

Abstract

The cross-coupling reaction of aryl bromides and iodides with aliphatic and aromatic thiols catalyzed by palladium complexes of the bisphosphine ligand CyPF-tBu (1) is reported. Reactions occur in excellent yields, broad scope, high tolerance of functional groups and with turnover numbers that exceed those of previous catalysts by two or three orders of magnitude. These couplings of bromo- and iodoarenes are more efficient than the corresponding reactions of chloroarenes and could be conducted with less catalyst loading and/or milder reaction conditions. Consequently, limitations regarding scope and functional group tolerance previously reported in the coupling of aryl chlorides are now overcome.
1. Introduction
Palladium-catalyzed cross-coupling reactions are fundamental organometallic transformations and have become a principal method of forming carbon-carbon and carbon-heteroatom bonds.1-3 Among this class of reactions, processes that form aromatic amines and ethers from aryl halides or pseudo-halides have been extensively studied and developed into practical methodology during the last decade.4-12 The analogous reactions forming carbon-sulfur bonds have received less attention,13 even though aryl thioethers are valuable synthetic intermediates frequently found in biologically and pharmaceutically active molecules. Indeed, a number of aromatic thioethers have shown potential clinical applications.14-18
Migita and co-workers first reported the coupling of iodo and bromoarenes with thiols catalyzed by Pd(PPh3)4.19,20 More recently, these reactions have been conducted with catalyst systems containing bidentate phosphines or dialkylphosphine oxide ligands.21-28 However, even recent catalysts react with short lifetimes and are, therefore, limited in their ability to form biologically active thioethers or precursors of biologically active compounds in a practical fashion. Nickel-, copper-, and more recently, cobalt- and iron-catalyzed coupling of thiols with aryl halides has also been reported.29-32 However, these processes require high temperatures or high catalyst loadings and have typically been conducted with aryl iodides.33-36
The limitations of this coupling reaction could result from a sensitivity of late metal catalysts to substrates containing reactive sulfur functionality. Although palladium thiolates are easily formed and undergo relatively fast reductive eliminations with aryl groups,37-39 the lifetime and concentrations of the catalysts used for the coupling of aryl halides with thiols is likely to be limited by the formation of species that lie off of the catalytic cycle.38 Thus, a more reactive catalyst for the coupling of thiolates might contain a bisphosphine that binds the metal strongly enough to prevent formation of anionic or bridging thiolate complexes, while simultaneously promoting oxidative addition and reductive elimination.
Based on this hypothesis, we considered that the particularly restricted backbone conformation, severe steric hindrance, and strong electron donation of the commercially available Josiphos ligand CyPF-tBu (1-dicyclohexylphosphino-2-di-tert-butylphosphinoethylferrocene, 1 in Figure 1) could create practical catalysts for the coupling of thiols with aryl halides with high turnover numbers. We have recently shown that palladium complexes generated from this hindered bisphosphine ligand overcome some of the current limitations on catalyst activity and scope for the amination of aryl halides and, therefore, could be considered a fourth-generation catalyst for forming carbon-heteroatom bonds.40 In particular, this system efficiently catalyzed the coupling of aryl and heteroaryl halides and pseudo-halides with nitrogen nucleophiles including primary amines,41-44 ammonia and lithium amide.45,46
Figure 1.
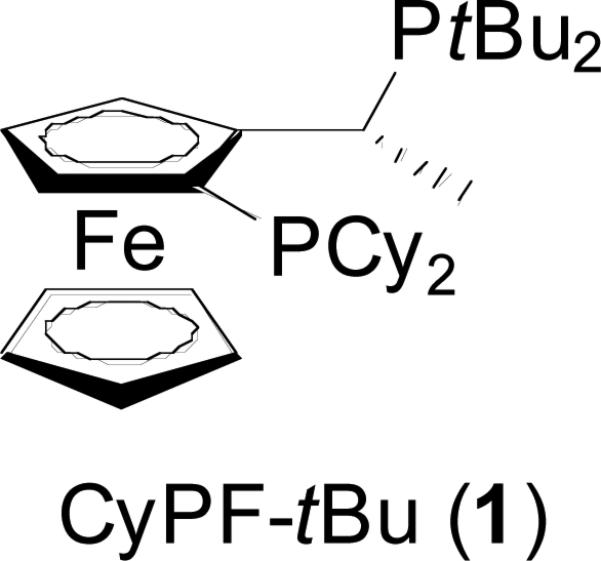
Josiphos CyPF-tBu ligand
We previously communicated47 that the combination of a palladium precursor and CyPF-tBu catalyzes the coupling of a wide range of thiols with aryl halides and pseudo-halides with turnover numbers typically two or three orders of magnitude higher than those of previous catalysts. The reactions of chloroarenes were then studied in more depth,48 and these reactions occurred with broad scope and high tolerance for functionality. However, reactions of aromatic thiols with hindered aryl chlorides and chloroarenes containing carboxaldehyde functionality or enolizable hydrogens proceeded to partial conversion and/or formed significant amounts of side products. Herein, we report a detailed study of the couplings of aryl bromides and iodides using this catalyst system. The reactions of aryl bromides and iodides occur with lower catalyst loadings and/or milder conditions than the reactions of aryl chlorides and overcome the limitations previously reported for the reactions of chloroarenes. Moreover, reactions of aryl iodides with thiols, unlike reactions of aryl iodides with amines, occur with high turnover numbers and broad scope under mild conditions.
2. Results and Discussion
2.1 Establishment of reaction conditions
We selected the coupling of 4-tolyl bromide and iodide to assess the catalyst activity and determine the optimum reaction conditions. We previously showed that the combination of Pd(OAc)2 and CyPF-tBu as catalyst NaOtBu as base, DME as solvent at 110 °C led to the coupling of aliphatic thiols with chloroarenes,47,48 but produced undesired symmetrical sulfides in a 5–10% combined yield for reaction of aromatic thiols with chloroarenes. Therefore, couplings of aryl chlorides with aromatic thiols were be conducted with toluene solvent, KOtBu as base, and Pd(dba)2 as precursor to obtain the desired diaryl sulfide in high yield without formation of appreciable amounts of side products. In the current studies we showed that the conditions for reactions of chlorines with aliphatic thiols were suitable for the coupling reactions of bromo- and iodoarenes with both aliphatic and aromatic thiols. Thus, the reaction of 4-bromotoluene and 4-iodotoluene with 1-octanethiol and thiophenol led to the corresponding aryl sulfides in excellent yields and short reaction times (< 2 h) in the presence of just 0.5 mol % of catalyst (Scheme 1).
Scheme 1.

Coupling reactions of bromo- and iodoarenes with aliphatic and aromatic thiols using CyPF-tBu ligand.
