

Abstract
Butyrylcholinesterase is a promiscuous enzyme that displays complex kinetic behavior. It is toxicologically important because it detoxifies organophosphorus poisons (OP) by making a covalent bond with the OP. The OP and the butyrylcholinesterase are both inactivated in the process. Inactivation of butyrylcholinesterase has no adverse effects. However inactivation of acetylcholinesterase in nerve synapses can be lethal. OP-inhibited butyrylcholinesterase and acetylcholinesterase can be reactivated with oximes provided the OP has not aged. Strategies for preventing the toxicity of OP include a) treatment with an OP scavenger, b) reaction of nonaged enzyme with oximes, c) reactivation of aged enzyme, d) slowing down aging with peripheral site ligands, and e) design of mutants that rapidly hydrolyze OP. Option (a) has progressed through phase I clinical trials with human butyrylcholinesterase. Option (b) is in routine clinical use. The others are at the basic research level. Butyrylcholinesterase displays complex kinetic behavior including activation by positively charged esters, ability to hydrolyze amides, and a lag time (hysteresis) preceding hydrolysis of benzoylcholine and N-methyl indoxyl acetate. Mass spectrometry has identified new OP binding motifs on tyrosine and lysine in proteins that have no active site serine. It is proposed, but not yet proven, that low dose exposure involves OP modification of proteins that have no active site serine.
Keywords: bioscavenger, mass spectrometry, phosphylation, dehydroalanine, aryl acylamidase, hysteresis
Introduction
Humans have two cholinesterases: acetylcholinesterase (AChE, accession #gi:177975) and butyrylcholinesterase (BChE, accession # gi:116353). Acetylcholinesterase in nerve synapses terminates nerve impulse transmission by hydrolyzing the neurotransmitter acetylcholine. Butyrylcholinesterase acts as a backup for acetylcholinesterase, and as a scavenger for poisons that might inhibit acetylcholinesterase activity. Butyrylcholinesterase was of little interest to research scientists until the US Department of Defense allocated millions of dollars for the mass production of pure human butyrylcholinesterase to use for protection against the toxicity of nerve agents. Animal studies had demonstrated that pretreatment with butyrylcholinesterase gave complete protection from up to 5 LD50 of nerve agents [1]. Chemical warfare agents were used by Iraq against Kurdish civilians in the war between Iraq and Iran (1980–1988), and by Aum Shinrikyo cult members in the 1995 sarin attack in the Tokyo subway, and in Matsumoto city the previous year.
Nerve agents are a threat because they are extremely toxic at low doses. Their acute toxicity is due to inhibition of acetylcholinesterase. Nerve agents are organophosphorus esters, similar in structure (Figure 1) and mechanism of toxicity to organophosphorus pesticides.
Figure 1.

Structures of organophosphorus agents. Paraoxon, chlorpyrifos oxon, dichlorvos, and malaoxon are the active metabolites of pesticides. Echothiophate has been used in eyedrops to treat glaucoma. Tabun, VX, soman, sarin, and cyclosarin are nerve agents developed for use as chemical warfare agents. FP-biotin and diisopropylfluorophosphate are research reagents.
This review focuses on human butyrylcholinesterase and unanswered questions about butyrylcholinesterase. New mass spectrometry evidence suggests that proteins other than the cholinesterases may be involved in disorders that arise from low dose exposure.
Function of butyrylcholinesterase
The human body has ten times more butyrylcholinesterase than acetylcholinesterase [2]: about 680 nanomoles of butyrylcholinesterase and 62 nanomoles of acetylcholinesterase. Butyrylcholinesterase protein or mRNA has been found in almost every tissue including plasma, liver, brain, muscle, saliva, kidney, heart, lining of blood vessels, skin, colon, small intestine, spleen, and lung (see AceView in the NCBI database). According to AceView, the BCHE gene is expressed at very high levels, 4.0 times the average gene. The sequence of the BCHE gene is defined by 439 GenBank accessions from 433 cDNAclones. Certain neurons in the thalamus of the human brain express butyrylcholinesterase exclusively, whereas others express both butyrylcholinesterase and acetylcholinesterase as shown by specific activity staining [3].
Acetylcholine hydrolysis
The high amount of butyrylcholinesterase present in nearly every tissue suggests that butyrylcholinesterase has a function. The neurotransmitter acetylcholine is an excellent substrate for butyrylcholinesterase; butyrylcholinesterase hydrolyzes acetylthiocholine at a rate just two fold lower than it hydrolyzes butyrylthiocholine [4]. Butyrylcholinesterase does not appear to have a significant role in acetylcholine hydrolysis under normal conditions as shown in muscle preparations where complete inhibition of butyrylcholinesterase activity has no effect on muscle contraction. However, butyrylcholinesterase does have a role in neurotransmission in mice that have no acetylcholinesterase. The AChE−/− mice have normal levels of butyrylcholinesterase activity. Treatment of AChE−/− mice with OP results in inhibition of butyrylcholinesterase activity and lethality at concentrations well below those that cause lethality in wild-type mice [5,6]. This suggests that butyrylcholinesterase performs the function of the missing acetylcholinesterase in these mice by hydrolyzing acetylcholine.
Additional evidence of a role for butyrylcholinesterase in terminating neurotransmission comes from studies of the Gly117His transgenic mouse. This mouse expresses low levels of the butyrylcholinesterase mutant G117H in all tissues [7]. The G117H mutant is resistant to inhibition by OP and is capable of hydrolyzing acetylcholine in the presence of OP. Live G117H mice treated with DFP survive even though their acetylcholinesterase is inhibited. Under the same conditions wild-type mice die. The survival of the G117H mice is attributed to the ability of G117H to hydrolyze acetylcholine in the presence of DFP. Though the G117H mutant hydrolyzes OP, the rate of OP hydrolysis is slow, so that OP hydrolysis does not explain survival. If OP hydrolysis were significant, then the level of acetylcholinesterase and butyrylcholinesterase inhibition should be lower in the G117H mouse than in the wild-type mice treated with DFP. This was not the case. The levels of inhibition were similar. It was concluded that G117H hydrolysis of acetylcholine, rather than hydrolysis of OP, explained survival of these mice.
