodeficiency (SCID) mice [9]. This process was dependent upon co-injection of human leucocyte antigen (HLA)-matched antigen-presenting cells and the relevant autoantigenic epitope and was amplified by β cell distress following pretreatment of recipient mice with low-dose streptozotocin. These data imply that islet autoreactive T cells isolated from the circulation of (pre)diabetic subjects may bear relevance to insulitis and possibly to the β cell destruction process.
Kent et al. have described oligoclonality of CD4 T cells in the pancreas-draining lymph nodes of two long-standing type 1 diabetes patients [10]. This report was the first to describe immune phenotype and reactivity in draining lymphoid tissue that may reflect autoimmune reactivities associated with the type 1 diabetic lesion, albeit that in the two reported cases, both insulitis and target β cells were lacking. The authors suggested further that some of these T cells responded to insulin peptide. While there is compelling evidence that insulin serves as a major autoantigen in animal models of type 1 diabetes [11–14], similar evidence of immunodominant T cell responses to insulin, rather than other candidate islet autoantigens, in clinical type 1 diabetes is circumstantial [6,15,16]. Nevertheless, this seminal study set the stage for studies on T cell autoreactivity in pancreas-associated tissues.
In this study we present four cases where whole pancreas and some pancreas-draining lymph nodes were obtained from recent-onset type 1 diabetic patients, including one case of viral infection of pancreatic β cells. Two of these patients died accidentally, the other two died of brain oedema as a complication of diabetic ketoacidosis. We examined these tissues for the presence of islet-specific expression of proinflammatory molecules involved in lymphocyte recruitment and for the presence and function of autoreactive T cells in one case where we obtained pancreas-draining lymph nodes.
Methods
The pancreatic tissues were handled and processed according to the recommendations of the Pisa Ethics Committee. The first whole pancreas and pancreas-draining lymph nodes were obtained from a 24-year-old type 1 diabetic Caucasoid male donor expressing HLA-A3, A29, B7, B24, DR7 and DR13 (Table 1). Type 1 diabetes was diagnosed 10 months prior to the car accident that caused his death. At the time of diagnosis, as well as at the time of the accident, the patient displayed autoantibodies against GAD, but not against IA-2. One month prior to the accident, he was in good metabolic control [glycated haemoglobin (HbA1c) 6·1%], with a low insulin need (a total of 16 units/day) and with basal circulating C-peptide level of 1·8 ng/ml. He had no family history of type 1 or type 2 diabetes. Retrospective studies revealed a selective infection of pancreatic β cells by enterovirus impairing β cell function. To test whether our observation was in common with, or distinct from, non-viral autoimmune insulitis, we tested an additional series of pancreatic tissue of new-onset type 1 diabetic cases without evidence of virus contributing to their β cell destruction [17]. Whole pancreas was obtained from a 14-year-old female donor expressing HLA-A2, A25, B8, DR3/3 and DQ2 who died in an accident 8 months after being diagnosed with type 1 diabetes (Table 1). At diagnosis, which was accompanied by diabetic ketoacidosis, she was tested positive for islet cell cytoplasmic antibodies (ICA) [160 Juvenile Diabetes Foundation (JDF) units], anti-GAD and anti-IA2 autoantibodies. Glycaemic control was fairly well maintained with HbA1c levels of less than 7·5% by approximately 0·4 units/kg of insulin daily.
Table 1.
Patient characteristics.
| Donor | Age | Sex | HLA typing |
|---|---|---|---|
| 1 | 24 | Male | A3,29; B7,44; DR7,13 |
| 2 | 14 | Female | A2,25; B8; DR3,3; DQ2 |
| 3 | 5 | Male | A1,24; B8,60; DR3,4 |
| 4 | 4 | Female | Unknown |
HLA, human leucocyte antigen.
The third whole pancreas was obtained from a 5-year-old male donor (HLA-A1, A24, B8, B60, DR3 and DR4) who died due to severe brain oedema developed after diabetic ketoacidosis. He was tested anti-GAD, anti-IAA and anti-IA2 autoantibody-positive. The last whole pancreas was obtained from a 4-year-old female donor, who also died due to severe brain oedema which developed after diabetic ketoacidosis. She was tested positive for anti-IA2 and anti-insulin autoantibodies.
