

Abstract
Anthrax receptor (ATR) shares similarities with molecules relevant to haematopoiesis. This suggests that anthrax proteins might bind to these mimicking molecules and exert non‐specific haematopoietic effects. The haematopoietic system is the site of immune cell development in the adult. As such, ATR ligand, protective antigen (PA) and the other anthrax proteins, lethal factor, edema factor, could be significant to haematopoietic responses against Bacillus anthracis infection. Because haematopoiesis is the process of immune cell development, effects by anthrax proteins could be relevant to vaccine development. Here, we report on effects of anthrax proteins and toxins on early and late haematopoiesis. Flow cytometry shows binding of PA to haematopoietic cells. This binding might be partly specific because flow cytometry and Western blots demonstrate the presence of ATR1 on haematopoietic cell subsets and the supporting stromal cells. Functional studies with long‐term initiating cell and clonogenic assays determined haematopoietic suppression by anthrax toxins and stimulation by monomeric proteins. The suppressive effects were not attributed to cell death, but partly through the induction of haematopoietic suppressors, interleukin (IL)‐10 and CCL3 (MIP‐1α). In summary, anthrax proteins affect immune cell development by effects on haematopoiesis. The type of effect, stimulation or suppression, depend on whether the stimulator is a toxin or monomeric protein. The studies show effects of anthrax proteins beginning at the early stage of haematopoiesis, and also show secondary mediators such as IL‐10 and CCL3. The roles of other cytokines and additional ATR are yet to be investigated.
Keywords: cytokines, bone marrow, anthrax proteins, haematopoiesis
Introduction
Bacillus anthracis (B. anthracis) is a virulent spore‐producing bacterium. The need to protect the civilian population was heightened by the anthrax mail attacks of 2001. The threat of bioterrorism and the potential use of biological weapons against both military and civilian populations have become a reality. Although immediate treatment could clear early anthrax infection, the subtle effects of an infection on the emerging immune system has not been examined [1]. The use of B. anthracis as a lethal biological weapon has provoked renewed research interest. Although there is a rapid movement to develop effective vaccines against B. anthracis, there is no effective vaccine [2]. The promise of an effective vaccine and host response rely on a functional emerging immune system, which encompass the early period of immune cell development up to the generation of mature immune cells. The complexity of anthrax toxins and the presence of its receptors in different cells provide challenges for vaccine development [3]. This study addresses one area of the immune system, the early developmental process, namely haematopoietic system.
B. anthracis releases three monomeric proteins, lethal factor (LF), edema factor (EF) and protective antigen (PA) [4, 5]. PA facilitates the biological activities of LF and EF by forming lethal toxin (LT) and edema toxin (ET), respectively [5, 6, 7, 8]. Once intracellular, PA is disassociated to release LT and ET where they mediate intracellular responses with pathophysiological effects, including septicaemia [9, 10].
The anthrax vaccine immunization program, which was initially developed in the 1970s, uses a PA‐based cell‐free subunit, also known as ‘anthrax vaccine absorbed’ (AVA) [11]. The AVA method requires multiple subcutaneous injections, mostly every 2 months up to 18 months, followed by annual boosts [12, 13]. The vaccine has been reported to exert local and systemic adverse reactions with contamination by ET and LT [3, 14]. The recognized need for large‐scale vaccine development and administration would not only require efficacy and safety, but also other scientific effects such as possible confound on the emerging immune system. It is possible that anthrax proteins in vaccines could have negative effects on the same cells and organs that are required to mediate protection against anthrax. The consensus for an improved vaccine has promoted interest in alternative strategies, such as live organism delivery systems, DNA vaccines and purified recombinant PA [11, 15].
Haematopoiesis is the process by which immune and other blood cells are produced from a finite number of haematopoietic stem cells (HSCs) [16]. These stem cells reside in the bone marrow (BM) and are sources of life‐long immune cell replacement. Insults to BM may affect all areas of the haematopoietic network, including effects on HSCs, their progenies and other microenvironmental cells and structures [17, 18]. Thus, haematopoietic responses by anthrax would recapitulate the immune effects [16]. In this regard, an understanding of the effects by anthrax proteins and/or toxins on haematopoiesis might provide insights on vaccine responses.
The BM is a complex organ with molecules that can bind to PA [5, 19, 20]. Because PA is required for interactions with EF and LF, the latter two molecules could interact specifically with haematopoietic cells that express anthrax receptor (ATR). At this time it is unclear if EF and LF could bind non‐specifically to BM cells. Such information is important in light of a case study reporting on BM failure following anthrax vaccine administration [8, 21]. We sought to determine the effects of anthrax monomeric proteins and toxins on haematopoiesis. This central question has been addressed with healthy BM aspirates in an in vitro culture system in the presence or absence of purified anthrax proteins. The goal is to obtain insights into effects in the event of anthrax infection and expectations following vaccinations. The premise is that an understanding of anthrax toxins on the emerging immune system may lead to the prevention of organ damage by anthrax infection, and also aid in the development of interventional strategies for safe and effective vaccines.
Methods
Reagents, antibodies, cytokines and chemokine
The following were purchased from Sigma (St. Louis, MO, USA): Polymyxin B, Ficoll‐Hypaque, phosphate buffered saline, pH 7.4 (PBS), RNase A, Propodium Iodide, protein G Sepharose, α‐minimum essential media (MEM), Iscoves, Dulbecco’s minimum essential media (DMEM) with high glucose, Roswell Park Memorial Institute (RPMI) 1640, bovine serum albumin (BSA), mouse anti‐β‐actin, non‐immune murine Immunoglobulin gamma (IgG) and non‐immune rabbit IgG. Foetal calf sera (FCS) and horse sera were purchased from HyClone Laboratories (Logan, UT, USA). The following were purchased from R&D Systems (Minneapolis, MN, USA): Recombinant human (rh) graulocyte‐macrophage‐colony stimulating factor (GM‐CSF), rhEpo, stem cell factor (SCF), interleukin (IL)‐3 and anti‐IL‐10 mAb, IL‐10 and C‐C motif ligand 3 (CCL3). Rabbit polyclonal anti‐CCL3 was generously provided by the Immunology Department of Genetics Institute (Cambridge, MA, USA).
Antibodies that were ordered from BD Biosciences (San Jose, CA, USA) follow: Purified mouse anti‐human monoclonal antibody CD38; rabbit anti‐human caspase‐3; Phycoerythrin (PE) mouse anti‐human CD34; PE‐Cy5 mouse anti‐human: CD117, CD7, CD41a, CD38, CD71; Allophycocyanin (APC) mouse anti‐human: CD38, CD79a, CD32, CD45RA, CD235a; Peridinin chlorophyll protein (PerCP) mouse anti‐human CD19; fluorescein siothiocyanate (FITC)‐mouse IgG isotype; PE mouse IgG isotype; APC mouse IgG2b isotype control; PerCP mouse IgG isotype. PE‐Cy5 rat anti‐human CD127 and PE‐Cy5 Rat IgG2a isotype was purchased from eBioscience (San Diego, CA, USA). Rabbit anti‐human ATR1 was purchased from Alpha Diagnostics, Inc. (San Antonio, TX, USA). Mouse anti‐human Von Willebrand Factor (vWF) and CD31 were purchased from DAKO (Carpinteria, CA, USA).