Acetylcholine levels in the hippocampus of live AChE−/− mice were measured by microdialysis followed by HPLC and electrochemical detection [8]. Infusion of a selective butyrylcholinesterase inhibitor (bambuterol, tolserine, or bis-norcymserine) through the microdialysis probe caused a 5-fold increase in acetylcholine levels in AChE−/− mice, but not in AChE+/+ mice. It was concluded that in the absence of acetylcholinesterase, the levels of extracellular acetylcholine in the brain are controlled by the activity of butyrylcholinesterase.
Neurons in the human thalamus were stained specifically for acetylcholinesterase or butyrylcholinesterase activity. In some nuclei, for example the anteroventral nucleus, virtually all neurons stained positive for butyrylcholinesterase activity and none stained positive for acetylcholinesterase activity. In contrast, some nuclei for example the anterodorsal nucleus, had only acetylcholinesterase positive neurons. It was concluded that the distinct distribution of butyrylcholinesterase in neurons is consistent with an important role for butyrylcholinesterase in neurotransmission in the human nervous system [3].
The action of acetylcholine on bronchial airway smooth muscle is prolonged following inhibition of butyrylcholinesterase, indicating that butyrylcholinesterase has a role in acetylcholine hydrolysis in this tissue [9,10].
Butyrylcholine hydrolysis
Butyrylcholine is the optimum substrate for human butyrylcholinesterase. Butyrylcholine has been found in bovine corneal epithelium [11] and in bovine brain [12] where its function is unknown. Local application of butyrylcholine to intrinsic cardiac neurons increases neuronal activity [13], suggesting that butyrylcholine can act as a neurotransmitter.
Protection from neurotoxins
It is generally agreed that butyrylcholinesterase functions to protect from man-made and naturally occurring poisons. The man-made poisons include OP nerve agents, OP pesticides, carbamate pesticides, and the Alzheimer drugs donepezil and rivastigmine [14,15]. Naturally occurring poisons include physostigmine (also called eserine) in the calabar bean, cocaine from the Erythroxylum coca plant, solanidine in green potatoes, huperzine A from the club moss Huperzia serrata, and anatoxin-a(S) an OP in blue-green algae [16]. Butyrylcholinesterase inactivates these poisons by reversible or irreversible binding or by hydrolysis. These poisons inhibit the activity of butyrylcholinesterase, but inhibition of butyrylcholinesterase has no known adverse effects. Many of these poisons are acetylcholinesterase inhibitors. By scavenging the poisons, butyrylcholinesterase protects acetylcholinesterase from inhibition.
Gaps in knowledge of butyrylcholinesterase deficiency
It is suspected but not proven that people with butyrylcholinesterase deficiency are more susceptible to the toxicity of organophosphorus pesticides, as well as to anti-Alzheimer drugs, and recreational doses of cocaine. Butyrylcholinesterase deficiency is due to genetic variation as well as to malnutrition and liver disease [17]. The activity of butyrylcholinesterase genetic variants ranges from about 70% of normal in the K variant, to 0% of normal in the silent variant. People with zero butyrylcholinesterase activity have normal health, are fertile, and live to old age [2]. However, they are paralyzed for 2 hours after treatment with a dose of succinylcholine that paralyzes most people for only 3 min.
The presence of neurons in the brain that express butyrylcholinesterase exclusively suggests that butyrylcholinesterase in those neurons may be acting on a neurotransmitter substrate that is hydrolyzed specifically by butyrylcholinesterase. A logical substrate would be butyrylcholine, but this has not been demonstrated. Mice completely deficient in butyrylcholinesterase have normal health and behavior [18], suggesting that other enzymes compensate for the missing butyrylcholinesterase activity.
Purification of butyrylcholinesterase and specific activity
Butyrylcholinesterase is purified from up to 100 liters of outdated human plasma by ion exchange chromatography at pH 4.0, followed by affinity chromatography on procainamide-Sepharose, and ion exchange at pH 7.4 [19]. Other protocols are also in the literature [20,21]. All use chromatography on procainamide-Sepharose as one of the steps. Recombinant human acetylcholinesterase is also purified by binding to procainamide-Sepharose [22].
The specific activity of pure human butyrylcholinesterase is 720 μmoles per min per mg assayed at pH 7.0 in 0.1 M potassium phosphate, 25°C with 1 mM butyrylthiocholine. Protein concentration is measured by absorbance at 280 nm where a 1 mg/ml solution of butyrylcholinesterase has an absorbance of 1.8.
Butyrylcholinesterase has a nonlinear response in plots of 1/activity versus 1/butyrylthiocholine concentration. The phase from 0.01 to 0.1 mM butyrylthiocholine yields a kcat of 24,000 min−1. The substrate activation phase from 0.4 to 40 mM yields a kcat value that is 3.2 times higher, 76,800 min−1 [23] measured at pH 7.0 and 25°C. At pH 8.0 the values are kcat = 45,500 min−1 with butyrylthiocholine, and kcat = 30,600 min−1 with acetylthiocholine for the substrate range 0.02 to 0.1 mM [20].
The specific activity of pure human acetylcholinesterase is 6000 μmoles per min per mg assayed at pH 8.0, 25°C with 1 mM acetylthiocholine, which corresponds to a kcat value of 400,000 min−1 [24].
Gaps in knowledge of purification method
Procainamide was selected as a ligand for affinity column purification because the inhibition constant for procainamide (Ki = 9×10−6 M) [25,26] is tight enough to give good binding, but not so tight as to make it impossible to release the bound butyrylcholinesterase. The procainamide affinity column does not purify butyrylcholinesterase in a single step. Two additional chromatography steps are required before the butyrylcholinesterase is pure.
Protocols with multiple chromatography steps are not ideal for sample preparation for mass spectrometry where the goal is to identify the OP-labeled peptide in a small clinical sample. What is needed is a one-step purification method. A one-step purification on procainamide-Sepharose is inadequate for identification of the OP-labeled active site tryptic peptide of butyrylcholinesterase, because this affinity gel does not yield highly purified butyrylcholinesterase. Ion suppression by contaminating peptides prevents the OP-labeled peptide from ionizing in the mass spectrometer. A more useful affinity column would bind butyrylcholinesterase with such high affinity that almost all contaminating proteins could be washed off. It would not be necessary to elute the butyrylcholinesterase from the affinity column because the bound butyrylcholinesterase could be digested with trypsin using protocols similar to those used to digest proteins embedded in acrylamide gel slices. Beads that do not dissolve in 50% acetonitrile would be optimal, because it would allow extraction of peptides with 50% acetonitrile.