2.2. Comparison of the reactivity of aryl chlorides, bromides and iodides in the Pd-catalyzed thioetherification reaction
To compare the reactivity of the aryl halides containing different halogens, we studied the coupling of 4-methyl substituted haloarenes with 1-octanethiol as a model system. As typically observed for palladium-catalyzed cross couplings, aryl bromides were more reactive than the corresponding aryl chlorides. However, like many C–C coupling processes and unlike most C–N coupling processes,40,42,49-52 reactions of aryl iodides occurred faster than those of aryl chlorides. As shown in Scheme 2, in the presence of 0.05 mol % of catalyst, full conversion of the aryl bromide and iodide was observed in 30 minutes or less, while full conversion of the corresponding chloride required 5 h. Moreover, using five times less catalyst, the reactions of 4-iodotoluene and 4-bromotoluene occurred to full conversion and high yield in 30 minutes and 2 h, while less than 50% conversion of the corresponding chlorides occurred in 5 h. The order of reactivity for the thioetherification reaction catalyzed by the complex derived from Pd(OAc)2 and CyPF-tBu ligand is then ArI > ArBr > ArCl. This order contrasts with the order of reactivity for the amination of aryl halides, which is typically ArBr > ArCl > ArI.40 We recently reported that the amination of aryl iodides with octylamine required up to 10 times more of the CyPF-tBu-Pd catalyst than reaction of the corresponding aryl bromides and chlorides. Competition studies revealed an inhibitory effect of the alkali metal iodide generated as byproduct, rather than a reversal of the typical rate of oxidative addition of haloarenes.42 Apparently this inhibiting effect of the iodide is not operative in the coupling of aryl iodides with thiols.
Scheme 2.

Comparison of the rates of the coupling of haloarenes with 1-octanethiol
To gain additional information on the order of reactivity of the different aryl halides, a set of competition experiments were conducted (Scheme 3). In the first experiment, equimolecular amounts of 4-iodotoluene and 4-bromotoluene were allowed to react with 1-octanethiol (1 equiv) in the presence of 0.1 mol % of the catalyst system (eq 1 of Scheme 3). After 1 h of heating at 110 °C more than 95% of the iodide was consumed, while less than 5% conversion of the bromide was consumed. Similar results were obtained from the related coupling of thiophenol. Analogous experiments with 4-bromotoluene and 4-chlorotoluene confirmed the expected high selectivity for reaction of a bromoarene over a chloroarene (eq 2 of Scheme 3). Consistent with these data, the reactions of 1-bromo-4-iodobencene with 1-octanethiol and thiophenol at 110 °C for 1 h occurred to high conversion and yield to afford 4-bromophenyl octyl sulfide and 4-bromophenyl phenyl sulfide (eq 3 of Scheme 3). The corresponding 4-iodophenyl octyl and phenyl sulfides were not detected by GC/MS. A related experiment on the coupling of 1-bromo-4-chlorotoluene with 1-octanethiol and thiophenol showed the formation of 4-chlorophenyl octyl sulfide and 4-chlorophenyl phenyl sulfide as the exclusive products (eq 4 of Scheme 3). Thus, this catalyst system is fully selective for thioetherification aryl iodides over aryl bromides and for thioetherification of aryl bromides over aryl chlorides.
Scheme 3.

Comparison of the reactivity of iodoarenes vs bromoarenes and bromoarenes vs chloroarenes in the thioetherification of aliphatic and aromatic thiols in the presence of Pd(OAc)2 and CyPF-tBu (1:1)
2.3. Comparison of the catalyst efficiency for haloarenes with different halides
As described in the above analysis, the coupling of aryl bromides and iodides is even faster than the analogous coupling of chlorides and, as a result, the amount of catalyst could be reduced. Consequently, reactions of p-bromo- and p-iodotoluene with octanethiol and benzenethiol in the presence of only 10 to 100 ppm of catalyst afforded high yields of sulfide product with turnover numbers at least one order of magnitude greater than those for reactions of aryl chlorides (Table 1, entries 1, 4 vs 2–3 and 5–6). Thus, couplings of p-bromotoluene with 1-octanethiol and thiophenol occurred with turnover numbers of 99,000 and 9,800, whereas reactions of the related iodoarenes occurred with turnover numbers of 84,000 and 82,000. These values are two or three orders of magnitude higher than those for the analogous couplings of thiols with unactivated bromo- or iodoarenes catalyzed by palladium complexes derived from DiPPF or DPEphos ligands (< 50 turnovers).24,27 Reactions under the standard conditions, but without catalyst, formed mostly disulfides, and less than 5% of the aryl sulfide was formed.
Table 1.
Relative reactivity of chloroarenes, bromoarenes and iodoarenes in the palladium catalyzed coupling of thiols with bromoarenes and iodoarenes vs. chloroarenes with CyPF-tBu as ligand.a
 | ||||
|---|---|---|---|---|
| Entry | X | R | Cat. [mol %] | Yield [%] |
| 1 | Cl | octyl | 0.05 | 97 |
| 2 | Br | octyl | 0.001 | 99 |
| 3 | I | octyl | 0.001 | 84b |
| 4c | Cl | Ph | 0.1 | 95 |
| 5 | Br | Ph | 0.01 | 98 |
| 6 | I | Ph | 0.001 | 82b |
All experiments were conducted with a 1:1 ratio of metal to ligand, 1 mmol of both ArX and thiol, and 1.1 equiv of NaOtBu in DME (1.5 mL), requiring 12–24 h to complete.
~90% conversion.
Reaction performed in toluene using Pd(dba)2 and 1.1 equiv of KOtBu as base.
2.4 Coupling of aryl bromides and iodides under milder conditions
The extraordinary efficiency of the catalyst system toward C–S bond forming coupling reactions between thiols and aryl bromides and iodides prompted us to explore milder reaction conditions that could potentially expand the methodology to thermally unstable compounds or to substrates containing functional groups incompatible with a nucleophilic alkoxide base.
2.4.1 Influence of temperature on the thioetherification of aryl bromide and iodides
Studies of the coupling process to fine-tune the catalyst loading and temperature are summarized in Table 2. These studies revealed that reactions of bromo- and iodoarenes with alkyl thiols occurred at 90 °C or 70 °C in the presence of only 100 to 500 ppm of catalyst (Table 2, entries 1–3). These loadings are equal or up to one order of magnitude lower than those in the related coupling of choloroarenes conducted at 110 °C. Parallel studies on the coupling of thiophenol with bromo- and iodoarenes showed that reactions conducted with catalyst loadings equivalent to those for coupling of aliphatic thiols required higher temperatures of 110 °C for aryl bromides and 90 °C for aryl iodides (Table 2, entries 4–5).
Table 2.
Influence of temperature on the coupling of thiols with aryl halides catalyzed by Pd(OAc)2 and CyPF-tBu.a
 | ||||||
|---|---|---|---|---|---|---|
| Entry | R1 | X | R | Cat. [mol %] | T [°C] | Yield [%] |
| 1 | Me | Br | octyl | 0.05 | 90 | 96 |
| 2 | Me | I | octyl | 0.01 | 90 | 97 |
| 3 | Me | I | octyl | 0.05 | 70 | 96 |
| 4 | Me | Br | Ph | 0.01 | 110 | 98 |
| 5 | Me | I | Ph | 0.01 | 90 | 97 |
| 6 | Me | Br | octyl | 1.0 | 50 | 97 |
| 7 | H | Br | sec-butyl | 1.0 | 50 | 91 |
| 8 | Me | Br | cyclohexyl | 1.0 | 50 | 93 |
| 9 | Me | Br | Ph | 1.0 | 50 | 91 |
| 10 | Me | Cl | octyl | 2.0 | 70 | 97 |
| 11b | Me | Cl | Ph | 3.0 | 70 | 83c |
| 12 | H | I | sec-butyl | 0.5 | 25 | 93 |
| 13 | Me | I | cyclohexyl | 0.5 | 25 | 99 |
| 14 | Me | I | Ph | 0.5 | 25 | 97 |
All experiments were conducted with a 1:1 ratio of metal to ligand, 1 mmol of both ArX and thiol, and 1.1 equiv of NaOtBu in DME (1.5 mL), requiring 2–48 h of heating at the appropriate temperature to complete.