Anthrax toxin and endotoxin‐free system
All commercial sources of media contained low endotoxin levels. Buffers are prepared with water from a MilliQ system with a filter that eliminates endotoxin. The following reagents were obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: Anthrax Protective Antigen, Lot #9819: Anthrax Edema Factor, Lot #9820: Anthrax Lethal Factor, Lot #9821. The factors were received as lyophilized and then reconstituted in 1.0 ml of BSA at 1 mg/ml, diluted in endotoxin‐free distilled water. Upon reconstitution, aliquots of 1 μl were stored in siliconized tubes and then stored at −80°C. The total amount of endotoxin present within the highest concentration of each protein (8 ng) was based on the level of endotoxin units (EU) provided on the data sheet: 10−4 ng EU for PA; 1.3 × 10−4 ng EU for LF and 1.9 × 10−3 for EF. To ensure that the functions are not due to endotoxin, we have boiled the proteins at 1 μg/ml for 30 min. immediately before each assay. The levels of endotoxin were determined in triplicates with samples before and after boiling, using the QCL‐1000 chromogenic LAL end‐point assay kit (Lonza, Inc., Allendale, NJ, USA). The levels differed by ±0.01.
Participants
BM aspirates were taken from the iliac crests of healthy participants between ages 20 to 30 years. Aspirates were taken in syringes with preservative‐free heparin diluted in Iscove’s media. The protocol for participants adhered to guidelines outlined by the institutional review board of the University of Medicine and Dentistry of New Jersey (Newark Campus, NJ, USA).
BM stromal cell culture
BM stromal cells cultures were established with unfractionated BM aspirates as previously described [22]. Briefly, BM aspirates were placed in stromal media: α‐MEM containing 12.5% FCS, 12.5% horse serum, 10−7 M hydrocortisone, 10−4 M 2‐ME and 1.6 mM glutamine. After 3 days at 33°C, RBC and granulocytes were removed by Ficoll‐Hypaque density gradient centrifugation, and cultures were re‐incubated with weekly replacement of 50% stromal media until confluent growth. Flow cytometry shows that the stromal cells are negative for CD14 (macrophage marker), CD31 (endothelial marker), SH2 (mesenchymal stem cell marker), but positive for α‐smooth muscle actin.
BM endothelial culture
Endothelial cell cultures were established with unfractionated BM aspirates in media prepared with Endothelial Cell EGM‐2 system (Cambrex, East Rutherford, NJ, USA). Bone marrow mononuclear cells (BMNCs) were isolated by Ficoll‐Hypaque density gradient and then placed in basal media, containing components of the SingleQuot Kit, which is part of the EGM‐2 system. A purified population of BM endothelial cells was determined if flow cytometry showed >99% positive for CD31 and vWF. This generally occurs after three passages.
Conjugation of PA to FITC
Direct labelling of PA to FITC was performed with EZ‐Label FITC Protein Labeling Kit (Pierce Biotechnology, Rockford, IL, USA). PA was diluted in borate buffer and then incubated for 1 hr with FITC reagent at a ratio of 1:24. Excess fluorochrome was removed by dialysis in PBS for 1 hr using the Slide‐A‐Lyzer MINI Dialysis Unit. The conjugated PA was titrated by flow cytometry with human endothelial cells. The concentration that showed >95% positive for PA was selected as optimum.
Immunofluorescence
Whole BM aspirates were studied for FITC‐PA binding in four‐colour flow cytometry. Red blood cells were first lysed with 1× BD Lyse then followed by fixing of the cells with 2% paraformaldehyde. The combinations of fluorochrome‐conjugated antibodies and the expected outcome for specific cell subset follow: Myeloid–lymphoid lineages, CD34+/CD117+/CD38−/lo Common lymphoid progenitors, CD34+/CD7+/CD38−; Megakaryocytes, CD34+/CD41a+/CD38+; B‐cell precursor, CD34+/CD127+/intracellular CD79a+; Mature B‐cells, CD34+/CD19+/CD38+. Early myeloid progenitors, CD34+/CD32−/lo/CD38+/−; Primitive stem cells were identified with CD34+/CD45RA−; Committed myeloid or lymphoid stem cells were identified with CD34+/CD45RA+; Early erythroid progenitors, CD34+/CD235a+; Mature erythroid progenitors, CD34+/CD71+/CD45RA−; Mature granulocyte/monocyte progenitors, CD34+/CD71−/CD45RA+. None of the aforementioned antibodies was conjugated to FITC because the fourth, PA, was conjugated to FITC. Non‐specific binding was studied in parallel with isotype control IgG conjugated to the appropriate fluorochrome. Compensation tubes were set up in parallel to correct for possible overlap among the different fluorochromes.
Isolation of CD34+ cell subsets
BMNCs were isolated by Ficoll‐Hypaque density gradient and then used to positively select CD34+ cells. Selection was done with Dynal CD34 Progenitor Cell Selection System (Dynal, Inc., Lake Success, NY, USA). BMNCs were incubated with Dynabeads M‐450 CD34 for 30 min. at 4°C. Cells were released from the magnetic particles by incubating for 45 min. at room temperature with DETACHaBEAD.
The retrieved CD34+ cells were subdivided into two subsets based on the expression of CD38: CD34+/CD38+ versus CD34+/CD38−. CD38+ cells were selected with CELLection Pan Mouse IgG Kit (Dynal, Inc.). CD34+ cells were resuspended at 106/ml and then incubated with CD38 monoclonal antibody at 1 μg/ml or 10 min. at 4°C. After this, cells were washed and then incubated with CELLection Pan Mouse IgG Dybnabeads for 20 min. at 4°C. Cells were released from the beads by gentle rotation for 15 min. at room temperature with DNase I Releasing Buffer. Immunofluorescence showed >90% cell purity.
Preparation of cell extracts
Cell membrane extracts were obtained from CD34+/CD38−, CD34+/CD38+ and BM stromal cells as previously described [23]. Cells were incubated with 400 μl of 1× lysis buffer (Promega, Madison WI, USA) for 15 min. at room temperature. Cell lysates were pelleted by centrifugation at 10,000 ×g for 15 min. at 4°C. The pellets with membrane fractions were resuspended in 300 μl PBS and then vortex. The total protein concentration was determined by BioRad protein assay (Bio‐Rad, Hercules, CA, USA). Supernatants containing cytosolic fractions were similarly analysed. Whole‐cell extracts were obtained by four cycles of freezing and thawing in 400 μl 1× lysis buffer followed by centrifugation at 10,000 ×g for 15 min. at 4°C. Supernatants containing the whole cell extracts were analysed for total protein levels.