In addition, pancreatic specimens were obtained from five non-diabetic multi-organ donors (age: 33·2 ± 14·4 years; three male/two female; body mass index: 24·9 ± 1·3 kg/m2).
Histochemical studies
Pancreatic specimens were formalin-fixed and paraffin-embedded for immunohistochemical investigations. Specifically, islet infiltration by CD3 expressing leucocytes and insulin content was analysed by immunohistochemistry using mouse monoclonal antibody against CD3 (Dako Corporation) and guinea pig polyclonal antibody against insulin (Sigma, St. Louis, MO, USA), employing a labelled streptavidin–biotin (Dako Italy S.p.A., Milan, Italy) peroxidase method. In addition, monoclonal mouse anti-human CXCR3 (R&D Systems Europe Ltd, Abingdon, UK), polyclonal rabbit anti-human CXCL10 (Dako) and monoclonal mouse anti-human Fas (CD95) (CH11, mouse IgM; Upstate Biotechnology, Lake Placid, NY, USA) antibodies were employed to characterize further ongoing islet inflammation.
Immunology
Immune reactivity to candidate islet autoantigens insulin (Sigma), GAD65 (Diamyd AS, Stockholm, Sweden), IA-2 (kindly provided by Dr John Elliott, University of Alberta, Edmonton, Canada) as well as a synthetic peptide of the insulin B9-23 epitope was tested in leucocytes isolated from pancreas-draining lymph nodes from donor 1 by T cell proliferation, as described elsewhere [18] (concentration of antigens 10 µg/ml). Corresponding cytokine production was measured by the cytometric bead assay [interleukin (IL)-2, IL-4, IL-5, IL-10, interferon (IFN)-γ, tumour necrosis factor (TNF)-α; Becton Dickinson Biosciences, San Jose, CA, USA)], following the manufacturer's instructions. Data are given as the mean of triplicates with standard deviations.
Results
Histology
Immunohistochemical investigations of donor 1 revealed the presence of insulitis as well as intact islets containing insulin-positive β cells at the time of death (Fig. 1). Insulitis was present in 44% of islets studied (n = 75) at the time of death and was characterized by CD3 expressing T cells (Fig. 1) and natural killer cells (data not shown); β cells could be demonstrated in the vast majority of pancreatic islets analysed (86%, n = 150).
Fig. 1.
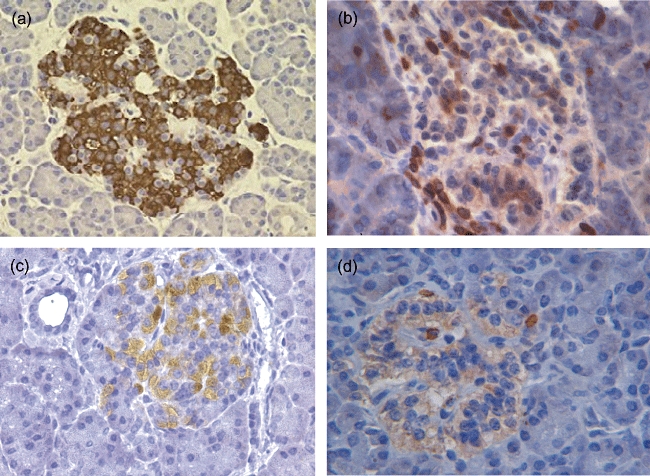
Histology of islets of Langerhans in pancreatic tissue obtained from a recent-onset type 1 diabetes patient with virally infected β cells during clinical remission. Sections show the presence of insulin-positive cells (a) and infiltration of islets with CD3-positive leucocytes (b). Active leucocyte recruitment is visualized by expression of the inflammation-induced chemokine CXCL10 by stromal cells (c) and its corresponding receptor CXCR3 on infiltrating leucocytes (d).