Reaction performed in toluene using Pd(dba)2 and 1.1 equiv of KOtBu as base.
~90% conversion
Reactions of aryl bromides and iodides with both aliphatic and aromatic thiols in the presence of higher catalysts loadings occurred at lower temperatures. Thus, reactions of bromoarenes with various aliphatic thiols and thiophenol occurred in high yield at 50 °C using 1 mol % catalyst, whereas related reactions of chloroarenes required higher temperatures and catalyst loadings (Table 2, entries 6–9 vs 10–11). The corresponding couplings of iodoarenes even occurred at room temperature with just 0.5 mol % catalyst (Table 2, entries 12–14).
2.4.2 Influence of base on the thioetherification of aryl bromides and iodides
The same model systems were used to evaluate the influence of base on the catalytic process. The experiments were conducted in DME at 110 °C or 90 °C using equimolecular amounts of Pd(OAc)2 and ligand (0.01 to 0.1 mol %) and 1.1 equivalents of base (Table 3). As in the related coupling of chloroarenes, NaOtBu was found to be the most effective base for the coupling of thiols with both aryl bromides and iodides. High conversions and yields were obtained after 12–24 h of heating at 110 °C in the presence of only 10 to 100 ppm of catalyst system (see Table 1, entries 2, 3, 5 and 6). Alternatively, full conversion in shorter reaction times (< 2 h) could be achieved by increasing the amount of catalyst to 0.01 mol % (Table 3 entries 1, 8, 12, and 16).
Table 3.
Influence of base on the coupling of thiols with bromoarenes and iodoarenes catalyzed by Pd(OAc)2 and CyPF-tBu.a
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | X | R | Base | Cat. [mol %] | T [°C] | t [h] | Conversion [%] | Yield [%] |
| 1 | Br | octyl | NaOtBu | 0.01 | 110 | 2 | 100 | 97 |
| 2 | Br | LiHMDS | 0.05 | 110 | 2 | 100 | 95 | |
| 3 | Br | NaHMDS | 0.05 | 110 | 2 | 100 | 95 | |
| 4 | Br | Cs2CO3 | 0.05 | 110 | 5 | 100 | 96 | |
| 5 | Br | Na2CO3 | 0.05 | 110 | 14 | 70 | - | |
| 6 | Br | K2CO3 | 0.05 | 110 | 14 | 86 | - | |
| 7 | Br | K3PO4 | 0.05 | 110 | 14 | 73 | - | |
| 8 | I | NaOtBu | 0.01 | 90 | 2 | 100 | 97 | |
| 9 | I | LiHMDS | 0.01 | 90 | 18 | 96 | 93 | |
| 10 | I | Cs2CO3 | 0.01 | 90 | 18 | 76 | - | |
| 11 | I | Cs2CO3 | 0.05 | 90 | 14 | 100 | 99 | |
| 12 | Br | Ph | NaOtBu | 0.01 | 110 | 2 | 100 | 98 |
| 13 | Br | LiHMDS | 0.01 | 110 | 8 | 96 | 93 | |
| 14 | Br | LiHMDS | 0.05 | 110 | 2 | 100 | 97 | |
| 15 | Br | Cs2CO3 | 0.05 | 110 | 14 | lowb | - | |
| 16 | I | NaOtBu | 0.01 | 90 | 2 | 100 | 95 | |
| 17 | I | NaOtBu | 0.1 | 70 | 8 | 100 | 93 | |
| 18 | I | LiHMDS | 0.1 | 90 | 2 | 100 | 94 | |
| 19 | I | Cs2CO3 | 0.05 | 110 | 14 | lowb | - | |
All experiments were conducted with a 1:1 ratio of metal to ligand, 1 mmol of both ArX and thiol, and 1.1 equiv of base in DME (1.5 mL) at the appropriate temperature.
Disulfide formation was observed.
On the other hand, and in contrast with aryl chlorides, reactions occurred to high conversions with low loadings of catalyst with bases other than alkoxides. Thus, the coupling of bromoarenes and aliphatic thiols in the presence of LiHMDS, NaHMDS and even Cs2CO3 proceeded to full conversion and high yields in short reaction times with 0.05 mol % catalyst (Table 3, entries 2–4). Lower temperatures or lower loadings of catalyst led to moderate conversions (not included in Table 3). Reactions with other carbonates and phosphates occurred with moderate to good conversions (Table 3, entries 5–7).
Similar results were observed for the coupling of iodoarenes with LiHMDS and Cs2CO3 as base (Table 3, entries 9–11). Reactions of aryl iodides could be conducted at lower temperatures (70–90 °C) with the same or lower amounts of catalyst as the reactions of aryl bromides. Coupling of aromatic thiols with both aryl bromides and iodides occurred in high yields with LiHMDS as base (Table 3, entries 13–14 and 18). Remarkably, no symmetrical thioethers were detected in these reactions of aryl bromides and iodides, whereas the coupling of thiophenol with 4-anisyl chloride in the presence of this base produced 44% of such byproducts.48
In contrast to the coupling reactions of aliphatic thiols, the coupling reactions of thiophenol with both bromo- and iodoarenes in the presence of Cs2CO3 formed large amounts of diphenyldisulfide and only traces of the desired aryl sulfide (Table 3, entries 15 and 19). Nevertheless, reactions of functionalized haloarenes conducted with this base and described later in this paper were valuable when a higher 0.1 mol % to 1 mol % catalyst was used.
These studies of reaction conditions showed that the coupling of aryl bromides and iodides with both aliphatic and aromatic thiols were most efficient at 110 °C with an alkoxide base. However, these studies also showed that reactions could be performed at moderate temperatures (25 to 90 °C) using loadings of catalyst in the range of 0.01 mol % to 1.0 mol % or with LiHMDS or Cs2CO3 as base in the presence of catalyst loadings between 0.01 and 0.1 mol %.
2.5. Scope of the thioetherification of aryl halides: coupling of unactivated aryl bromides and iodides
Reactions of a series of aryl bromides and iodides with aliphatic and aromatic thiols catalyzed by the combination of Pd(OAc)2 and Josiphos ligand 1 are summarized in Tables 4 and 5. A majority of the reactions were assembled in a drybox. However, identical catalyst activity was observed for reactions performed under inert atmosphere using common Schlenk techniques (Tables 4 and 5 compare entries 1 and 2). Reactions of aliphatic thiols with aryl bromides were conducted at 90 °C, and reactions of aryl iodides were conducted at 70 °C. Under these conditions, high activity of the catalyst was observed, although reactions did occur with turnover numbers one order of magnitude greater at 110 °C (compare Table 4 entries 1 and 3 vs Table 1 entries 2 and 3). Primary, secondary, and tertiary aliphatic thiols reacted to form the corresponding thioethers in excellent yields within short reaction times (typically < 6 h) using only 0.05 mol % catalyst. In general, reactions of aryl iodides conducted at 70 °C occurred with turnover numbers comparable to those obtained from reactions of aryl bromides conducted at 90 °C.
Table 4.