Immunoprecipitation and Western blot
ATR was immunoprecipitated from cell membrane and cytosolic extracts obtained from CD34+/CD38− and CD34+/CD38+ cells. Cell extracts were incubated with rabbit anti‐human ATR (1/1000) at 4°C overnight. After this, samples were incubated with protein G Sepharose at 4°C for 6 hrs. Reactions were centrifuged at 4°C, 10 000 ×g for 30 min. The pellets were washed once with PBS, resuspended in sample buffer, and electrophoresed in 12% SDS‐PAGE. Proteins were transferred to polyvinylidene fluoride (PVDF) membranes (Perkin Elmer, Wellesley, MA, USA), and ATR was detected by overnight incubation at 4°C with rabbit anti‐human ATR at 1/1000 dilution. After this, membranes were washed and incubated with horseradish peroxidase (HRP)‐conjugated goat anti‐rabbit IgG (1/2000) for 1 hr at 4°C. HRP was developed with chemiluminescence Western blot detection reagents (Perkin Elmer). The molecular weights were determined by comparing to Kaleidoscope pre‐stained standards (BioRad). Although the total proteins per sample in the immunoprecipitation assay were similar, we nonetheless performed Western blots in parallel for β‐actin. Caspase‐3 was not immunoprecipitated, but was detected by similar methods using rabbit anti‐human caspase‐3 at 1/1000 dilution with BMNC whole cell extracts.
Cytokine analyses
Cytokine production in BM stroma used two methods: (i) Microarray III (Ray Biotech, Norcross, GA, USA) and (ii) ELISA kits for MIP‐1α and IL‐10, purchased from R&D Systems. The array studies were analysed according to the manufacturer’s instructions, as previously described [24]. Membranes were incubated for 30 min. in 1× blocking buffer provided with the kit, followed by the addition of 1 ml sera‐free conditioned media obtained from confluent stroma. The membranes were incubated at room temperature for 1 hr then subjected to three 5‐min. washes with 1× Wash Buffer I, followed by two 5‐min. washes with 1× Wash Buffer II, also provided with the kit. The membranes were incubated for 1 hr with biotin‐conjugated antibodies and then developed for 1 hr with HRP‐conjugated streptavidin and detection reagent mix provided in the kit. ELISA for IL‐10 and CCL3 were done with the same samples used for the array studies.
BM clonogenic assay
Short‐term BM clonogenic assays were performed with BMNCs as previously described [25]. Briefly, BMNCs (105/ml) were assayed in the presence or absence of LF, EF, PA or combinations, each at 1, 2 and 8 ng/ml. Control cultures contained vehicle. The assays were performed in methylcellulose matrices for colony forming unit‐granulocytic/monocytic (CFU‐GM) and CFU‐granulocytic/erythrocytic/megakarocytic/monocytic (CFU‐GEMM).
Cultures for CFU‐GM contained 3 U/ml recombinant human granulocytic/monocytic (rhGM)‐CSF and CFU‐GEMM assays contained 3 U rhGM‐CSF, 3 U/ml rhIL‐3, 10 ng/ml SCF and 2 U/ml rhEpo. CFU‐GM colonies were counted at day 8 by a blind observer (PR). Colonies with >20 cells were included. CFU‐GEMM colonies were counted at day 21. Random colonies of CFU‐GEMM were placed on slides and then stained by Wright’s to verify mixed cell lineages.
Modified long‐term culture initiating cell (LTC‐IC) cultures
LTC‐IC cultures were performed as previously described [26]. Confluent stromal cells were cultured in 25‐cm2 flasks and then subjected to 150 Gy, delivered by a caesium source. BMNCs were added to the stromal cultures at 107 cells/flask. Cultures were performed in the presence or absence of anthrax proteins at 1 and 2 ng/ml. Each experimental point was performed in duplicate. One set was studied at week 5 for CFU‐GEMM progenitors and the other at week 12.
Cell apoptosis detection with TUNEL
Terminal deoxynucleotidyl transferase mediated dUTP nick end labeling (TUNEL) assays were performed with the DeadEnd Fluorometric TUNEL System (Promega), following manufacturer’s recommendation for suspension cells. BMNCs were incubated for different times in the presence or absence of LF, EF, PA, added singly, or in combinations, each at 2 ng/ml. Positive controls were incubated overnight with 5 μg/ml cycloheximide and 10 ng/ml TNF‐α. For the analyses, BMNCs were resuspended in PBS at 2 × 107 cells/ml. Aliquots of cells, 100 μl, were added to superfrost slides and then fixed with 0.4% paraformaldehyde for 25 min. at 4°C. Slides were washed twice in PBS for 5 min. at room temperature, permeabilized for 5 min. and then incubated with Equilibration Buffer for 10 min. After this step, the DNA strand breaks were labelled with fluorescein‐12‐dUTP for 1 hr at 37°C in a humidified chamber. The reaction was terminated with 2× Sodium Chloride‐Sodium Citratein (SSC) for 15 min. at room temperature. The samples were then washed thrice with PBS and VECTASHIELD mounting media anti‐fading reagent with DAPI (300 nM) to stain the nuclei. The total number of TUNEL positive cells was determined by fluorescence microscopy with >200 cells were counted for each experiment.
Cell cycle analysis
BM cells were treated with 2 ng/ml EF, LF, PA or combinations for 4 hrs. After this, cells were washed, treated with RNase A (1 mg/ml) and then fixed in 70% ethanol. Cells were stained with 20 μg/ml Propodium Iodide. DNA analysis was done by FACScan (BD Biosciences) and the analysis was performed with CellQuest software (BD Biosciences).
Semi‐quantitative RT‐PCR
Total RNA was extracted from BM mononuclear cells and 2 μg was reverse transcribed and then subjected to PCR with ATR1 primers (Accession number NM_032208), spanning +329/+569, 5′ cca gaa agt tct gcc agg ag 3′ (forward) and 5′ gcc agc tgt gtc tca ttg aa 3′ (reverse). The cycling profile for ATR1 was 35 cycles with 95°C for 45 sec., 55°C for 30 sec., 72°C for 1 min. 30 sec. and extension for 7 min. The same cDNA was normalized with primers for fl‐actin (Accession number NM_001101) spanning +842/+1037, 5′ tgc cct gag gca ctc ttc 3′ (forward) and 5′ gtg cca ggg cag tga tct 3′ (reverse). The cycling profile for β‐actin was 30 cycles, 95°C for 45 sec., 55°C for 30 sec., 72°C for 1 min. 30 sec. and extension for 7 min.
Data analyses
Statistical evaluations of the data were done with analysis of variance and Tukey–Kramer multiple comparisons test. A P‐value of <0.05 was considered significant.
Results
ATR expression on BM cells
The first set of studies determined whether anthrax toxin receptor 1 (ATR1) is expressed on two major BM cell subsets: CD34+/CD38−, which comprises mostly HSCs and CD34+/CD38+ (mostly haematopoietic progenitors). ATR1 was selected because it is the uncleaved form of the three ATR subtypes [27]. The cell subsets were isolated from freshly obtained BM aspirates from healthy donors. The low frequency of HSCs could compromise sensitivity. We therefore employed combinations of immunoprecipitation and Western blots. Parallel Western blots were performed for β‐actin to ascertain equal amounts of proteins in the starting samples.