Structure of the butyrylcholinesterase tetramer
The butyrylcholinesterase tetramer consists of 4 identical subunits, each containing 574 amino acids. The C-terminal 40 amino acids comprise the tetramerization domain. Assembly of butyrylcholinesterase subunits into a tetramer requires a polyproline rich peptide derived from lamellipodin [27]. A crystal of the native butyrylcholinesterase tetramer has not yet been obtained. Crystallization is difficult because of the flexible tetramerization domain which protrudes from the tetramer like a stem from a flower. A model of the butyrylcholinesterase tetramer including its tetramerization domain interleaved with a polyproline-rich peptide has been constructed and is shown in Figure 2. The model is supported by biochemical data that indicate the tetramerization domain can be cleaved off with proteases without affecting the catalytic properties of the enzyme. Knowledge of the tetramer organization is important: a) it will confirm the presence of a polyproline peptide within the tetramerization domain; b) it will reveal the relationship of the tetramerization domain to the subunit assembly; c) it will provide information about the structure of membrane-anchored butyrylcholinesterase; d) it will reveal the relative orientation of subunits in the tetramer and might tell whether cooperativity exists between subunits, e) it will help in design of butyrylcholinesterase-based bioscavengers with longer residence times in the circulation;
Figure 2.
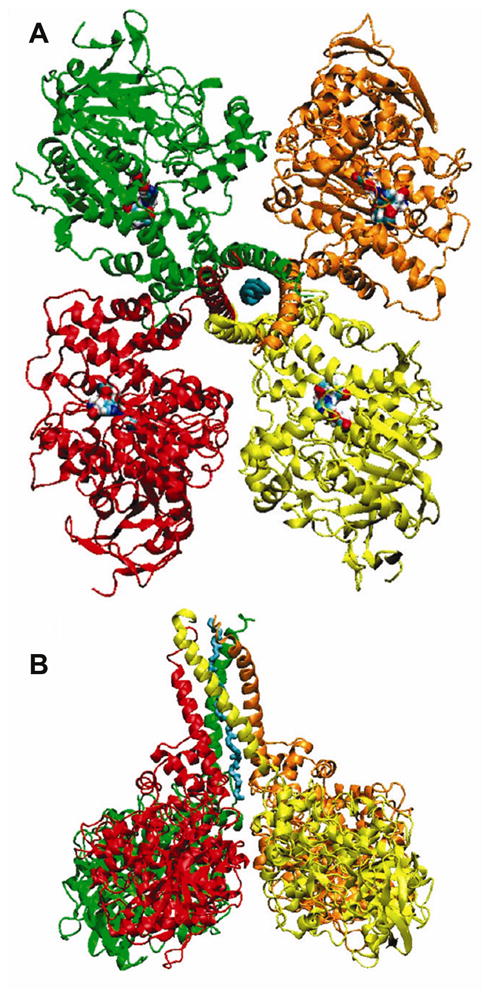
Model of the human butyrylcholinesterase tetramer. Four subunits assemble through the tetramerization domain at the C-terminus. The tetramerization domain has four parallel alpha helices wrapped around a single antiparallel polyproline helix. The polyproline peptide derives from lamellipodin. The tetramer is a dimer of dimers. Dimers have an interchain disulfide bond at Cys571. A) Viewed from the top, with the tetramerization domain and the polyproline peptide in the center. Two of the active sites are exposed to solvent, while two face the central cavity of the butyrylcholinesterase tetramer. B) Viewed from the side. Reproduced from [185].
Catalytic issues
Site-directed mutagenesis studies, the crystal structure of butyrylcholinesterase and molecular dynamic studies support the formal minimum mechanistic model (Scheme 1) proposed from steady-state kinetic analysis of cholinesterases [28]. This minimum model implies a conformational link between residues located at the rim of the gorge, the peripheral anionic site, and the active center located at the bottom of the gorge. Conformational linkage occurs through motion of the omega loop that connects Asp70, the main component of the peripheral anionic site, and Trp82, the main component of the “choline”-binding site in the catalytic center.
Scheme 1.

The non-Michaelian behavior of butyrylcholinesterase with excess positively charged substrates (b<1 or b>1) results from the binding of a second substrate molecule (Sp) on the peripheral anionic site. Neutral substrates in excess do not bind to the peripheral anionic site, thus butyrylcholinesterase follows the Michaelis-Menten model (b =1) with neutral substrates. For most esters, the acylation and deacylation steps are partially rate limiting, so that the turnover number, kcat, is determined by the magnitude of both rate constants (kcat =k2k3/(k2+k3)). The charge of the substrate is important for binding. The Km values for positively charged esters are of the order of microM, while for neutral and negatively charged esters, Km values are 3 orders of magnitude higher. After substrate is bound, electric charge is not the determining factor of catalytic efficiency. A negatively charged substrate like aspirin (kcat=6 200 min−1) is hydrolyzed at a rate only 2 times lower than the positively charged acetylcholine (13 000 min−1) and 8 times lower than its related neutral ester phenylacetate (32 000 min−1). Because the active site pocket of butyrylcholinesterase is larger than that of acetylcholinesterase (≈500 Å3 against 300Å3) [29], butyrylcholinesterase is less specific and can accommodate bulkier substrates. The catalytic efficiency of butyrylcholinesterase to hydrolyze esters is in fact mostly dependent on the size and enantiomeric configuration of the acyl or alcohol moieties. For instance, kcat for (+)cocaine is 7500 min−1 while it is 3.9 min−1 for (−)cocaine [30]. The substrate slides down the gorge to form a first enzyme-substrate complex, ES1, that is non productive. Then ES1 rotates to a position favorable for nucleophilic attack by Ser198. This second complex, ES2, is the productive Michaelis complex [23,31].
The promiscuity of butyrylcholinesterase and its hysteretic behavior with certain substrates raise interesting questions related to putative catalytic functions.