Coupling of bromo- and iodoarenes with aliphatic thiols catalyzed by Pd(OAc)2 and CyPF-tBu.a
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Ar | X | R | Cat. [mol %] | T [°C] | t [h] | Yield [%] |
| 1 | 4-Tol | Br | octyl | 0.05 | 90 | 2 | 96 |
| 2b | Br | octyl | 0.05 | 90 | 2 | 98 | |
| 3 | I | octyl | 0.05 | 70 | 2 | 96 | |
| 4 | Ph | Br | octyl | 0.05 | 90 | 4 | 98 |
| 5 | I | octyl | 0.05 | 70 | 4 | 98 | |
| 6 | 4-anisyl | Br | octyl | 0.05 | 90 | 3 | 98 |
| 7 | I | octyl | 0.05 | 70 | 3 | 96 | |
| 8 | Ph | Br | 2-methylbutyl | 0.05 | 90 | 5 | 90 |
| 9 | I | 2-methylbutyl | 0.05 | 70 | 6 | 96 | |
| 10 | 3-anisyl | Br | 2-methylbutyl | 0.05 | 90 | 6 | 96 |
| 11 | I | 2-methylbutyl | 0.05 | 70 | 6 | 96 | |
| 12 | Ph | Br | sec-butyl | 0.05 | 90 | 4 | 90 |
| 13 | I | sec-butyl | 0.05 | 70 | 4 | 93 | |
| 14 | 4-Tol | Br | cyclohexyl | 0.05 | 90 | 6 | 94 |
| 15 | I | cyclohexyl | 0.05 | 70 | 6 | 99 | |
| 16 | Br | tert-butyl | 0.05 | 90 | 5 | 93 | |
| 17 | 4-trifluoromethylphenyl | Br | tert-butyl | 0.05 | 90 | 3 | 79 |
| 18 | 1-naphthyl | Br | octyl | 0.05 | 90 | 8 | 96 |
| 19 | I | octyl | 0.05 | 90 | 12 | 93 | |
| 20 | 2-anisyl | Br | octyl | 0.05 | 110 | 4 | 99 |
| 21 | I | octyl | 0.05 | 90 | 12 | 96 | |
| 22 | 2-Tol | Br | octyl | 0.01 | 110 | 12 | 99 |
| 23 | I | octyl | 0.05 | 70 | 12 | 98 | |
| 24 | 2,5-dimethylphenyl | I | tert-butyl | 0.05 | 90 | 12 | 93 |
| 25 | 2,6-dimethylphenyl | Br | tert-butyl | 0.25 | 110 | 4 | 92 |
| 26 | I | tert-butyl | 0.1 | 110 | 8 | 94 | |
All experiments were conducted with a 1:1 ratio of metal to ligand, 1 mmol of both ArX and thiol, and 1.1 equiv of NaOtBu in DME (1.5 mL) at the appropriate temperature.
Reaction performed without using a drybox.
Table 5.
Coupling of bromo- and iodoarenes with aromatic thiols catalyzed by Pd(OAc)2 and CyPF-tBu.a
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Ar | X | Ar' | Cat. [mol %] | T [°C] | t [h] | Yield [%] |
| 1 | 4-Tol | Br | Ph | 0.01 | 110 | 2 | 98 |
| 2b | Br | Ph | 0.01 | 110 | 2 | 91 | |
| 3 | Br | Ph | 0.1 | 90 | 5 | 99 | |
| 4 | I | Ph | 0.01 | 90 | 2 | 97 | |
| 5 | Ph | Br | Ph | 0.01 | 110 | 2 | 97 |
| 6 | I | Ph | 0.05 | 90 | 5 | 98 | |
| 7c | Br | 4-tol | 0.1 | 110 | 5 | 93 | |
| 8c | I | 4-tol | 0.05 | 90 | 2 | 97 | |
| 9 | 4-anisyl | Br | Ph | 0.01 | 110 | 2 | 97 |
| 10 | I | Ph | 0.05 | 90 | 5 | 94 | |
| 11 | Br | 4-anisyl | 0.01 | 110 | 2 | 98 | |
| 12 | I | 4-anisyl | 0.05 | 90 | 5 | 98 | |
| 13 | 4-Tol | Br | 4-anisyl | 0.01 | 110 | 2 | 94 |
| 14 | I | 4-anisyl | 0.05 | 90 | 5 | 94 | |
| 15 | 3-anisyl | Br | 4-anisyl | 0.01 | 110 | 4 | 94 |
| 16 | I | 4-anisyl | 0.05 | 90 | 5 | 93 | |
| 17 | 4-trifluoromethylphenyl | Br | Ph | 0.1 | 110 | 2 | 93 |
| 18 | 1-naphthyl | Br | Ph | 0.05 | 110 | 2 | 92 |
| 19 | I | Ph | 0.05 | 110 | 2 | 87 | |
| 20 | 2-anisyl | Br | Ph | 0.1 | 110 | 2 | 99 |
| 21 | I | Ph | 0.05 | 110 | 2 | 99 | |
| 22 | 4-Tol | Br | 2-iso-propylphenyl | 0.1 | 110 | 2 | 86d |
| 23 | Br | 2-iso-propylphenyl | 0.5 | 90 | 12 | 95 | |
| 24 | I | 2-iso-propylphenyl | 0.05 | 110 | 5 | 91 | |
| 25 | 2-Tol | Br | 2-iso-propylphenyl | 0.1 | 110 | 2 | 88d |
| 26 | Br | 2-iso-propylphenyl | 0.5 | 90 | 12 | 97 | |
| 27 | I | Ph | 0.05 | 110 | 2 | 75e | |
| 28 | I | Ph | 0.25 | 70 | 24 | 99 | |
| 29 | 2,6-dimethylphenyl | Br | Ph | 0.5 | 110 | 2 | 93f |
| 30 | I | Ph | 0.25 | 110 | 8 | 94f | |
All experiments were conducted with a 1:1 ratio of metal to ligand, 1 mmol of both ArX and thiol, and 1.1 equiv of NaOtBu in DME (1.5 mL) at the appropriate temperature.
Reaction performed without using a drybox.
1.1 equiv of LiHMDS were used as base.
~10% of desproportionation byproducts were also observed.
~20% of desproportionation products were also observed.
< 5% of Ph2S was also detected.
Sterically demanding ortho-substituted substrates also coupled in high yield in the presence of the same catalyst loadings, although longer reaction times and/or higher temperatures were required (Table 4, entries 18–24). Even di-ortho-substituted aryl halides reacted with a hindered tertiary thiol in excellent yields in less than 8 h in the presence of 0.1–0.25 mol % catalyst (Table 4, entries 25–26). The equivalent reaction of 2,6-dimethylchlorobenzene occurred in lower yield (77%) and conversion (82%) in the presence of 3.0 mol % catalyst at extended reaction times (36 h).
Studies on the scope of the coupling of arene thiols are summarized in Table 5. Again, very high yields in short reaction times (frequently 2–5 h) were obtained using one or two orders of magnitude less catalyst loading than was needed for analogous couplings with prior catalyst systems and one order of magnitude lower than the corresponding coupling of chloroarenes with the same catalyst. Reactions of aromatic thiols with unhindered aryl bromides at 110 °C and with aryl iodides at 90 °C in the presence of 0.01–0.1 mol % catalyst occurred without formation of side products.
A slight increase in the loading and/or the temperature was necessary for the coupling of sterically demanding substrates. The reactions of 1-naphthyl and 2-anisyl derivatives with thiophenol at 110 °C in the presence of 0.05–0.1 mol % catalyst occurred in excellent yields (Table 5, entries 18–21) without formation of side products, such as the symmetrical thioethers formed in some cases with aromatic thiols and hindered chloroarenes.