Ongoing islet inflammation and active recruitment of leucocytes was confirmed in all donors by in situ detection of the proinflammatory chemokine CXCL10 (20 of 42 positive islets, Fig. 1c) and its ligand CXCR3 (Fig. 1d). Using immunohistochemistry, electron microscopy, whole-genome ex vivo nucleotide sequencing, cell culture and immunological studies, we have demonstrated previously Coxsackie B4 enterovirus infection, specifically in β cells of donor 1 [17]. Insulitic lesions of three new-onset type 1 diabetes patients without evidence of virally infected β cells showed similar combinations of CXCL10 production by insulitic β cells and CXCR3 expression by pancreas-infiltrating lymphocytes that were absent in pancreatic sections of non-diabetic organ donors (Fig. 2).
Fig. 2.
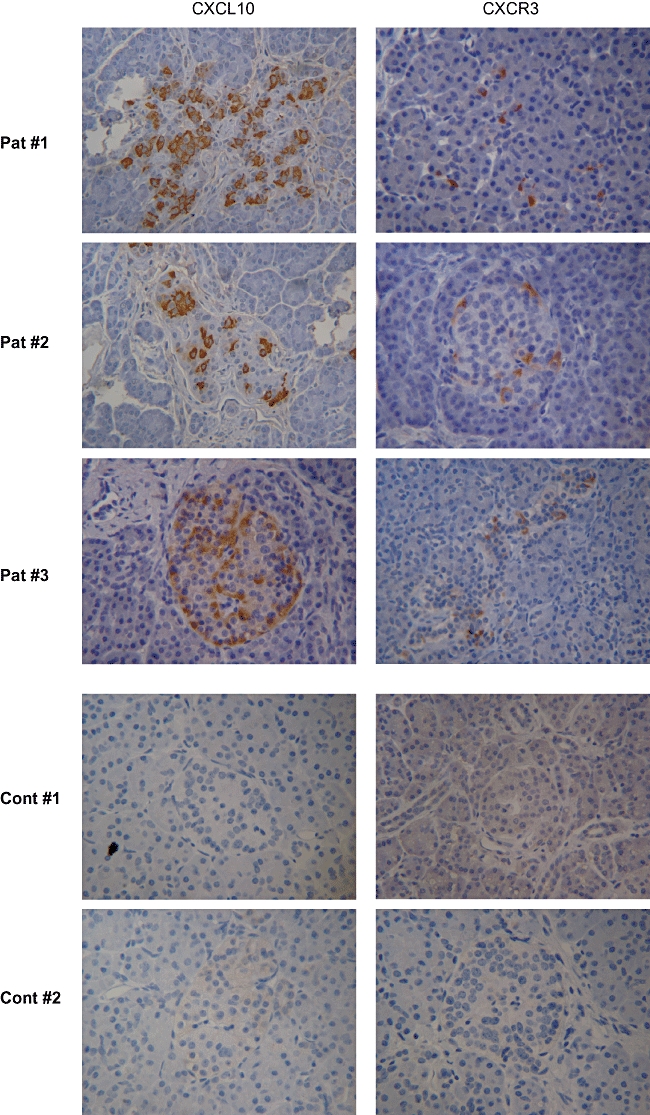
CXCL10 and CXCR3 expression in histology of pancreatic islets of three new-onset type 1 diabetes patients and two non-diabetic control subjects without evidence of viral infection of β cells.
Immunological studies were performed on freshly isolated and unseparated lymph node cells of the case with viral infection to study islet autoreactivity in pancreas-draining lymph nodes. Cellular autoimmune responses as defined by proliferation and cytokine production were measured against the candidate islet autoantigens insulin, GAD65 and IA-2 (Fig. 3). In addition, a synthetic peptide of the insulin B-chain (aa9-23), that was shown previously to be an immunodominant epitope of insulitic T cells in NOD mice, was tested [19]. Increased proliferation of autoreactive T cells isolated from pancreas-draining lymph nodes was measured directly ex vivo in response to GAD65 compared to medium alone (P = 0·0006), and to a lesser extent to insulin peptide (P = 0·012), but not to IA-2 or insulin protein. Islet autoantigens acquired by antigen-presenting cells in vivo and residing in the pancreas-draining lymph nodes are conceivably presented to local T cells, which may result in elevated baseline reactivity as demonstrated by relatively increased medium values (Fig. 3a).