Aryl acylamidase activity
Like acetylcholinesterase, butyrylcholinesterase displays a genuine aryl acylamidase activity with o-nitroacetanilide, by hydrolyzing an amide bond [32]. See Figure 3. This activity reflects the conformational plasticity of the active site to adjust to different types of substrates [33]. Clear evidence has been provided that the aryl acylamidase activity of butyrylcholinesterase is fully explained by its single active site serine (Ser198) [34,35]. Echothiophate completely inhibits both aryl acylamidase and esterase activities. Titration with echothiophate shows that echothiophate inhibits both aryl acylamidase and esterase activities with the same bimolecular rate constant. Plasma from a silent butyrylcholinesterase genetic variant with a frame shift at amino acid 117 (FS117) has neither aryl acylamidase nor esterase activity. Mutation of the active site serine to cysteine or aspartate yields butyrylcholinesterase with neither aryl acylamidase nor esterase activity. It is concluded that Ser198 is the active site for both aryl acylamidase and esterase activity.
Figure 3.

Hydrolysis of o-nitroacetanilide by acetylcholinesterase and butyrylcholinesterase.
Study of highly purified monomers and tetramers of butyrylcholinesterase showed that the aryl acylamidase activity of butyrylcholinesterase does not depend on quaternary structure [34]. Yet, it was reported that enzyme molecular forms isolated by sucrose gradient ultracentrifugation differ in aryl acylamidase activity, the monomer being more active than the tetramer [36,37]. The discrepancy could be explained by contamination of butyrylcholinesterase monomer fractions by tissue-specific carboxylesterase-amidase isozymes in gut and kidney [38,39] or albumin since carboxylesterase-amidases and albumin have aryl acylamidase activity [40].
The question of the possible physiological significance of the aryl acylamidase activity of cholinesterases has repeatedly been addressed [32,41,42]. With the exception of melatonin, no endogenous aryl acylamide compound has been isolated so far, and it should be noted that melatonin is not hydrolyzed by cholinesterases (Masson and Froment, unpublished result). Thus, the hypothetical function of butyrylcholinesterase and acetylcholinesterase in the metabolism of putative endogenous aryl acylamide substrates remains to be demonstrated.
Moreover, we should point out that the aryl acylamidase activity of butyrylcholinesterase is very low, with acylation as the rate-limiting step (k2≪k3), and that the Km with neutral aryl acylamide substrates is high. Therefore, under Michaelis-Menten conditions (enzyme concentration much less than substrate concentration), hydrolysis of aryl acylamide substrates at non-lethal concentrations is pseudo-first order. The neutral aryl acylamides, propanilil (fungicide), acetaminophen and phenacetin (drugs), and the negatively charged aryl acylamide, N-acetylanthranilic acid (drug precursor), are not hydrolyzed by butyrylcholinesterase under these conditions. These results suggest that it is unlikely that plasma butyrylcholinesterase plays a role in the metabolism of exogenous aryl-acylamide drugs and xenobiotics [34,35].
Hysteretic behavior
The establishment of a steady state for butyrylcholinesterase-catalyzed hydrolysis of certain substrates is preceded by a long induction phase. This phenomenon was first described with N-methylindoxyl acetate as the substrate where a lag of several minutes is needed to reach steady state [43]. Actually, “chaotic” lags preceding steady state have been observed for decades by investigators using benzoylcholine as the substrate, but the nature of this phenomenon had not been investigated. Investigators using benzoylcholine just recorded hydrolysis kinetics after establishment of “equilibrium”. Kinetic analysis of benzoylcholine hydrolysis showed that the pre-steady state phase can be described by damped oscillations that superimpose on a mono-exponential lag [44]. The dependence of induction time on substrate concentration was interpreted according to hysteretic models developed by Frieden [45]. Accordingly, the enzyme exists in two interconvertible states, E and E′ in slow equilibrium (Scheme 2). Substrate can bind on E and/or on E′. Depending on the affinity of E and E′ for S, (KS and K’S are dissociation constants of enzyme-substrate complexes), and whether ES and E’S make products (P) or not, induction time will be a lag or a burst. With N-methylindoxyl acetate, only E′ binds S and makes products, while with benzoylcholine, E and E′ bind S, but only E’S makes products. With both substrates induction times are lags. With an anilide, 3-(acetamido) N,N,N-trimethylanilinium both enzyme states bind substrate and make products, but K’S<KS and E’S is catalytically less active than ES. In that case, a burst is observed [34,46]. Damped oscillations with benzoylcholine and long-alkyl chain homologues of benzoylcholine [44,47] were the result of an additional time-dependent constraint: the slow formation of a suitable substrate molecule due to multiple slow equilibria between different forms of the substrate (conformers, oligomers and micelles) [46].
Scheme 2.

The molecular mechanism for the hysteretic behavior of butyrylcholinesterase and acetylcholinesterase [46,48,49] is still unkown. Site-directed mutagenesis, pH-dependence studies, and occurrence of hysteresis with both cholinesterases suggest that hysteresis originates in the function of the catalytic triad. Effects of temperature, hydrostatic pressure, organic solvents and lyotropic salts indicate that hysteresis is related to enzyme hydration change. Water in the active site gorge of enzyme form E′ appears to be more structured than in the gorge of the unprimed form E. A preliminary kinetic crystallography study indicates that water structure change in the gorge may induce a flip in the position of the catalytic histidine (Colletier, unpublished work).
The physiological relevance of the hysteretic behavior of the cholinesterases is questionable because acetylcholinesterase-catalyzed hydrolysis of acetylcholine does not require hysteresis. However, affinity and reaction with inhibitors may differ between E and E′. Thus, it can be hypothesized that hysteresis could act as a “retarder” mechanism in natural defenses against toxicity of anticholinesterase agents. In fact, ligand binding and inhibition studies provided evidence for slow isomerization between two cholinesterase forms differing in affinity for ligands of the peripheral site [50,51]. Also, biphasic inhibition kinetics of butyrylcholinesterase by the carbamylating agent N-methyl-N-(2-nitrophenyl)carbamoyl chloride was explained by a mechanism in which both E and E′ forms have the same affinity for the inhibitor but are inactivated at different rates (k’carb=10 kcarb) [52]. Therefore, if reaction with certain irreversible inhibitors is faster for E′ than for E as in slow-binding inhibition of type c [53], then it may be hypothesized that hysteresis could play a protective role in damping the cholinergic response to these agents.