We performed the analyses with membrane extracts because cytoplasmic retention of ATR1 would indicate non‐responsiveness to exogenous anthrax proteins. Membrane extracts from the two major cell subsets were subjected to combined immunoprecipitation/Western blots with anti‐ATR1. Western blot analyses showed bands at molecular weights at the expected size of ATR1, ∼50–60 kD, for both cells subsets. Figure 1A shows a representative of six experiments. Each lane was studied with extracts from a different donor. The detection could not be due to cytosolic contamination because there was undetectable band in lanes with cytosolic extracts (Fig. 1A).
Figure 1.

ATR1 and PA‐binding sites on CD34+ BM cell subsets. (A) Immunoprecipitation/Western blots for ATR1 were performed with membrane extracts from CD34+/CD38− and CD34+/CD38+ cells from different donors. Negative controls used cytoplasmic extracts. The CD34+ cell subsets were separated by immunoaffinity methods. Each analysis was done with BM cells from four different healthy donors. Each lane was done with extracts from a different donor. In parallel studies, Western blots were done for β‐actin with each extract. (B) and (C) BM aspirates were analysed by four‐colour flow cytometry using FITC‐PA, PE‐CD34 and PC5‐CD38 and APC‐CD32 (B) or, PC5‐CD117 and APC‐CD38 (C). The results represent four different donors. The analyses were done in gated populations that were CD34+/PA+ (R1).
PA‐binding sites on haematopoietic cells
Computer analyses showed similarities among ATR1 and haematopoietic‐relevant molecules (Fig. 2). Thus, it is possible that PA could bind to different subsets of haematopoietic cells, through interactions with molecules other than ATR1. This question was addressed in four‐colour flow cytometry with fluorochrome labelled PA and antibodies specific for different haematopoietic cell subsets [28]. Antibodies to CD34, CD117 (c‐kit), CD32 (FcR) and CD38 were used to differentiate between myelo‐lymphoid progenitors (CD34+/CD117+/CD38lo/−) and early myeloid progenitors (CD34+/CD32lo/CD38+/−) [29, 30]. The CD34+/PA+ subset (represented in Fig. 1B, middle panel) showed 46 ± 6% dim/low fluorescence for CD38, and negative to dim for the myeloid marker, CD32 (Fig. 1B, right panel). This indicates binding of PA to early myeloid progenitors. We further determined if CD34+/PA+ cells comprised CD117 (c‐kit), which is expressed on the most primitive haematopoietic cells. As expected, most CD38+ cells were negative for CD117 (Fig. 1C, lower right panel), verifying a mature haematopoietic phenotype. A small subset of immature haematopoietic cells that bind PA was detected, 1.4 ± 0.01% CD38−/CD117+ (Fig. 1C, right panel). The results described in this section show PA interaction with both immature and mature CD34+ haematopoietic cells.
Figure 2.

Computer‐assisted alignment of ATR1 with molecules relevant to haematopoiesis. The boxed sections show regions of similarities.
Effects of monomeric anthrax toxins on haematopoietic progenitors
Western blots and immunofluorescence indicated expressions of ATR1, and PA‐binding to both primitive and mature haematopoietic progenitors (Fig. 1). We therefore asked whether PA binding is significant to haematopoietic functions. We first examined mature progenitors of granulocyte–monocyte lineage (CFU‐GM) in short‐term clonogenic assays. The assays were performed with BMNCs from seven different healthy donors, in the presence or absence of monomeric LF, EF and PA at 1, 2 or 8 ng/ml. Dose–response curves were performed with proteins ranging between 10−4 and 103 ng/ml. For each protein, the effects were similar between 1 and 8 ng/ml. Studies with combined proteins contained equal amounts of each. This ratio differed from other studies that added 5× more LF and EF to PA [31]. This difference might be attributed to differences in the experimental systems.
The haematopoietic response of each protein is concentration dependent (Fig. 3A). Compared to baseline colonies (media alone), LF caused no significant (P > 0.05) proliferation at 1 and 2 ng/ml, but significance (P < 0.05) at 8 ng/ml (Fig. 3A, left diagonal bars). In the case of EF, we observed a bell‐shaped effect with significant increase at 2 ng/ml (Fig. 3A, hatched bars). Cultures with PA showed dose‐dependent suppression on CFU‐GM, with significant (P < 0.05) increases at 1 and 2 ng/ml, and significant (P < 0.05) suppression at 8 ng/ml. In summary, each monomeric anthrax toxin showed a specific effect on CFU‐GM.
Figure 3.

Effects of anthrax toxins on mature and primitive haematopoietic progenitors. Short‐term clonogenic CFU‐GM assays were performed in the presence or absence of separate (A) or combined (B) anthrax toxins at 1, 2 and 8 ng/ml. The results are presented as mean CFU‐GM ± S.D., n= 7. LTC‐IC assays at 6 and 12 weeks (C) to determine the effects of anthrax toxins on immature progenitors, were established in the presence or absence of individual or combined anthrax toxins, each at 1, 2 and 8 ng/ml. The end‐points of LTC‐IC assays were clonogenic assays at weeks 6 and 12 for CFU‐GEMM. The results are shown as the mean ± S.D. of total CFU‐GEMM, n= 7. Studies repeated in the presence of polymyxin B showed similar outcome. In contrast, boiled samples showed outcome similar to baseline/media alone. A. *P > 0.05 versus baseline; **P < 0.05 versus baseline. B. *P < 0.05 versus experimental groups. C. *P < 0.05 versus individual toxins.
We next verified that the effects of anthrax proteins are not due to endotoxin. This question was addressed by repeating the assays with the denatured proteins, which was achieved by boiling for 20 min. The results showed no significant (P > 0.05) change in CFU‐GM (Fig. 4), indicating that the outcome of Fig. 3A cannot be explained by endotoxin.
Figure 4.

Apoptotic effects of anthrax monomeric proteins/toxins on bone marrow mononuclear cells (BMNCs). TUNEL assays were performed with BMNC incubated for 1, 2, 4, 8 and 12 weeks, in the presence or absence of 2 or 8 ng/ml of each protein. Baseline cultures contained only media. (diagonal bar). The results, (±S.D., n= 7, each with a different BM donor) are presented for 12‐week cultures with 8 ng/ml of anthrax protein(s). Positive controls were treated overnight with 5 μg/ml cyclohexamide and 10 ng/ml TNF‐α (open bar). *P > 0.05 versus baseline cultures.