Fig. 3.
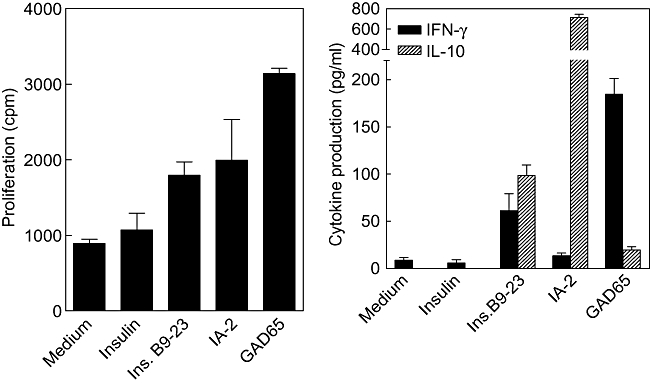
Immune reactivity to candidate islet autoantigens was tested in leucocytes isolated from pancreas-draining lymph nodes by T cell proliferation (left) and cytokine production (right).
T cell autoreactivity was accompanied by production of IFN-γ (GAD65) or IL-10 (IA-2) or both (insulin B9-23), possibly reflecting pathogenic as well as regulatory immune autoreactivity to islets [6]. The insulin A-chain (aa1-14) epitope, claimed recently to be recognized dominantly by T cells from pancreas-draining lymph nodes of long-standing type 1 diabetes patients, was not yet known at the time of the patient's death [10], and was therefore not tested.
Discussion
It is conceivable that pancreas-draining lymph nodes contain islet immune components that bear relevance to insulitis and islet destruction. Preliminary evidence of oligoclonality and reactivity to insulin peptide in two cases of long-standing type 1 diabetes exists [10]. The T cell response to insulin in that study was detected by IL-13 production in response to high doses (in the millimolar range) of insulin peptide, but not to whole insulin or proinsulin. In view of the lack of remaining β cells or insulitis in the latter donors, it remains unresolved whether the immune reactivity to insulin described is relevant to the disease onset. Given the clinical heterogeneity of type 1 diabetes, other candidate antigens such as GAD65, IA-2 or as yet unidentified β cell proteins should still be considered [16].
Our first case expressed an HLA genotype that does not particularly predispose to development of type 1 diabetes [20]. Diagnosis of this disease was, however, corroborated by the detection of autoantibodies against GAD65 [21]. However, this patient presented unexpectedly with enteroviral infection of pancreatic β cells that may contribute to loss of immunological tolerance and impaired β cell function [17]. Despite the presence of intact β cells and insulitis in our patient, it is not yet possible to determine the degree of representation of this case in defining immune responses that are associated with the onset of inflammatory lesions in the islets of genetically predisposed patients. Furthermore, studies were performed at a time that the patient's blood glucose could be regulated by modest doses of exogenous insulin, implying that our patient was either in remission (‘honeymoon’) at the time of his accidental death, or suffering from a syndrome referred to as latent autoimmune diabetes in adults (LADA) [22]. There is no reason to believe that the pathology of LADA differs from type 1 diabetes in terms of disease mechanism and manifestation [23]. In fact, the low insulin requirement and good metabolic control were accompanied by islet autoreactivity composed of pro- as well as anti-inflammatory immune responses. We propose that the immune response described could bear relevance to disease regulation [6] similar to the pre-onset (peri-insulitis) stage in NOD mice.