Inhibition by OP and aging of the inhibited enzyme
Phosphylation of cholinesterases
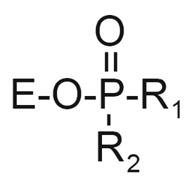
Organophosphorus compounds are progressive irreversible inhibitors of acetylcholinesterase and butyrylcholinesterase. After stereoselective formation of the enzyme-OP complex, the OP makes a covalent bond with the catalytic serine and simultaneously releases the leaving group (Figure 4). The stereochemistry of the groups attached to phosphorus is inverted. The catalytic histidine remains protonated, so that water-mediated dephosphylation is very slow or zero. After aging (dealkylation of the organophosphate), the positively charged catalytic histidine stabilizes the negatively charged aged OP adduct. Bimolecular rate constants for phosphylation of cholinesterases are 107 to 1010 M−1min−1. Fast phosphylation of acetylcholinesterase fully explains the acute lethal toxicity of OP [54]. However, the sub-lethal toxicity of OP with no inhibition of acetylcholinesterase, but with chronic effects can be explained by inhibition of other enzymes [15] or phosphylation of other protein targets.
Figure 4.

Reaction of butyrylcholinesterase and acetylcholinesterase with OP. 1) formation of reversible complex; 2) phosphylation of the active site serine, with departure of the leaving group X and inversion of the stereochemistry of the OP; 3) reactivation of phosphylated enzyme by oximes or by 2 M potassium fluoride; 4) aging to produce a negatively charged OP stabilized by interaction with the positively charged histidine of the catalytic triad. Inset. When the OP is soman, R1 is pinacolyl alcohol. Dealkylation does not yield pinacolyl alcohol, but instead yields 3 rearranged products [75].
Reactivation of phosphylated cholinesterases
The term “phosphylated” denotes both phosphorylation and phosphonylation. Phosphylated cholinesterases can be reactivated by nucleophilic compounds such as fluorides, hydroxamates and oximates. The pyridinium oximes 2-PAM and obidoxime (toxogonin) are currently used as antidotes to OP poisoning. The more potent oximes HI-6 and MMB-4 are under advanced development. The structures of oximes are in Figure 5. The efficacy of oximes to reactivate phosphylated cholinesterases depends on the type of enzyme, and the chemical structure of the organophosphorus agent bound to the catalytic serine. Phosphylated human butyrylcholinesterase is reactivated less efficiently than phosphylated human acetylcholinesterase. Cholinesterases inhibited by tabun are resistant to most oximes at concentrations compatible with medical use [55]. Hydroxamates and fluoride ions release OP in vitro at conditions (2 M potassium fluoride pH 4) incompatible for therapeutic use in vivo, but well suited for biomonitoring of exposure [56].
Figure 5.

Oxime structures
Aging of OP-inhibited cholinesterases
Reactivation of phosphylated enzymes is complicated by post-inhibitory reactions. Phosphylated cholinesterases as well as the serine proteases trypsin and chymotrypsin [57] may become progressively resistant to reactivators. On the other hand, phosphylated serine enzymes like carboxylesterases [58] and phospholipase A2[59] can be reactivated. Carboxylesterase spontaneously reactivates.
Progressive loss of reactivatability of phosphylated enzymes is called “aging”. Aging is the functional consequence of unimolecular dealkylation of the OP adduct. Aging is catalyzed by residues in the active site, His 438 and Glu 197 [36]. The rate of aging depends on both the enzyme structure and the OP structure. The rate of aging is modulated by temperature, pH, and ligand binding. Aging can be halted by denaturing the protein. The half time of aging ranges from a few minutes to several days (Table 1). Treatment of OP poisoning may be dramatically impaired when there is rapid aging. In particular, in the case of poisoning by soman, aging of phosphonylated acetylcholinesterase is so fast that treatment with a potent oxime like HI-6 is ineffective. In the case of poisoning by sarin, which has a slow aging rate, treatment by pralidoxime (2-PAM) effectively reactivates acetylcholinesterase as was observed in Tokyo casualties after release of sarin in the subway [60].
Table 1.
Half-time of aging for human acetylcholinesterase and human butyrylcholinesterase inhibited by organophosphorus compounds
 | ||||||
|---|---|---|---|---|---|---|
| Human AChE | ||||||
| OP | R1 | R2 | t (°C) | pH | t1/2 | Reference |
| soman * | (CH3)3C(CH3)CHO | CH3 | 27 | 8.0 | 6.3 min | [61] |
| sarin | (CH3)2CHO | CH3 | 37 | 8.0 | 3.9 h | [62] |
| cyclosarin | cyclohexyl-O | CH3 | 37 | 7.4 | 8.7 h | [63] |
| tabun | (CH3)2N | CH3CH2O | 37 | 7.4 | 13.6 h | [64] |
| DFP | (CH3)2CHO | (CH3)2CHO | 37 | 7.8 | 4.4 h | [65] |
| paraoxon-ethyl | CH3CH2O | CH3CH2O | 37 | 41 h | [63] | |
| paraoxon-methyl | CH3O | CH3O | 37 | 7.4 | 3.7 h | [66] |
| Human BChE | ||||||
| OP | R1 | R2 | t (°C) | pH | t1/2 | |
| soman* | (CH3)3C(CH3)CHO | CH3 | 25 | 8.0 | 9 min | [67] |
| sarin | (CH3)2CHO | CH3 | 37 | 7.4 | 5.8–6.4 h | [64] |
| cyclosarin | cyclohexyl-O | CH3 | 37 | 7.4 | 2.2 h | [63] |
| tabun | (CH3)2N | CH3CH2O | 37 | 7.4 | 6.1–6.4 h | [64] |
| DFP | (CH3)2CHO | (CH3)2CHO | 37 | 7.8 | 28 min | [65] |
| paraoxon-ethyl | CH3CH2O | CH3CH2O | 20 | 8 | 11.6 h | [63] |
| paraoxon-methyl | CH3O | CH3O | 37 | 7.4 | 3.9 h | [66] |
racemic soman
Beta-elimination to form dehydroalanine
When the pH of a solution of phosphylated butyrylcholinesterase is raised to 11 the organophosphorus agent and the active site serine are both lost during beta-elimination (Figure 6). As a result the active site serine is converted to dehydroalanine. Both aged and nonaged phosphylated butyrylcholinesterase undergo beta-elimination. The dehydroalanine peptide is detectable by mass spectrometry. For human butyrylcholinesterase, the masses of the tryptic peptides are 2910.5 m/z for the dehydroalanine form of the active site peptide, 2928.5 for the unlabeled active site peptide, 3006.5 for aged sarin, soman, and cyclosarin peptide, and 3090.5 for the soman-labeled peptide before aging. Dehydroalanine also forms at pH 7.4 though the rate of formation is much slower than at pH 11.