Effects of monomeric anthrax proteins and toxins on mature haematopoietic progenitors
Anthrax toxins are formed when LF and/or EF binds to PA with subsequent endocytosis of heptamers and the releases of LF and LT [7]. The CFU‐GM cultures were therefore repeated with combinations of anthrax proteins. There was significant (P > 0.05) increases in CFU‐GM for LF + EF (Fig. 3B, left diagonal bars), but not for EF + PA or LF+PA (Fig. 3B, right diagonal and square bars). In the presence of EF + LF + PA, there were significant (P < 0.05) suppression (Fig. 3B, hatched bars). In summary, this section shows varied effect of combined proteins. However, anthrax toxin caused suppression on CFU‐GM.
Effects of anthrax toxin on immature haematopoietic progenitors
Western blots and flow cytometry studies indicate that primitive haematopoietic progenitors express ATR1 and can also bind PA (Fig. 1). We therefore studied the effects of proteins at 1, 2 and 8 ng/ml on primitive haematopoietic progenitors using LTC‐IC assays. An LTC‐IC cell is indicated if the cell can generate mixed progenitors along granulocytic, erythrocytic and megarkaryocytic lineages (CFU‐GEMM). We selected 2 ng/ml of anthrax toxin (combinations of LF, EF and PA) for the LTC‐IC assay, based on the outcome shown in Fig. 3B. We observed suppression on LTC‐IC for both 6‐ and 12‐week cultures (Fig. 3C). Week 12 outcome has physiological significance because this time period recapitulates the responses expected of early haematopoietic progenitors that are close to the maturation of HSCs. There was significant (P < 0.05) reduction in CFU‐GEMM for monomeric and combined proteins at both 6‐ and 12‐week cultures (Fig. 3C). In summary, this section shows suppressive effects of anthrax proteins on CFU‐GM and CFU‐GEMM progenitors.
Despite lack of evidence for endotoxin in the anthrax protein samples, we verified that the effects on LTC‐IC were not due to endotoxin. LTC‐IC studies were repeated with boiled samples and the results were similar to cultures with media alone (Fig. 3C). Similarly, when the assay was repeated in the presence of the endotoxin inhibitor, polymyxin B (12.5 μg/ml) [32], the results were similar to cultures with anthrax proteins alone, ±0.02 (Fig. 3C), further indicating that the effects by anthrax proteins on LTC‐IC are not due to endotoxin.
Effects of anthrax toxins on apoptosis of BM cells
The section describes studies to determine whether the suppressive effects of anthrax proteins could be explained by cell death. Firstly, BMNCs were incubated with or without anthrax toxins. In one set of experiment, the media were not changed. In parallel experiment, there were biweekly media changes with fresh anthrax proteins. In both cases, trypan blue exclusion indicated <5% cell death by week 12. Flow cytometry for the total number of CD34+ cells showed no change by week 12. Because there was no Further verification was done with TUNEL assay and Western blot for active caspase‐3. BMNCs from seven different donors were incubated with various combinations of anthrax proteins, each at 2 or 8 ng/ml. At weeks 1, 2, 4, 8 and 12, the cells were analysed for percentages of TUNEL positive cells. Baseline cultures (no anthrax proteins) showed 2 ± 0.55%, n= 7. Because the total TUNEL positive cells were similar for all time periods, the data are shown for 12‐week cultures. Except for studies with EF, LF and PA, the total TUNEL positive cells were not significantly different from baseline studies (Fig. 4). Positive controls performed with cycloheximide and TNF‐α showed approximately 50% TUNEL staining. Western blots for caspase‐3 showed undetectable bands (not shown). Together, the data indicate that the suppressive effects of anthrax proteins and toxins on haematopoiesis cannot be explained by cell death. Because B. anthracis and other bacterial agents have been linked to macrophage death involving caspase‐1 [33], this mediator might be involved as a secondary mechanism in macrophages. However, we do not have any evidence of its involvement in the CD34+ cell population. Caspase‐1 has been shown to precede caspase‐3 in apoptosis of haematopoietic cells [34]. In this case, we have not detected caspase‐3, suggesting that caspase‐1 might not be involved, at least for haematopoietic progenitors.
Cytokine production in BM stroma
This section describes studies to determine whether anthrax toxins can affect the major cellular support of haematopoiesis, stroma [35]. To address this question, we focused on cytokine production because of their major roles in haematopoiesis [36]. We first determine whether BM stroma express ATR1. Immunoprecipitation/Western blots with membrane extracts from stroma of four different healthy donors showed single dense bands (Fig. 5A). Because ATR1 is undetectable in cytoplasmic extracts of CD34+/CD38+ cells (Fig. 1A), this served as negative control (Fig. 5A, left lane). Parallel Western blots were done for β‐actin for the purpose of normalization (Fig. 5A, top lane).
Figure 5.
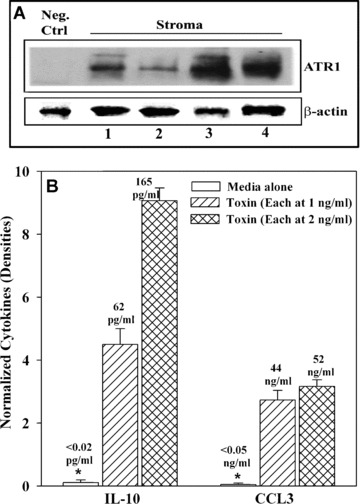
Effects of anthrax toxins on IL‐10 and CCL3 production in BM stroma. (A) Membrane extracts from four different donors were analysed for ATR1 by combination of immunoprecipitation and Western blots. Negative control used cytoplasmic extracts from CD34+/CD38+. In parallel studies, Western blots were done for β‐actin. (B) Stromal cells were stimulated with 1 or 2 ng/ml of anthrax toxins and after 16 hrs, the culture media and cell extracts were analysed by cytokine microarrays. The densities of internal positive controls were assigned values of 10 and then used as reference for unknown densities. The results represent three experiments, mean ± S.D. The array studies are presented as bar graphs and the ELISA results are placed immediately above each graph.
We next asked if anthrax toxin can induce the production of cytokines and chemokines by protein arrays. Stromal cells were incubated with LF + EF + PA, each at 2 ng/ml. After 16 hrs, the analyses were done with culture media and whole cell extracts. The results are shown for normalized densities of two haematopoietic suppressors, IL‐10 and MIP‐1α (CCL3) (Fig. 5B). In both cases, there were significant (P < 0.05) increases in IL‐10 and CCL3 by anthrax toxins as compared to unstimulated stroma. We also observed a twofold increase for SDF‐1α (CXCL12) production with LF + EF + PA, each at 2 ng/ml. This increase is intriguing because CXCL12 could cause the release of HSCs from their niche within BM [37]. IL‐10 and MIP‐1α levels were quantitated by ELISA and the values were placed at the top of the bar graphs of the array results (Fig. 5B).