The absence of such side products is likely due to the lower temperatures (70 °C–90 °C) of reactions conducted with 0.5 mol % catalyst. The reaction of p-bromotoluene and o-bromotoluene with 2-isopropylbenzenethiol in the presence of only 0.1 mol % catalyst formed the corresponding coupled products in good yield, but about 10% of symmetrical sulfides formed as side products (Table 5, entries 22 and 25). Formation of the undesired side product was avoided by conducting the reaction at 90 °C and increasing the loading to 0.5 mol % (Table 5, entries 23 and 26). Reaction of p-iodotoluene with 2-isopropylbenzenethiol occurred in excellent yield at 110 °C using only 500 ppm of catalyst and without formation of side products (Table 5, entry 24). However, the corresponding reaction of 2-iodotoluene with benzenethiol under the same reaction conditions produced about 20% of side products. By performing this coupling of 2-iodotoluene at 70 °C in the presence of 0.25 mol % catalyst (Table 5, compare entries 27 and 28) the desired thioether was formed in 99% yield. Furthermore, the reactions of 2,6-dimethylphenyl bromide and 2,6-dimethylphenyl iodide with thiophenol occurred in excellent yields in the presence of 0.25–0.5 mol % catalyst. Only small amounts of diphenylsulfide were detected (Table 5, entries 29–31). In contrast, the corresponding reactions of 2-chlorotoluene with thiophenol and with 2-isopropylbenzenthiol did not occur at temperatures below 110 °C with this catalyst. Therefore, the coupled products were formed in 70% yield with about 20% of side products. Moreover, coupling of 2,6-dimethylphenyl chloride with thiophenol did not occur, even at 110 °C and in the presence of 3.0 mol % catalyst.
2.6. Scope of the thioetherification of aryl halides: coupling of functionalized aryl bromides and iodides
As described in section 2.4.2, the coupling of aryl bromides and iodides occurred in the presence of bases other than alkoxides. To address issues of functional group compatibility, we evaluated reactions of functionalized aryl bromides and iodides catalyzed by a combination of Pd(OAc)2 and CyPF-tBu with LiHMDS or Cs2CO3 as base. Our previously reported couplings of thiols with chloroarenes using the present catalyst system and an alkoxide base occurred with a tolerance for functionality that far surpassed that of previous methodologies for C-S coupling. Although some reactions of aliphatic thiols occurred with weaker carbonate bases, reactions of aromatic thiols did not occur with these weaker bases. As a result, the coupling of arene thiols with aryl chlorides containing certain functional groups, such as an aldehyde or enolizable keto functionality, was unsuccessful.
The coupling of a wide range of aryl bromides and iodides containing potentially reactive functionality with primary, secondary, and tertiary aliphatic thiols is summarized in Table 6. Reactions of aryl bromides were conducted at 110 °C, and the corresponding couplings of aryl iodides were performed at 90 °C. Aryl halides containing functional groups, such as phenol, free aliphatic alcohol, protected and free amine, nitrile, carboxylic acid, ester, aldehyde or enolizable ketone coupled to form the corresponding aryl sulfide in good to excellent yields. The catalyst loadings for some of these reactions are extremely low, and even reactions of electron-rich and/or hindered substrates occurred with loadings at or below 0.25 mol %.
Table 6.
Coupling of functionalized aryl bromides and iodides with aliphatic thiols catalyzed by Pd(OAc)2 and CyPF-tBu.a
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Aryl halide | R | Base | Cat. [mol %] | T [°C] | Yield [%] |
| 1 |  |
tert-butyl | LiHMDS | 0.1 | 110 | 91 |
| 2 | tert-butyl | LiHMDS | 0.1 | 110 | 93b | |
| 3 | 2-methylbutyl | LiHMDS | 0.1 | 110 | 90 | |
| 4 |  |
tert-butyl | LiHMDS | 0.1 | 90 | 90 |
| 5 | 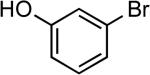 |
2-methylbutyl | LiHMDS | 0.1 | 110 | 93 |
| 6 | 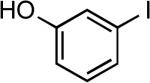 |
2-methylbutyl | LiHMDS | 0.1 | 90 | 96 |
| 7 | 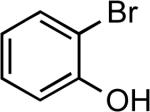 |
Octyl | LiHMDS | 0.25 | 110 | 88 |
| 8 | 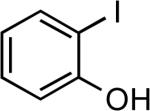 |
Octyl | LiHMDS | 0.1 | 90 | 89 |
| 9 | 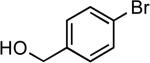 |
2-methylbutyl | LiHMDS | 0.05 | 110 | 87 |
| 10 | 2-methylbutyl | Cs2CO3 | 0.1 | 110 | 92 | |
| 11 | 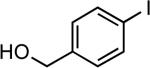 |
2-methylbutyl | LiHMDS | 0.1 | 90 | 89 |
| 12 | 2-methylbutyl | Cs2CO3 | 0.1 | 90 | 88 | |
| 13 | 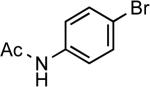 |
2-methylbutyl | LiHMDS | 0.005 | 110 | 92 |
| 14 | 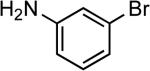 |
tert-butyl | LiHMDS | 0.05 | 110 | 92 |
| 15 | 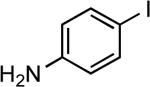 |
cyclohexyl | LiHMDS | 0.25 | 90 | 97 |
| 16 | 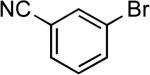 |
tert-butyl | LiHMDSc | 0.1 | 110 | 78 |
| 17 | tert-butyl | NaOtBuc | 0.05 | 90 | 91 | |
| 18 | 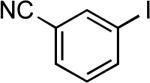 |
tert-butyl | LiHMDSc | 0.05 | 90 | 80 |
| 19 | tert-butyl | NaOtBuc | 0.05 | 70 | 89 | |
| 20 | 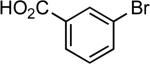 |
sec-butyl | LiHMDS | 0.05 | 110 | 73 |
| 21 | 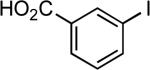 |
sec-butyl | NaOtBu | 0.01 | 90 | 97 |
| 22 | 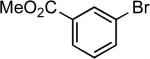 |
cyclohexyl | Cs2CO3c | 0.005 | 110 | 92 |
| 23 | 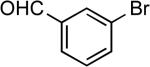 |
tert-butyl | Cs2CO3c | 0.005 | 110 | 90 |
| 24 | 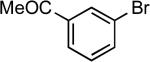 |
sec-butyl | Cs2CO3c | 0.05 | 110 | 95 |
| 25 | 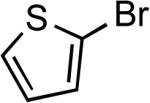 |
octyl | Cs2CO3c | 0.1 | 110 | 98 |
| 26 | 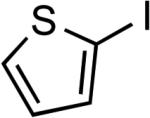 |
octyl | Cs2CO3c | 0.25 | 110 | 82 |
| 27 | 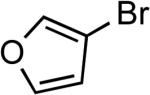 |
octyl | Cs2CO3c | 0.1 | 110 | 95 |
| 28 | 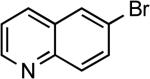 |
octyl | LiHMDSc | 0.25 | 110 | 98 |
All experiments were conducted with a 1:1 ratio of metal to ligand, 1 mmol of both ArX and thiol, and 2.4 equiv of base, unless otherwise stated, in DME (1.5 mL), requiring 2–24 h of heating at the appropriate temperature to complete.
Yield of O–TMS protected aryl sulfide.
1.1 equiv of base were employed.