Figure 6.

Beta-elimination. OP-labeled serine loses the OP and a molecule of water in the beta-elimination reaction. The active site serine is converted to dehydroalanine.
Another way to generate dehydroalanine is to bombard the phosphylated peptide with high energy during acquisition of MS/MS fragmentation data [68]. The dehydroalanine form of the peptide is among the most intense peaks in the MS/MS spectrum indicating that beta-elimination occurs more easily than fragmentation of the backbone bonds in the peptide. OP-inhibited butyrylcholinesterase undergoes beta-elimination regardless of the structure of the OP.
The curious mass spectrometry observation is that a dehydroalanine peptide is also observed in MS spectra. A peak at 2910.5 m/z is observed in MS spectra acquired on the MALDI-TOF mass spectrometer (Figure 7). 2910.5 is the mass of the dehydroalanine form of the human butyrylcholinesterase tryptic peptide. MS spectra acquired on the QTRAP tandem quadrupole ion trap mass spectrometer also have the 2910.5 peptide. Fragmentation shows that the sequence of the 2910.5 peptide is SVTLFGEΔAGAASVSLHLLSPGSHSLFTR where the symbol Δ represents dehydroalanine. The energy for acquisition of MS spectra is low so that the energy of ionization is not likely to be the cause of beta-elimination. The beta-elimination could have occurred during sample preparation or it could reflect spontaneous formation of dehydroalanine under mild conditions. It is not known whether dehydroalanine forms spontaneously under physiological conditions from OP-inhibited butyrylcholinesterase and OP-inhibited acetylcholinesterase. Dehydroalanine forms of the cholinesterases cannot be reactivated by treatment with oximes.
Figure 7.

MS spectrum of tryptic digest of soman-labeled human butyrylcholinesterase. Peptides were separated by HPLC and 1 ml fractions collected. A 0.5 microliter aliquot of each fraction was analyzed in the MALDI-TOF mass spectrometer. Panel A shows that fraction 35 contains the aged soman-labeled peptide at 3006.5 m/z and a small amount of unlabeled peptide at 2928.5 m/z. Panel B shows that fraction 36 contains the dehydroalanine form of the peptide at 2910.5 m/z, unlabeled peptide at 2928.5 m/z, and a small amount of aged-soman labeled peptide at 3006.5 m/z. The active site serine of human butyrylcholinesterase is Ser198 (accession # gi:116353).
Gaps in knowledge of rate of beta-elimination to make dehydroalanine
It is not certain that phosphylated cholinesterases undergo beta-elimination under physiological conditions. The possibility that artifacts are introduced during preparation of samples for analysis in the mass spectrometer needs to be ruled out. It is not known whether a substantial amount of dehydroalanine forms in vivo. The rate of formation of dehydroalanine under physiological conditions has not yet been determined. It is assumed that phosphylated acetylcholinesterase undergoes the same beta-elimination as phosphylated butyrylcholinesterase, but this has not been demonstrated. Since the dehydroalanine form of cholinesterases cannot be reactivated, it is important to know how much of an aged acetylcholinesterase preparation is in the dehydroalanine form when attempting to evaluate oxime drugs for their reactivation efficacy.
Mechanism of dealkylation of phosphylated cholinesterase
Dealkylation of phosphylated cholinesterase is an SN1 reaction. The reaction has been thoroughly investigated for aging of soman-inhibited cholinesterase where it involves a carbocation-like transition state and then scission of the P-O-C chain to form a carbocation on the pinacolyl chain. For organophosphates like echothiophate and DFP, and organophosphonates like sarin and soman, phosphylation and aging of human butyrylcholinesterase were carried out in 18O-water and compared to aging in 16O-water. Analysis of tryptic peptides by MALDI-TOF mass spectrometry provided evidence that dealkylation occurs by breaking of the O-C bond and not the P-O bond [69]. For the phosphoramidate, tabun, mass spectrometry and x-ray structure analyses provided evidence that aging of cholinesterase occurs through O-dealkylation, not N-dealkylation [70]. For the phosphorodithioate, isomalathion, aging is more complex, involving O-C, P-S and S-C cleavages, depending on the enantiomer that reacted with the enzyme [69].
Chemical modification of histidine by diethylpyrocarbonate implicated the active site histidine in aging [71]. Studies of pH dependence and site-directed mutagenesis provided evidence for participation of the catalytic histidine and the glutamate vicinal to the catalytic serine in the mechanism of dealkylation [72]. Other residues in the active center of cholinesterase have been found to play a role in the aging process. In particular, tryptophan (W82 in human butyrylcholinesterase), the main component of the choline-binding pocket, has been found to stabilize the developing carbocationic chain that will be released [61].
Identification of products released by dealkylation of the pinacolyl chain of soman bound to acetylcholinesterase [73–75] as well as pH profiles and solvent isotope effects support a “push-pull” mechanism for aging in which tryptophan (W82 in human butyrylcholinesterase) and glutamate (E197 in human butyrylcholinesterase) exert electrostatic and steric “push”, and histidine (H438 in human butyrylcholinesterase) and the oxyanion hole act as “pullers” [76,77]. This mechanism involves migration of methyl from Cβ to Cα in the pinacolyl chain in the dealkylation transition state. Another proposed mechanism supported by mutagenesis and structural data involves protonation of the pinacoloxy oxygen by the protonated catalytic histidine, and then scission of the O-C bond [61]. More recently, the high-resolution crystal structure of non-aged and aged soman-acetylcholinesterase conjugates led to a critical reexamination of both models, highlighting the role of a conserved water molecule in dealkylation [78].