Role of CCL3 and IL‐10 on the suppression of LTC‐IC cells
CCL3 and IL‐10 can mediate haematopoietic suppression [38, 39]. Because there is no compelling evidence of cell death by anthrax proteins, we asked whether haematopoietic suppression could be partly explained by secondary production of IL‐10 and CCL3 (Fig. 5B). We performed LTC‐IC assays in the presence or absence of LF + EF + PA, each at 2 ng/ml (toxin), in the presence or absence of anti‐IL‐10 and/or anti‐CCL3, each ranging between 1 and 1000 ng/ml. Control cultures contained similar concentrations of non‐immune species IgG. CFU‐GEMM colonies, the end‐points of the LTC‐IC assays, were similar between 5 and 100 ng/ml of antibodies. We have determined specificity of the antibodies because non‐immune IgG showed total CFU‐GEMM similar to cultures with media alone. Although neither antibody was able to fully reverse the suppressive effects of toxins, the degree of reversal was significant (P < 0.05) as compared to cultures with toxins alone (Fig. 6).
Figure 6.
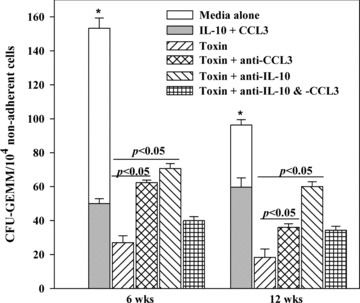
Role of anthrax toxin‐induced IL‐10 and CCL3 on LTC‐IC cells. LTC‐IC assays were established as for Fig. 3C, in the presence or absence of 2 ng/ml each of EF, LF and PA (Toxin). Parallel cultures contained toxin and 20 ng/ml of anti‐IL‐10 and/or anti‐CCL3. Specificity was tested in parallel with similar amounts of non‐immune murine IgG. Positive control cultures were done by adding 50 pg/ml each, IL‐10 and CCL3, in baseline cultures (inset solid bar). The results are shown as mean ± S.D. (n= 7). Each experiment was performed with cells from a different BM donor.
Positive controls were established with media (baseline), in the presence of 50 pg/ml each, IL‐10 and CCL3. Because these are not the only haematopoietic suppressors, their combined effects, although significantly suppressive, did not totally blunt the formation of CFU‐GEMM (Fig. 6, solid inserted bar). The concentrations of IL‐10 and CCL3 were selected based on the total levels induced by the anthrax proteins (Fig. 5B). In addition, IL‐10 and CCL3 with 5 ng/ml of the respective antibody blunted the ability of these two factors to exert haematopoietic suppression to revert to levels observed in cultures with media alone (open bar). These studies supported the neutralization effects of the antibodies. In summary, the results show CCL3 and IL‐10 as partial mediators in anthrax‐induced suppression on LTC‐IC cells.
Cell cycle responses to anthrax toxins
We observed differences in BM functions depending on the combinations of anthrax toxins (Fig. 3). Cytokine analyses also correlated with the suppressive effects of anthrax proteins on haematopoiesis (Fig. 6). We therefore studies BM cells with anthrax proteins to determine if the suppressive effects could be explained by blocking of specific regions of the cell cycle. To this end, we incubated whole BM cells with 2 ng/ml of single or combined anthrax proteins. After 4 hrs, the cells were analysed by FACScan. The results showed significant changes for cells incubated with the three proteins (Fig. 7). Because the incubation period was relatively short, the results suggest that anthrax proteins might rapidly affect haematopoietic cell cycle when by three combinations that are required to comprise a toxin.
Figure 7.
Cell cycle analysis of bone marrow mononuclear cells (BMNCs) with anthrax proteins. BMNCs were incubated with 2 ng of various combinations of EF, LT and PA for 4 hrs. After this the cells were labelled with Propidium Iodide and then immediately analysed by the FACS analysis. The results represent three experiments, each with different donor. The mean percentages ± S.D. of cells in S‐phase are inserted in each graph.
Discussion
This study reports on haematopoietic alterations by anthrax proteins and toxins, which could occur directly and/or indirectly through the productions of cytokines and chemokines. We focused on haematopoietic suppression because significant effects were observed for the most primitive haematopoietic progenitors, which were based on LTC‐IC assays. The most interesting observation of this study is the fact that anthrax toxins show a non‐canonical effect as opposed to the classical report of toxins formed by EF, LF and PA. The binding of PA to the two subsets of CD34+ indicate that there is the potential for direct effects on haematopoiesis (Fig. 1). In addition, the effects on haematopoiesis could occur through molecular mimicry with PA interacting with other homologous molecules that are relevant to haematopoiesis (Fig. 2). The alignment of homologous molecules with relevance to haematopoiesis, although not addressed in functional assays might explain the direct effects of PA. These are exciting observations. Current studies are underway to knockdown ATR1 and/or CMG2 to determine if PA binds non‐specifically to the homologous molecules shown in Fig. 2.
Of interest are the functional differences between 1 and 2 ng/ml of toxins. We interpret these differences to indicate that the cells are sensitive to small changes in the levels of anthrax toxins. Future plans are in place to explore these small changes because the haematopoietic system could be sensitive to vaccine administration and also, to low levels of anthrax exposure. The latter could be a scenario if the individuals have competent immune systems, or in cases where there is rapid responses to antibiotics. Another interesting observation is that conversion of anthrax proteins to molar ratio did not follow the expected optimal effects by ratios of 2:1. This further verifies that the effects of anthrax proteins on haematopoiesis is non‐canonical and require in‐depth studies.
The effects of anthrax proteins and toxins are different on the most primitive and the more mature haematopoietic progenitors. These differences are shown in Fig. 3B and C, which studied myeloid progenitors and cells close to the maturational stage of HSCs, respectively. The partial subunits with EF or LF and PA show minimal inhibition as compared to significant inhibition in studies with the entire complex of EF, LF and PA (Fig. 3B). This suggests that the complex could determine the outcome that might be direct or indirectly mediated through other non‐progenitor cells. The interesting observation in Fig. 3B is the stimulator effect of LF and EF. Regardless of the mechanism, it is evident that these factors show varying effects on progenitors and that the outcome depends on small changes in each and combined toxins.
Anthrax toxins showed significant suppression on LTC‐IC cells (Fig. 3C). Because these cells represent the most primitive haematopoietic cells, in particular the 12‐week cultures, it is possible that the toxins could be protective and prevent cycling, or reduce the total numbers of these primitive cells. In the case of the latter, the toxins could lead to BM ablation and exposures to the toxins could cause possible haematopoietic failure.
Although two of the identified factors, IL‐10 and CCL3, have been shown to have secondary effects on LTC‐IC cells, other mechanisms appear to be operative. This premise is based on studies that show failure to totally reverse the suppressive effects of anthrax toxins despite cytokine neutralization (Fig. 6). Ongoing studies are designed to fully understand the mechanisms by which anthrax toxins mediate haematopoietic suppression through cytokine and/or chemokine production. Also, ongoing studies are directed towards an understanding of the intracellular changes caused by anthrax toxins. An unanswered question is to identify the effects of the toxins on candidate subsets within the myelo‐lymphoid lineages. These studies, combined with ongoing and future studies would provide detailed insights on the mechanisms by which anthrax infection and vaccines affect adult haematopoiesis.