Reactions of haloarenes bearing aromatic or aliphatic hydroxyl groups occurred in the presence of LiHMDS to form the corresponding sulfide in which the hydroxyl group is capped as the silyl ether. These aryl sulfides containing the silyl ether were isolated in high yield (Table 6, entry 2), or the aryl sulfide containing a free hydroxyl group was isolated after removal of the silyl group with acid prior to the chromatographic purification. The coupling in the presence of nitrile and carboxylic acid groups afforded the coupled product with yields in the range of 70–80%; however, the reactions occurred in higher yields when using an alkoxide base (Table 6, compare entries 16, 18 and 20 vs 17, 19 and 21).
Reactions of aryl bromides containing ester or aldehyde functionalities did not proceed to high conversions and yields in the presence of LiHMDS base (results not reported in the table), however, these couplings occurred in high yield in the presence of the weaker Cs2CO3 base and only 50 ppm of catalyst (Table 6, entries 22–23). In addition, reaction of an aryl bromide possessing enolizable keto functionality, using the same protocol, occurred in excellent yield with only 500 ppm catalyst. Couplings of the corresponding chloroarenes did not give the coupled product. Coupling reactions of heteroaryl bromides and iodides also occurred in good to excellent yields in the presence of LiHMDS or Cs2CO3 bases with catalyst loadings up to 0.25 mol % (Table 6, entries 25–28).
The coupling of a wide range of aryl bromides and iodides containing potentially reactive functionality with aromatic thiols is summarized in Table 7. Reactions were conducted with the combination Pd(OAc)2 and CyPF-tBu as catalyst and LiHMDS as base. A few coupling reactions were this combination failed to give high conversions or yields were performed in the presence of Cs2CO3 base. In general, reactions of aryl iodides conducted at 90 °C occurred with comparable turnover numbers to reactions of aryl bromides conducted at 110 °C. Like the reactions of aliphatic thiols, the coupling reactions conducted with these bases were tolerant of aromatic and aliphatic hydroxyl groups, protected and free amino groups, nitriles, and carboxylic acids. With one exception, these reactions occurred in good to excellent yield with 0.05 to 0.1 mol % catalyst for reactions of the electron-poor aryl halides and 0.25 to 0.5 mol % catalyst for the sterically demanding or electron-rich substrates. Again, haloarenes bearing nitrile or carboxylic acid functionalities occurred in higher yields when conducted with an alkoxide base (Table 7, entries 14–19). In addition, 1-bromo-2-fluorobenzene coupled with thiophenol in excellent yield without a competitive uncatalyzed background reaction at the C–F bond (Table 7, entry 20). Reactions of aryl bromides in the presence of weaker carbonate bases also occurred. In the presence of Cs2CO3 base, an aryl bromide possessing a formyl group reacted with thiophenol in good yield and conversion in the presence of 1 mol % catalyst (Table 7, entry 21). This catalyst system also leads to the coupling of heteroaryl bromides and iodides. Good to excellent yields of thioether products were obtained when the reactions were conducted with 0.1-0.5 mol % catalyst and LiHMDS or Cs2CO3 as base (Table 7, entries 22–24).
Table 7.
Coupling of functionalized aryl bromides and iodides with aromatic thiols catalyzed by Pd(OAc)2 and CyPF-tBu.a
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Aryl halide | Ar | Base | Cat. [mol %] | T [°C] | Yield [%] |
| 1 | 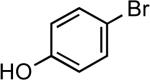 |
2-iso-propylphenyl | LiHMDS | 0.25 | 110 | 96 |
| 2 | 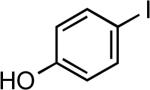 |
2-iso-propylphenyl | LiHMDS | 0.25 | 90 | 97 |
| 3 | 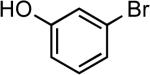 |
Ph | LiHMDS | 0.05 | 110 | 95 |
| 4 | 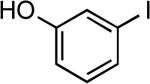 |
Ph | LiHMDS | 0.05 | 90 | 98 |
| 5 | 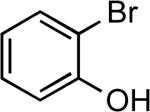 |
4-anisyl | LiHMDS | 0.25 | 110 | 90 |
| 6 | 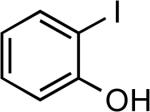 |
4-anisyl | LiHMDS | 0.25 | 110 | 98 |
| 7 | 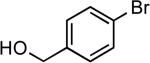 |
Ph | LiHMDS | 0.5 | 110 | 94 |
| 8 | Ph | Cs2CO3 | 0.5 | 110 | 91 | |
| 9 | 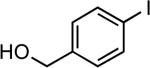 |
Ph | Cs2CO3 | 0.25 | 110 | 95 |
| 10 | 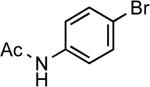 |
Ph | LiHMDS | 0.05 | 110 | 61 |
| 11 | Ph | NaOtBu | 0.05 | 110 | 76 | |
| 12 | 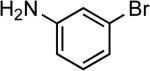 |
Ph | LiHMDS | 0.25 | 110 | 87 |
| 13 | 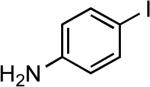 |
4-Tol | LiHMDS | 0.25 | 90 | 96 |
| 14 | 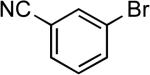 |
Ph | LiHMDSb | 0.25 | 110 | 79 |
| 15 | Ph | NaOtBub | 0.01 | 110 | 98 | |
| 16 | 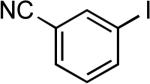 |
Ph | NaOtBub | 0.05 | 90 | 95 |
| 17 | 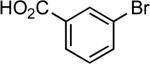 |
4-anisyl | LiHMDS | 0.05 | 110 | 67 |
| 18 | 4-anisyl | NaOtBu | 0.05 | 110 | 85 | |
| 19 | 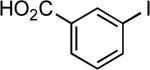 |
4-anisyl | NaOtBu | 0.05 | 110 | 77 |
| 20 | 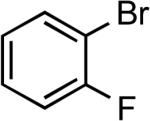 |
Ph | LiHMDSb | 0.1 | 110 | 89 |
| 21 | 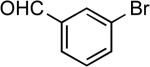 |
Ph | Cs2CO3b | 1 | 110 | 76c |
| 22 | 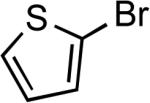 |
Ph | Cs2CO3b | 0.25 | 110 | 85 |
| 23 | 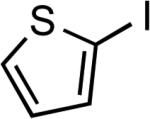 |
Ph | Cs2CO3b | 0.1 | 110 | 74 |
| 24 | 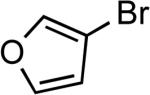 |
Ph | LiHMDSb | 0.5 | 110 | 95 |
All experiments were conducted with a 1:1 ratio of metal to ligand, 1 mmol of both ArX and thiol, and 2.4 equiv of base, unless otherwise stated, in DME (1.5 mL), requiring 2-24 h of heating at the appropriate temperature to complete.
1.1 equiv. of base were employed.
88% conversion.