Chemokines are key players in orchestrating lymphocyte traffic into sites of inflammation. In mice, the CXCL10-binding chemokine receptor CXCR3 was shown to play a crucial role in the recruitment of autoaggressive T cells to pancreatic islets [24,25]. CXCL10 is produced by β cells [24] and increasingly detectable in serum of newly diagnosed or prediabetic subjects [26]. Inhibition of CXCL10 homing to islets prevents autoimmune diabetes in experimental models [25,27]. CXCL10 production by islets of type 2 diabetes patients has been described and claimed to impair β cell function [28]. Our observed CXCL10 and CXCR3 expression in pancreatic islets of a new-onset type 1 diabetes patient with enterovirus infection in β cells is strikingly similar to a very recent report on fulminant diabetes and enterovirus infection [29]. A role for CXCL10 and CXCR3 was proposed in which enterovirus infection of the pancreas initiated co-expression of CXCL10 in β cells, attracting autoreactive T cells and macrophages to the islets via CXCR3. We described the expression of this particular chemokine receptor on human autoreactive T cell clones obtained from peripheral blood samples of (pre)diabetic individuals and demonstrated their capacity to home to pancreatic tissue of NOD/SCID mice after adoptive transfer [9]. In addition, recruited T cells were found to express CXCR3 in situ, suggesting that peripheral blood T cells display the proper homing receptors, which is an important check-point for participation in the process of insulitis and also perhaps in β cell destruction. Indeed, a type 1 diabetes patient-derived autoreactive CD8 T cell clone against preproinsulin, which was shown to kill human pancreatic β cells, selectively expressed CXCR3 [30]. The CXCL10–CXCR3 pathway facilitating leucocyte migration to pancreatic islets is active in all donors, but not in non-diabetic controls. This may provide the basis for the development of a novel therapeutic target in type 1 diabetes [24,25,27].
Our report underscores the value of extensive studies on human insulitis [31–34]. Indeed, the Juvenile Diabetes Research Foundation has launched an initiative to collect pancreatic tissue from diabetic donors to facilitate and drive such studies that are likely to bridge the gap in knowledge on immune as well as environmental factors contributing to β cell destruction in human type 1 diabetes (http://www.jdrfnpod.org/).
Acknowledgments
These studies were supported by the Juvenile Diabetes Research Foundation, the Dutch Diabetes Research Foundation, the Italian Ministries of Health, University and Research and the Italian Diabetes Society Research Foundation (FORISID). Printing of the colour graphs was supported by a donation from the Lugtenburg family.
Disclosure
The authors declare no conflict of interest.
References
- 1.Roep BO. The role of T-cells in the pathogenesis of Type 1 diabetes: from cause to cure. Diabetologia. 2003;46:305–21. doi: 10.1007/s00125-003-1089-5. [DOI] [PubMed] [Google Scholar]
- 2.Martin S, Wolf-Eichbaum D, Duinkerken G, et al. Development of type 1 diabetes despite severe hereditary B-lymphocyte deficiency. N Engl J Med. 2001;345:1036–40. doi: 10.1056/NEJMoa010465. [DOI] [PubMed] [Google Scholar]
- 3.Roep BO, Kallan AA, Duinkerken G, et al. T-cell reactivity to beta-cell membrane antigens associated with beta-cell destruction in IDDM. Diabetes. 1995;44:278–83. doi: 10.2337/diab.44.3.278. [DOI] [PubMed] [Google Scholar]
- 4.Lindley S, Dayan CM, Bishop A, Roep BO, Peakman M, Tree TI. Defective suppressor function in CD4(+)CD25(+) T-cells from patients with type 1 diabetes. Diabetes. 2005;54:92–9. doi: 10.2337/diabetes.54.1.92. [DOI] [PubMed] [Google Scholar]