Although we have examined the effects of anthrax toxin on the emerging immune system, namely haematopoiesis, the toxins have effects on mature cells – in particular B‐cells, whose proliferation and IgG production are inhibited by LT [9]. Although lethal toxins inhibit MAP kinase activity, this role on anthrax‐mediated effects on haematopoiesis is unclear [40, 41]. Septic shock, caused by anthrax infections cannot be explained by cytokine production as causative agents [42]. This underscore the need for dissected studies, such as the case presented in this report to understand the effects of anthrax proteins and toxins on haematopoiesis and other biological functions.
The results of 6‐week LTC‐IC cultures represent the more mature progenitors (Fig. 6). Anti‐MIP‐1α resulted in more pronounced reversal of CFU‐GEMM in the 6‐week cultures as compared to the 12‐week assays (Fig. 6). This suggests that CCL3 is important for the mature progenitors rather than the immature progenitors. IL‐10 effects were similar for both the 6‐ and 12‐week cultures suggesting that this cytokine could be acting at all levels. The biology of IL‐10 with respect to haematopoietic effects is complex. In some cases, IL‐10 could be stimulatory to haematopoiesis because it suppresses the activity of IFN‐γ, which could inhibit haematopoiesis at high levels [43]. The fact that neither anti‐CCL3 nor anti‐IL‐10 was able to reverse the haematopoietic effects of the anthrax toxin (Fig. 6) indicates that other mechanisms are important. Future studies are underway to address these mechanistic processes.
Although we could not detect evidence of cell death by single or combined anthrax proteins, prolonged exposures in 12‐week cultures to anthrax toxin (EF/LF/PA) showed a trend towards apoptosis, as compared to baseline cultures (Fig. 4). This finding could be important and might contribute to haematopoietic failure during infection. The question is whether a particular subset of BM cells might be sensitive to prolonged anthrax exposures. In addition, there was no evidence of cell death in the LTC‐IC cultures (Fig. 3C), suggesting that the most primitive haematopoietic cells could not account for apoptosis. Cell cycle analyses were quite remarkable. It is interesting that the combination of the two toxins shows an increase in cells in the S‐phase as compared to the combinations of the toxins EF/PA/LF, where the cells show a significant reduction in the S‐phase (Fig. 7). These results are consistent with reduced clonogenicity in the presence of combined toxins. The cell cycle analyses were performed during 4‐hr incubation to determine the relatively early response by BM cells. It is yet to be determined what chronic exposure of the anthrax toxin would show.
A recent report showed inhibitory effects on B‐cell proliferation and immunoglobulin production [44]. This indicates that the anthrax toxins are affecting both the haematopoietic primitive progenitors (Figs 3A and 7), and the mature B‐cells. Although the other haematopoietic lineages have not been examined, these questions are important and are the subject of ongoing studies. Our report advises that current and future anthrax vaccine development should consider BM dysfunction. This dysregulation, especially in the haematopoietic system, may lead to repercussions in the body’s ability to respond to anthrax infection as well as the ability to maintain vaccine‐based protection. It is understandable that in the current state of increased biological weapons threat, the quick production of an effective anthrax vaccine is favoured. However, certain concerns must be addressed to prevent potential harm to the general population, if given the anthrax vaccine. In addition, vaccine development for other biothreat agents may benefit from similar consideration.
In conclusion, this study shows haematopoietic effects by anthrax proteins and toxins. These effects could be directly mediated by ATR1 present on different haematopoietic cell subsets, including the most primitive type of cells. The effects could also be indirect through stromal cells. Specifically, this could occur by the toxins inducing cytokine production to modulate haematopoiesis. Anthrax toxin could also target actin filaments, destruction of which could lead to cellular instability [45]. The observed effects by EF and LF, in the absence of PA are of significance. It appears that these factors could be interacting with other molecules found in BM. These are intriguing findings and form the basis for current studies in the laboratory. These studies could lead to an understanding of health disorders associated with anthrax exposures, and could also be explored for efficient responses by BM to anthrax vaccines.
Acknowledgement
This work was supported by a grant awarded by the NIH, AI63578.
References
- 1. Malecki J, Wiersma S, Bresnitz E, et al . Investigation of bioterrorism‐related anthrax, 2001. JAMA . 2001; 286: 2662–3. [PubMed] [Google Scholar]
- 2. Weiss MM, Weiss PD, Weiss JB. Anthrax vaccine and public health policy. Am J Public Health . 2007; 97: 1945–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Banks DJ, Ward SC, Bradley KA. New insights into the functions of anthrax toxin. Expert Rev Mol Med . 2006; 8: 1–18. [DOI] [PubMed] [Google Scholar]
- 4. Brossier F, Mock M. Toxins of Bacillus anthracis . Toxicon . 2001; 39: 1747–55. [DOI] [PubMed] [Google Scholar]
- 5. Collier RJ, Young JAT. Anthrax toxin. Annu Rev Cell Dev Biol . 2003; 19: 45–70. [DOI] [PubMed] [Google Scholar]
- 6. Bradley KA, Young JAT. Anthrax toxin receptor proteins. Biochem Pharmacol . 2003; 65: 309–14. [DOI] [PubMed] [Google Scholar]
- 7. Scobie HM, Young JA. Interactions between anthrax toxin receptors and protective antigen. Curr Opin Microbiol . 2005; 8: 106–12. [DOI] [PubMed] [Google Scholar]
- 8. Young J, Collier J. Attacking anthrax. Sci Am . 2002; 286: 48–50. [DOI] [PubMed] [Google Scholar]
- 9. Friedlander AM. Anthrax: clinical features, pathogenesis, and potential biological warfare threat. Curr Clin Top Infect Dis . 2007; 20: 335–49. [PubMed] [Google Scholar]
- 10. Gao M, Schulten K. Onset of Anthrax Toxin Pore Formation. Biophys J . 2006; 90: 3267–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang JY, Roehrl MH. Anthrax vaccine design: strategies to achieve comprehensive protection against spore, bacillus, and toxin. Med Immunol . 2005; 4: 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brey RN. Molecular basis for improved anthrax vaccines. Adv Drug Delivery Rev . 2005; 57: 1266–92. [DOI] [PubMed] [Google Scholar]