Conclusions
In summary, we have shown that palladium complexes generated from the Josiphos ligand CyPF-tBu (1) are general, highly efficient catalysts for the coupling of bromo- and iodoarenes with thiols. The ability to conduct reactions with low catalyst loadings illustrates that the catalyst containing CyPF-tBu as ligand resists decomposition from unproductive reactions with the thiol or thiolate. Reactions catalyzed by the complexes generated in situ from Pd(OAc)2 and ligand 1 in most cases occur with turnover numbers that are two or three orders of magnitude higher than those of related couplings by catalysts containing other ligands. The process occurs with broad scope and includes the coupling of thiols with heteroaryl halides. The reaction tolerates fluoro, cyano, keto, free carboxylate, amido, carboalkoxy, carboxaldehyde, aromatic and aliphatic hydroxyl and amino functionalities. The relative rates for reaction of different halides followed the conventional trend of ArI > ArBr > ArCl, instead of the order ArBr > ArCl > ArI for reactions of amines with aryl halides. The superior efficiency of reactions of bromo- and iodoarenes were demonstrated by conducting the couplings with less catalyst, lower temperature or in the presence of bases other than alkoxides. Consequently, the most challenging coupling of substrates containing aldehyde or enolizable ketone functionalities and reactions with hindered aryl halides with aromatic thiols occur in excellent yields without the formation of side products.
Experimental Section
Preparation of stock solution A (1.0 × 10–2 M)
DME (1.0 mL) was added to a mixture of Pd(OAc)2 (2.2 mg) and CyPF-tBu (5.5 mg). The resulting orange solution was stirred at room temperature for 1 min before using.
Preparation of stock solution B (1.0 × 10–4 M)
10 μL of stock solution A was diluted to 1.0 mL with DME. The resulting pale yellow solution was stirred at room temperature for 1 min before using.
General procedure for the coupling of aryl bromides and iodides with aliphatic and aromatic thiols
The appropriate quantity of stock solution A or B was added to a 4 mL vial containing the aryl bromide or iodide (1.00 mmol) and NaOtBu (106 mg, 1.10 mmol) in DME (1.5 mL). The thiol (1.00 mmol) was then added, and the vial sealed with a cap containing a PTFE septum. The mixture was heated at the temperature indicated in Tables 4 and 5 until the aryl halide was consumed, as determined by GC. Silica gel (0.5 g) was added, and the solvents were evaporated under reduced pressure. The crude residue was purified by column chromatography on silica gel using hexane or a mixture of hexane and ethyl acetate as eluent. Aryl sulfides were isolated in the yields reported in Tables 4 and 5.
2-Methylbutyl 3-methoxyphenyl sulfide (Table 4, entries 10–11).47
Colorless liquid. 1H NMR (CDCl3): δ 7.07 (t, J = 7.9 Hz, 1H), 6.80-6.76 (m, 2H), 6.59-6.56 (m, 1H), 3.67 (s, 3H), 2.84 (dd, J = 12.5 Hz and 5.9 Hz, 1H), 2.64 (dd, J = 12.5 Hz and 7.6 Hz, 1H), 1.60-1.52 (m, 1H), 1.49-1.39 (m, 1H), 1.22-1.11 (m, 1H), 0.91 (d, J = 6.6 Hz, 3H), 0.81 (t, J = 7.1 Hz, 3H). 13C NMR (CDCl3): δ 159.6, 138.8, 129.4, 120.5, 113.7, 110.9, 55.0, 40.2, 34.3, 28.7, 18.8, 11.1.
General procedure for the coupling of functionalized aryl bromides and iodides with aliphatic and aromatic thiols
The appropriate quantity of stock solution A or B was added to a 4 mL vial containing the aryl bromide or iodide (1.00 mmol) and LiHMDS or Cs2CO3 (2.4 mmol) as base, unless otherwise stated, in DME (1.5 mL). The thiol (1.00 mmol) was then added, and the vial sealed with a cap containing a PTFE septum. The mixture was heated at the temperature indicated in Tables 6 and 7 until the aryl halide was consumed, as determined by GC. Silica gel (0.5 g) was added, and the solvents were evaporated under reduced pressure. The crude residue was purified by column chromatography on silica gel using hexane or a mixture of hexane and ethyl acetate as eluent. Aryl sulfides were isolated in the yields reported in Tables 6 and 7. Reactions of haloarenes bearing aromatic or aliphatic hydroxyl groups and LiHMDS as base initially form the corresponding sulfide with the hydroxyl group protected as the silyl ether. However the silyl group was removed by treatment with acid (HCl 1N) prior to the chromatographic purification.
3-Phenylsulfanylphenol (Table 7, entries 3–4)
Colorless liquid. 1H NMR (CDCl3): δ 7.33-7.28 (m, 2H), 7.24-7.14 (m, 3H), 7.05 (t, J = 7.8 Hz, 1H), 6.81-6.77 (m, 1H), 6.64-6.62 (m, 1H), 6.59-6.56 (m, 1H), 5.00 (bs, 1H). 13C NMR (CDCl3): δ 155.8, 137.7, 134.5, 131.8 (2C), 130.1, 129.2 (2C), 127.4, 122.6, 116.7, 113.8. Elem. Anal. Calcd for C12H10OS: C, 71.25; H, 4.98. Found: C, 71.01; H, 5.02.
Supplementary Material
Acknowledgment
We thank the NIH-NIGMS (GM-55382) for support of this work. M. F.-R. thanks the Ministerio de Educación y Ciencia (Spain) for a MEC/Fulbright fellowship.
Footnotes
Supporting Information Available: All experimental procedures and characterization data of new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Handbook of Organopalladium Chemistry for Organic synthesis. Vol. 1. Wiley Interscience; New York: 2002. [Google Scholar]
- 2.Cross-Coupling Reacions: A Practical Guide. Springer; Berlin: 2002. (Series Topics in Current Chemistry, 219) [Google Scholar]
- 3.Hassan J, Sévignon M, Gozzi C, Schulz E, Lemaire M. Chem. Rev. 2002;102:1359–1469. doi: 10.1021/cr000664r. [DOI] [PubMed] [Google Scholar]
- 4.Hartwig JF. Synlett. 2006:1283–1294. [Google Scholar]
- 5.Hartwig JF. In: Handbook of Organopalladium Chemistry for Organic synthesis. Negishi, editor. Vol. 1. Vol. 1. Wiley-Interscience; New York: 2002. pp. 1051–1106. [Google Scholar]
- 6.Hartwig JF. In: Modern Arene Chemistry. Austruc C, editor. Wiley-VCH; Weinheim, Germany: 2002. pp. 107–168. [Google Scholar]
- 7.Muci AR, Buchwald SL. Top. Curr. Chem. 2002;219:131–209. [Google Scholar]
- 8.Prim D, Campagne J-M, Joseph D, Andrioletti B. Tetrahedron. 2002;58:2041–2075. [Google Scholar]
- 9.Yang BH, Buchwald SL. J. Organomet. Chem. 1999;576:125–146. [Google Scholar]
- 10.Hartwig JF. Angew. Chem. Int. Ed. 1998;37:2046–2067. doi: 10.1002/(SICI)1521-3773(19980817)37:15<2046::AID-ANIE2046>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 11.Hartwig JF. Acc. Chem. Res. 1998;12:852–860. [Google Scholar]
- 12.Wolfe JP, Wagaw S, Marcoux J-F, Buchwald SL. Acc. Chem. Res. 1998;12:805–818. [Google Scholar]
- 13.Review: Kondo T, Mitsudo T.-a. Chem. Rev. 2000;100:3205–3220. doi: 10.1021/cr9902749.