- 5.Naik RG, Beckers C, Wentwoord R, et al. Precursor frequencies of T-cells reactive to insulin in recent onset type 1 diabetes mellitus. J Autoimmun. 2004;23:55–61. doi: 10.1016/j.jaut.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 6.Arif S, Tree TI, Astill TP, et al. Autoreactive T cell responses show proinflammatory polarization in diabetes but a regulatory phenotype in health. J Clin Invest. 2004;113:451–63. doi: 10.1172/JCI19585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tree TI, Duinkerken G, Willemen S, de Vries RR, Roep BO. HLA-DQ-regulated T-cell responses to islet cell autoantigens insulin and GAD65. Diabetes. 2004;53:1692–9. doi: 10.2337/diabetes.53.7.1692. [DOI] [PubMed] [Google Scholar]
- 8.Reijonen H, Novak EJ, Kochik S, et al. Detection of GAD65-specific T-cells by major histocompatibility complex class II tetramers in type 1 diabetic patients and at-risk subjects. Diabetes. 2002;51:1375–82. doi: 10.2337/diabetes.51.5.1375. [DOI] [PubMed] [Google Scholar]
- 9.van Halteren AG, Kardol MJ, Mulder A, Roep BO. Homing of human autoreactive T cells into pancreatic tissue of NOD-scid mice. Diabetologia. 2005;48:75–82. doi: 10.1007/s00125-004-1613-2. [DOI] [PubMed] [Google Scholar]
- 10.Kent SC, Chen Y, Bregoli L, et al. Expanded T cells from pancreatic lymph nodes of type 1 diabetic subjects recognize an insulin epitope. Nature. 2005;435:224–8. doi: 10.1038/nature03625. [DOI] [PubMed] [Google Scholar]
- 11.Wegmann DR, Norbury-Glaser M, Daniel D. Insulin-specific T cells are a predominant component of islet infiltrates in pre-diabetic NOD mice. Eur J Immunol. 1994;24:1853–7. doi: 10.1002/eji.1830240820. [DOI] [PubMed] [Google Scholar]
- 12.Thebault-Baumont K, Dubois-Laforgue D, Krief P, et al. Acceleration of type 1 diabetes mellitus in proinsulin 2-deficient NOD mice. J Clin Invest. 2003;111:851–7. doi: 10.1172/JCI16584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakayama M, Abiru N, Moriyama H, et al. Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice. Nature. 2005;435:220–3. doi: 10.1038/nature03523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moriyama H, Abiru N, Paronen J, et al. Evidence for a primary islet autoantigen (preproinsulin 1) for insulitis and diabetes in the nonobese diabetic mouse. Proc Natl Acad Sci USA. 2003;100:10376–81. doi: 10.1073/pnas.1834450100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schloot NC, Roep BO, Wegmann D, et al. Altered immune response to insulin in newly diagnosed compared to insulin-treated diabetic patients and healthy control subjects. Diabetologia. 1997;40:564–72. doi: 10.1007/s001250050716. [DOI] [PubMed] [Google Scholar]
- 16.Roep BO. T-cell responses to autoantigens in IDDM. The search for the Holy Grail. Diabetes. 1996;45:1147–56. doi: 10.2337/diab.45.9.1147. [DOI] [PubMed] [Google Scholar]
- 17.Dotta F, Censini S, van Halteren AG, et al. Coxsackie B4 virus infection of beta cells and natural killer cell insulitis in recent-onset type 1 diabetic patients. Proc Natl Acad Sci USA. 2007;104:5115–20. doi: 10.1073/pnas.0700442104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roep BO, Kallan AA, Duinkerken G, et al. T-cell reactivity to beta-cell membrane antigens associated with beta-cell destruction in IDDM. Diabetes. 1995;44:278–83. doi: 10.2337/diab.44.3.278. [DOI] [PubMed] [Google Scholar]
- 19.Wegmann DR. The immune response to islets in experimental diabetes and insulin-dependent diabetes mellitus. Curr Opin Immunol. 1996;8:860–4. doi: 10.1016/s0952-7915(96)80016-1. [DOI] [PubMed] [Google Scholar]
- 20.Koeleman BP, Lie BA, Undlien DE, et al. Genotype effects and epistasis in type 1 diabetes and HLA-DQ trans dimer associations with disease. Genes Immun. 2004;5:381–8. doi: 10.1038/sj.gene.6364106. [DOI] [PubMed] [Google Scholar]