- 13. Leppla SH, Robbins JB, Schneerson R, et al . Development of an improved vaccine for anthrax. J Clin Invest . 2002; 110: 141–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Boor AK. An antigen prepared in vitro effective for immunization against anthrax. I. Preparation and evaluation of the crude protective antigen. J Infect Dis . 1955; 97: 194–202. [DOI] [PubMed] [Google Scholar]
- 15. Baillie L. The development of new vaccines against Bacillus anthracis . J Appl Microbiol . 2001; 91: 609–13. [DOI] [PubMed] [Google Scholar]
- 16. Li Z, Li L. Understanding hematopoietic stem‐cell microenvironments. Trends Biochem Sci . 2006; 31: 589–95. [DOI] [PubMed] [Google Scholar]
- 17. Dorshkind K. Regulation of hemopoiesis by bone marrow stromal cells and their products. Annu Rev Immunol . 1990; 8: 111–37. [DOI] [PubMed] [Google Scholar]
- 18. Young NS. Acquired aplastic anemia. Ann Intern Med . 2002; 136: 534–46. [DOI] [PubMed] [Google Scholar]
- 19. Lacy DB, Wigelsworth DJ, Scobie HM, et al . Crystal structure of the von Willebrand factor A domain of human capillary morphogenesis protein 2: an anthrax toxin receptor. Proc Natl Acad Sci USA . 2004; 101: 6367–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Springer TA. Complement and the multifaceted functions of VWA and integrin I domains. Structure . 2006; 14: 1611–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shah C, Lemke S, Singh V, et al . Case reports of aplastic anemia after vaccine administration. Am J Hematol . 2004; 77: 204. [DOI] [PubMed] [Google Scholar]
- 22. Rameshwar P, Gascon P. Substance P (SP) mediates production of stem cell factor and interleukin‐ 1 in bone marrow stroma: potential autoregulatory role for these cytokines in SP receptor expression and induction. Blood . 1995; 86: 482–90. [PubMed] [Google Scholar]
- 23. Patel HJ, Ramkissoon SH, Patel PS, et al . Transformation of breast cells by truncated neurokinin‐1 receptor is secondary to activation by preprotachykinin‐A peptides. Proc Natl Acad Sci USA . 2005; 102: 17436–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Oh HS, Moharita A, Potian JG, et al . Bone marrow stroma influences transforming growth factor‐β production in breast cancer cells to regulate c‐myc activation of the preprotachykinin‐I gene in breast cancer cells. Cancer Res . 2004; 64: 6327–36. [DOI] [PubMed] [Google Scholar]
- 25. Rameshwar P, Gascon P. Induction of negative hematopoietic regulators by neurokinin‐A in bone marrow stroma. Blood . 1996; 88: 98–106. [PubMed] [Google Scholar]
- 26. Antoniou ES, Sund S, Homsi EN, et al . A theoretical simulation of hematopoietic stem cells during oxygen fluctuations: prediction of bone marrow responses during hemorrhagic shock. Shock . 2004; 22: 415–22. [DOI] [PubMed] [Google Scholar]
- 27. Bonuccelli G, Sotgia F, Frank PG, et al . ATR/TEM8 is highly expressed in epithelial cells lining Bacillus anthracis’ three sites of entry: implications for the pathogenesis of anthrax infection. Am J Physiol Cell Physiol . 2005; 288: C1402–10. [DOI] [PubMed] [Google Scholar]
- 28. Haylock DN, Nilsson SK. Stem cell regulation by the hematopoietic stem cell niche. Cell Cycle . 2005; 4: 1353–5. [DOI] [PubMed] [Google Scholar]
- 29. Gaipa G, Coustan‐Smith E, Todisco E, et al . Characterization of CD34+, CD13+, CD33– cells, a rare subset of immature human hematopoietic cells. Haematologica . 2002; 87: 347–56. [PubMed] [Google Scholar]
- 30. Lynch RG. Regulatory roles for FcgammaRIII (CD16) and FcgammaRII (CD32) in the development of T‐ and B‐lineage lymphoid cells. J Leukoc Biol . 2000; 67: 279–84. [DOI] [PubMed] [Google Scholar]
- 31. Comer JE, Galindo CL, Chopra AK, et al . GeneChip analyses of global transcriptional responses of murine macrophages to the lethal toxin of Bacillus anthracis . Infect Immun . 2005; 73: 1879–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rameshwar P, Gascon P. Release of interleukin‐1 and interleukin‐6 from human monocytes by antithymocyte globulin: requirement for de novo synthesis. Blood . 1992; 80: 2531–8. [PubMed] [Google Scholar]
- 33. Ting JPY, Willingham SB, Bergstralh DT. NLRs at the intersection of cell death and immunity. Nat Rev Immunol . 2008; 8: 372–9. [DOI] [PubMed] [Google Scholar]
- 34. Ali A, Mundle SD, Ragasa D, et al . Sequential activation of caspase‐1 and caspase‐3‐like proteases during apoptosis in myelodysplastic syndromes. J Hematother Stem Cell Res . 1999; 8: 343–56. [DOI] [PubMed] [Google Scholar]
- 35. Bianco P, Riminucci M, Gronthos S, et al . Bone marrow stromal stem cells: nature, biology, and potential applications. Stem Cells . 2001; 19: 180–92. [DOI] [PubMed] [Google Scholar]
- 36. Rameshwar P, Poddar A, Gascon P. Hematopoietic regulation mediated by interactions among the neurokinins and cytokines. Leuk Lymphoma . 1997; 28: 1–10. [DOI] [PubMed] [Google Scholar]
- 37. Zhong R, Law P, Wong D, et al . Small peptide analogs to stromal derived factor‐1 enhance chemotactic migration of human and mouse hematopoietic cells. Exp Hematol . 2004; 32: 470–5. [DOI] [PubMed] [Google Scholar]
- 38. Broxmeyer HE. Regulation of hematopoiesis by chemokine family members. Int J Hematol . 2001; 74: 9–17. [DOI] [PubMed] [Google Scholar]
- 39. Oehler L, Kollars M, Bohle B, et al . Interleukin‐10 inhibits burst‐forming unit‐erythroid growth by suppression of endogenous granulocyte‐macrophage colony‐stimulating factor production from T cells. Exp Hematol . 1999; 27: 217–23. [DOI] [PubMed] [Google Scholar]
- 40. Chopra AP, Boone SA, Liang X, et al . Anthrax lethal factor proteolysis and inactivation of MAPK kinase. J Biol Chem . 2003; 278: 9402–6. [DOI] [PubMed] [Google Scholar]
- 41. Pellizzari R, Guidi‐Rontani C, Vitale G, et al . Anthrax lethal factor cleaves MKK3 in macrophages and inhibits the LPS/IFN[gamma]‐induced release of NO and TNF[alpha]. FEBS Lett . 1999; 462: 199–204. [DOI] [PubMed] [Google Scholar]
- 42. Prince AS. The host response to anthrax lethal toxin: unexpected observations. J Clin Invest . 2003; 112: 656–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Asano Y, Shibata S, Kobayashi S, et al . Effect of interleukin 10 on the hematopoietic progenitor cells from patients with aplastic anemia. Stem Cells . 1999; 17: 147–51. [DOI] [PubMed] [Google Scholar]
- 44. Fang H, Xu L, Chen TY, et al . Anthrax lethal toxin has direct and potent inhibitory effects on B cell proliferation and immunoglobulin production. J Immunol . 2006; 176: 6155–61. [DOI] [PubMed] [Google Scholar]
- 45. During RL, Gibson BG, Li W, et al . Anthrax lethal toxin paralyzes actin‐based motility by blocking Hsp27 phosphorylation. EMBO J . 2007; 26: 2240–50. [DOI] [PMC free article] [PubMed] [Google Scholar]