- 14.De Martino G, Edler MC, La Regina G, Coluccia A, Barbera MC, Barrow D, Nicholson RI, Chiosis G, Brancale A, Hamel E, Artico M, Silvestri R. J. Med. Chem. 2006;49:947–954. doi: 10.1021/jm050809s. [DOI] [PubMed] [Google Scholar]
- 15.Alcaraz M-L, Atkinson S, Cornwall P, Foster AC, Gill DM, Humphries LA, Keegan PS, Kemp R, Merifield E, Nixon RA, Noble AJ, O'Beirne D, Patel ZM, Perkins J, Rowan P, Sadler P, Singleton JT, Tornos J, Watts AJ, Woodland IA. Org. Process Res. Dev. 2005;9:555–569. [Google Scholar]
- 16.De Martino G, La Regina G, Coluccia A, Edler MC, Barbera MC, Brancale A, Wilcox E, Hamel E, Artico M, Silvestri R. J. Med. Chem. 2004;47:6120–6123. doi: 10.1021/jm049360d. [DOI] [PubMed] [Google Scholar]
- 17.Meng CQ, Somers PK, Hoong LK, Zheng XS, Ye Z, Worsencroft KJ, Simpson JE, Hotema MR, Weingarten MD, MacDonald ML, Hill RR, Marino EM, Suen K-L, Luchoomun J, Kunsch C, Landers LK, Stefanopoulos D, Howard RB, Sundell CL, Saxena U, Wasserman MA, Sikorski JA. J. Med. Chem. 2004;47:6420–6432. doi: 10.1021/jm049685u. [DOI] [PubMed] [Google Scholar]
- 18.Liu G, Huth JR, Olejniczak ET, Mendoza R, DeVries P, Leitza S, Reilly EB, Okasinski GF, Fesik SW, von Geldern TW. J. Med. Chem. 2001;44:1202–1210. doi: 10.1021/jm000503f. [DOI] [PubMed] [Google Scholar]
- 19.Migita T, Shimizu T, Asami Y, Shiobara J, Kato Y, Kosugi M. Bull. Chem. Soc. Jpn. 1980;53:1385–1389. [Google Scholar]
- 20.Kosugi M, Shimizu T, Migita T. Chem. Lett. 1978:13–14. [Google Scholar]
- 21.Cai L, Cuevas J, Peng Y-Y, Pike VW. Tetrahedron Lett. 2006;47:4449–4452. [Google Scholar]
- 22.Mispelaere-Canivet C, Spindler J-F, Perrio S, Beslin P. Tetrahedron. 2005;64:5253–5259. [Google Scholar]
- 23.Itoh T, Mase T. Org. Lett. 2004;6:4587–4590. doi: 10.1021/ol047996t. [DOI] [PubMed] [Google Scholar]
- 24.Murata M, Buchwald SL. Tetrahedron. 2004;60:7397–7403. [Google Scholar]
- 25.Li GY, Zheng G, Noonan AF. J. Org. Chem. 2001;66:8677–8681. doi: 10.1021/jo010764c. [DOI] [PubMed] [Google Scholar]
- 26.Li GY. Angew. Chem. Int. Ed. 2001;40:1513–1516. doi: 10.1002/1521-3773(20010417)40:8<1513::aid-anie1513>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 27.Schopfer U, Schlapbach A. Tetrahedron. 2001;57:3069–3073. [Google Scholar]
- 28.Zheng N, McWilliams JC, Fleitz FJ, Armstrong JD, III, Volante RP. J. Org. Chem. 1998;63:9606–9607. [Google Scholar]
- 29.Nickel-catalyzed: Cristau HJ, Chabaud B, Chene A, Christol H. Synthesis. 1981:892–894.
- 30.For a review on cooper-catalyzed coupling, see: Ley SV, Thomas AW. Angew. Chem. Int. Ed. 2003;42:5400–5449. doi: 10.1002/anie.200300594.
- 31.Wong Y-C, Jayanth TT, Cheng C-H. Org. Lett. 2006;8:5613–5616. doi: 10.1021/ol062344l. [DOI] [PubMed] [Google Scholar]
- 32.Correa A, Carril M, Bolm C. Angew. Chem. Int. Ed. 2008;47:2880–2883. doi: 10.1002/anie.200705668. [DOI] [PubMed] [Google Scholar]
- 33.Nickel-catalyzed coupling (3-4 mol%) of bromoarenes with aromatic thiols has been recently described: Zhang Y, Ngeow KC, Ying JY. Org. Lett. 2007;9:3495–3498. doi: 10.1021/ol071248x.
- 34.For a recent report of the coupling of bromobenzene with benzenethiol catalyzed by CuI (5 mol%)/N,N-dimethyl glycine (20 mol %) see: Deng W, Zou Y, Wang Y-F, Liu L, Guo Q-X. Synlett. 2004:1254–1258.
- 35.Three examples of coupling of aryl bromides were reported using CuI (10 mol %) under microwave heating: Wu Y-J, He H. Synlett. 2003:1789–1790.
- 36.A recent report describes the coupling of bromoarenes with arylthiols in water catalyzed by CuCl (8.5 mol%) in the presence of trans-1,2-diaminocyclohexane (3.9 equiv.): Carril M, SanMartin R, Domínguez E, Tellitu I. Chem. Eur. J. 2007;13:5100–5105. doi: 10.1002/chem.200601737.
- 37.Mann G, Barañano D, Hartwig JF, Rheingold AL, Guzei IA. J. Am. Chem. Soc. 1998;120:9205–9219. [Google Scholar]
- 38.Louie J, Hartwig JF. J. Am. Chem. Soc. 1995;117:11598–11599. [Google Scholar]
- 39.Barañano D, Hartwig JF. J. Am. Chem. Soc. 1995;117:2937–2938. [Google Scholar]
- 40.Hartwig JF. Acc. Chem. Res. 2008;41:1534–1544. doi: 10.1021/ar800098p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shen Q, Shekhar S, Stambuli JP, Hartwig JF. Angew. Chem. Int. Ed. 2005;44:1371–1375. doi: 10.1002/anie.200462629. [DOI] [PubMed] [Google Scholar]
- 42.Shen Q, Ogata T, Hartwig JF. J. Am. Chem. Soc. 2008;130:6586–6596. doi: 10.1021/ja077074w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shen Q, Hartwig JF. Org. Lett. 2008;10:4109–4112. doi: 10.1021/ol801615u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ogata T, Hartwig JF. J. Am. Chem. Soc. 2008;130:13848–13849. doi: 10.1021/ja805810p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.For a Highlight on direct coupling of ammonia and lithium amide see: Willis MC. Angew. Chem. Int. Ed. 2007;46:3402–3404. doi: 10.1002/anie.200605071.
- 46.Shen Q, Hartwig JF. J. Am. Chem. Soc. 2006;128:10028–10029. doi: 10.1021/ja064005t. [DOI] [PubMed] [Google Scholar]
- 47.Fernández-Rodríguez MA, Shen Q, Hartwig JF. J. Am. Chem. Soc. 2006;128:2180–2181. doi: 10.1021/ja0580340. [DOI] [PubMed] [Google Scholar]
- 48.Fernández-Rodríguez MA, Shen Q, Hartwig JF. Chem. Eur. J. 2006;12:7782–7796. doi: 10.1002/chem.200600949. [DOI] [PubMed] [Google Scholar]
- 49.Urgaonkar S, Nagarajan M, Verkade JG. J. Org. Chem. 2003;68:452–459. doi: 10.1021/jo0205309. [DOI] [PubMed] [Google Scholar]
- 50.Ali M, Buchwald SL. J. Org. Chem. 2001;66:2560–2565. doi: 10.1021/jo0008486. [DOI] [PubMed] [Google Scholar]
- 51.Wolfe JP, Buchwald SL. J. Org. Chem. 1996;61:1133–1135. [Google Scholar]
- 52.See also references 1-12.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.