Dealkylation is accompanied by formation of a salt bridge between the negatively charged P-O− and the protonated catalytic histidine [79,80]. Crystal structures of aged butyrylcholinesterase (Figure 8) and other serine hydrolases confirm the existence of this strong salt bridge in the active center of aged conjugates [57,70,78,81].
Figure 8.
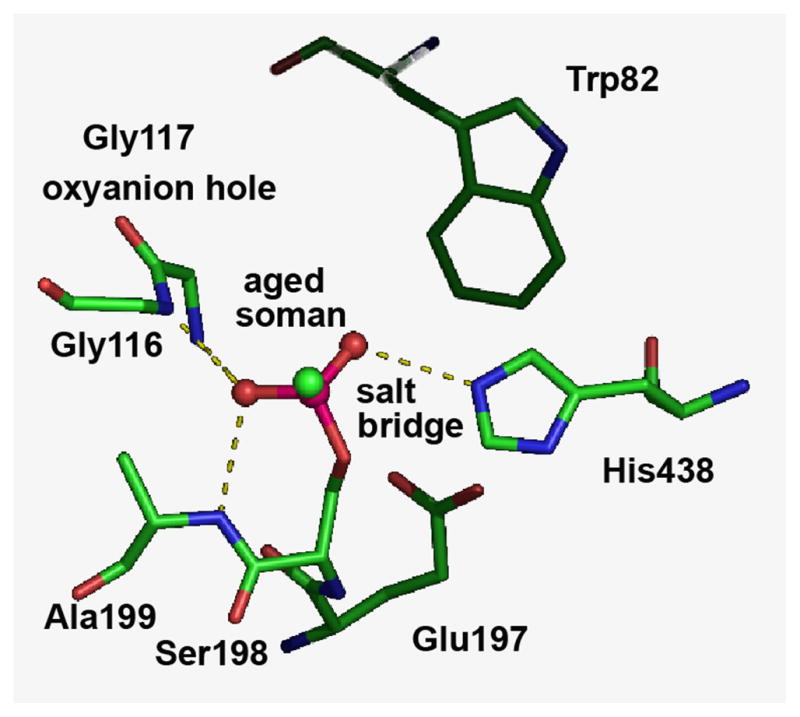
Salt bridge between the negatively charged aged soman bound to Ser198 and the positively charged catalytic His438 of human butyrylcholinesterase. The crystal structure is in pdb code 1p0q [81].
Consequence of aging on cholinesterase structure
Fluorescence decay of pyrenebutyl-containing organophosphates bound to acetylcholinesterase was found to have a lower quantum yield for non-aged conjugates than for aged ones [82]. This suggested that the pyrenebutyl group in aged conjugates is more deeply buried than in non-aged conjugates. Later, kinetic and equilibrium studies showed alteration in binding of peripheral anionic site ligands in aged acetylcholinesterase compared to non-aged acetylcholinesterase [83]. This was interpreted to be the result of a change in the topographic relationships between the active center and the peripheral anionic site.
Cross-linking of butyrylcholinesterase with dimethylimidates of different chain length showed that upon aging there was no change in the quaternary structure of butyrylcholinesterase or in the overall conformation of subunits [26]. However, enzyme denaturation studies showed that the conformational stability of aged cholinesterase is dramatically increased compared to non-inhibited and non-aged enzymes [84–87]. See Figure 9.
Figure 9.
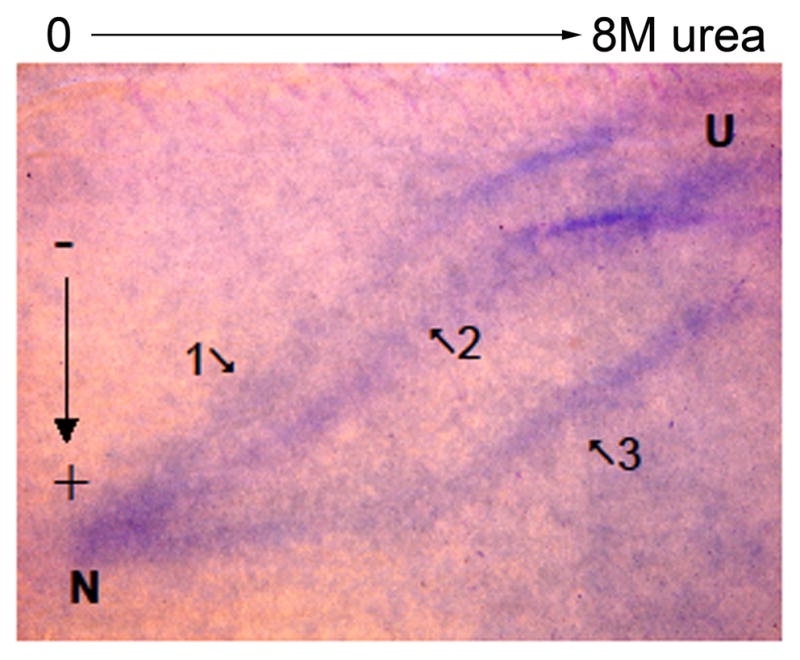
Soman-inhibited butyrylcholinesterase tetramer is resistant to unfolding. A mixture of native unlabeled butyrylcholinesterase, nonaged soman-butyrylcholinesterase, and aged soman-butyrylcholinesterase, 0.5 mg butyrylcholinesterase protein total, was layered on a polyacrylamide gel containing a 0–8 M transverse gradient of urea. After electrophoresis the gel was stained with Coomassie blue. Three well-separated curves of butyrylcholinesterase protein are seen. Curve 1) not labeled butyrylcholinesterase tetramer swells into an unfolded structure as the urea concentration increases from 0 to 8 M. The unfolded butyrylcholinesterase migrates more slowly through the gel. The urea concentration at the mid-point for unfolding is 3.5 M. 2) Nonaged soman-phosphylated butyrylcholinesterase begins to unfold at a higher urea concentration. 3) Aged soman-butyrylcholinesterase retains its folded structure up to 4 M urea and has a midpoint for unfolding at 6 M urea. N indicates the migration of normal folded tetrameric butyrylcholinesterase in the absence of urea. U indicates migration of unfolded butyrylcholinesterase in 8 M urea. The direction of migration is from minus (−) to plus (+).