- 21.Bingley PJ, Bonifacio E, Ziegler AG, Schatz DA, Atkinson MA, Eisenbarth GS. Proposed guidelines on screening for risk of type 1 diabetes. Diabetes Care. 2001;24:398. doi: 10.2337/diacare.24.2.398. [DOI] [PubMed] [Google Scholar]
- 22.Zimmet PZ, Tuomi T, Mackay IR, et al. Latent autoimmune diabetes mellitus in adults (LADA): the role of antibodies to glutamic acid decarboxylase in diagnosis and prediction of insulin dependency. Diabet Med. 1994;11:299–303. doi: 10.1111/j.1464-5491.1994.tb00275.x. [DOI] [PubMed] [Google Scholar]
- 23.Gale EA. Latent autoimmune diabetes in adults: a guide for the perplexed. Diabetologia. 2005;48:2195–9. doi: 10.1007/s00125-005-1954-5. [DOI] [PubMed] [Google Scholar]
- 24.Frigerio S, Junt T, Lu B, et al. Beta cells are responsible for CXCR3-mediated T-cell infiltration in insulitis. Nat Med. 2002;8:1414–20. doi: 10.1038/nm1202-792. [DOI] [PubMed] [Google Scholar]
- 25.Rhode A, Pauza ME, Barral AM, et al. Islet-specific expression of CXCL10 causes spontaneous islet infiltration and accelerates diabetes development. J Immunol. 2005;175:3516–24. doi: 10.4049/jimmunol.175.6.3516. [DOI] [PubMed] [Google Scholar]
- 26.Nicoletti F, Conget I, Di Mauro M, et al. Serum concentrations of the interferon-gamma-inducible chemokine IP-10/CXCL10 are augmented in both newly diagnosed Type I diabetes mellitus patients and subjects at risk of developing the disease. Diabetologia. 2002;45:1107–10. doi: 10.1007/s00125-002-0879-5. [DOI] [PubMed] [Google Scholar]
- 27.Baker MS, Chen X, Rotramel AR, et al. Genetic deletion of chemokine receptor CXCR3 or antibody blockade of its ligand IP-10 modulates posttransplantation graft-site lymphocytic infiltrates and prolongs functional graft survival in pancreatic islet allograft recipients. Surgery. 2003;134:126–33. doi: 10.1067/msy.2003.213. [DOI] [PubMed] [Google Scholar]
- 28.Schulthess FT, Paroni F, Sauter NS, et al. CXCL10 impairs beta cell function and viability in diabetes through TLR4 signaling. Cell Metab. 2009;9:125–39. doi: 10.1016/j.cmet.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 29.Tanaka S, Nishida Y, Aida K, et al. Enterovirus infection, CXC chemokine ligand 10 (CXCL10), and CXCR3 circuit: a mechanism of accelerated beta-cell failure in fulminant type 1 diabetes. Diabetes. 2009;58:2285–91. doi: 10.2337/db09-0091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Skowera A, Ellis RJ, Varela-Calvino R, et al. CTLs are targeted to kill beta cells in patients with type 1 diabetes through recognition of a glucose-regulated preproinsulin epitope. J Clin Invest. 2008;118:3390–402. doi: 10.1172/JCI35449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Analysis of islet inflammation in human type 1 diabetes. Clin Exp Immunol. 2009;155:173–81. doi: 10.1111/j.1365-2249.2008.03860.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spencer J, Peakman M. Post-mortem analysis of islet pathology in type 1 diabetes illuminates the life and death of the beta cell. Clin Exp Immunol. 2009;155:125–7. doi: 10.1111/j.1365-2249.2008.03864.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Planas R, Carrillo J, Sanchez A, et al. Gene expression profiles for the human pancreas and purified islets in Type 1 diabetes: new findings at clinical onset and in long-standing diabetes. Clin Exp Immunol. 2009 doi: 10.1111/j.1365-2249.2009.04053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Velthuis JH, Unger WWJ, Van der Slik AR, et al. Accumulation of auto-reactive effector T-cells and allo-specific regulatory T-cells in the pancreas allograft of a type 1 diabetic recipient. Diabetologia. 2009;52:494–503. doi: 10.1007/s00125-008-1237-z. [DOI] [PubMed] [Google Scholar]