Neutron scattering studies as a function of temperature up to 90°C showed that the increase in stability of aged butyrylcholinesterase correlated to decrease in molecular flexibility [88]. Formation of a salt bridge and partial dehydration of the active site gorge upon dealkylation may explain the stability and molecular dynamic changes of aged butyrylcholinesterase.
Resistance to oxime reactivation of aged cholinesterases
Shortly after the discovery of aging, Hobbiger suggested that a new bond was formed between aged adduct and the active center, thus preventing reactivation [89]. It was proposed that the presence of the negatively charged oxygen in the adducted phosphorus atom formed an electrostatic shield opposing nucleophilic attack by negatively charged oximates [90]. In fact, studies of reactions between different acylmethylphosphonates and neutral or negatively charged nucleophiles showed that the presence of a negative charge slows down the rate of nucleophilic displacement at phosphorus due to the electrostatic effect, but does not prevent reaction between compounds [91–93]. Conformational rigidity of the aged enzymes cannot explain by itself the loss of reactivation. Moreover, the crystal structure of soman-aged acetylcholinesterase, compared to native or non-aged acetylcholinesterase, revealed only a slight displacement of the catalytic histidine upon formation of a salt bridge at the bottom of the gorge [78]. An explanation for the resistance to reactivation was provided by the crystal structure of the complex of soman-aged acetylcholinesterase and 2-PAM. The oximate function of 2-PAM points away from the phosphorus atom, in an orientation that does not permit nucleophilic attack on the phosphorus atom. This provides a new explanation for the failure of 2-PAM to reactivate aged-soman acetylcholinesterase.
In the case of aged phosphoramidate- inhibited cholinesterase, resistance to reactivators results from the conjunction of steric factors imposed by the salt bridge, and electron delocalization along the P-N-R chain due to the nitrogen doublet [70,94].
Perspectives
Modulation of aging velocity
Because reactivation of aged cholinesterase is not yet possible, medical counter-measures could be improved by using drugs capable of slowing the aging process. The D70G mutation on the peripheral anionic site of human butyrylcholinesterase was found to slow the aging process, and molecular dynamic simulations indicated that the peripheral anionic site and the catalytic binding site are coupled via conformational change of the omega loop that connects both sites [95]. Therefore, occupation of the peripheral anionic site may affect the rate of aging by altering interactions between the adduct and the active center residues. In fact, the effects of several peripheral anionic site ligands of acetylcholinesterase, i.e. bispyridinium compounds like SAD-128, HH54 [96–98], and gallamine [99], acting as allosteric effectors have been investigated. Other ligands have been found to slow down the aging of phosphylated cholinesterases, e.g. ketamine [100] and tacrine [101]. None of these compounds was found to be of practical interest for slowing down aging in vivo in association with other medical countermeasures. However, new generations of peripheral anionic site and bifunctional ligands are emerging. Screening of libraries of tethered ligands and cyclic peptides [102], and bifunctional ligands produced by click chemistry [103] may be a strategy for discovery of such compounds. Alternatively, a computer-aided approach based on flexible docking in the crystal structure of cholinesterase appears to be a very promising strategy [104].
Is reactivation of aged cholinesterase possible?
Reactivation of aged cholinesterase is a challenge. Solving this major issue would greatly improve medical counter measures, in particular against acute poisoning by soman. Direct reactivation of aged conjugates using oximes has not been possible so far. Yet hundreds of oximes have been synthesized. However, most oximes have been designed without knowledge of the three-dimensional structure and molecular dynamics of phosphylated enzymes. Very few structures of complexes between oximes and phosphylated cholinesterase have been reported so far. The three-dimensional structure of complexes such as that of the non-aged tabun-acetylcholinesterase conjugate with the oxime Hlö-7 explains why reactivation of cholinesterase inhibited by tabun is so difficult [105]. Recent work of Sanson [78] provides a rationale to understand why non-aged soman-acetylcholinesterase is not reactivatable by 2-PAM, and why 2-PAM cannot reactivate the aged enzyme. Therefore, lessons from crystallography and molecular dynamics are expected to provide information for improving the orientation of the oxime function for effective attack on phosphorus. In silico design of new oximes, as well as other chemical strategies mentioned above, including click chemistry and combinational chemistry, should lead to a new generation of reactivators. Another strategy would be to realkylate the aged adduct by using an electrophilic molecule, and then to displace the phosphotriester by a nucleophilic compound.
Aging-resistant cholinesterase mutants as potential pseudocatalytic bioscavengers
Bioscavengers are proteins that neutralize nerve agents before they damage the nervous system. Bioscavenger-based cholinesterase is an alternative to prophylaxis against organophosphate poisoning [1], and administration of bioscavengers may improve efficacy of treatment of acute nerve agent poisoning. However, cholinesterases act as stoichiometric scavengers, so that efficacy of cholinesterase in OP poisoned humans needs administration of 200–300 mg of costly enzymes. Efforts to convert cholinesterases into efficient catalytic bioscavengers have not been successful so far [106]. Thus, selected mutants of cholinesterase non susceptible to aging, i.e, reactivatable, could be used in association with oximes as pseudo-catalytic bioscavengers. Several attempts have been made [107–109], and this approach looks promising.
Butyrylcholinesterase as a bioscavenger for protection against OP
OP poisoning is a major public-health concern. According to the World Health Organization, OP self-poisoning is responsible for 200 000 deaths a year in the world [110]. Though 188 countries are now part of the Chemical Weapons Convention, OP nerve agents still represent military and terrorist threats. The pharmacological approach for prophylaxis and treatment of OP poisoning has reached its limit [55,111–115]. Thus, alternative approaches have been considered. In particular, acute toxicity of OP can be countered by lowering OP concentration in the blood compartment. This prevents the transfer of OP molecules to physiological targets. The bioscavenger approach is based on the concept of inactivation of OP molecules in the blood stream before they reach their central and peripheral neuronal and neuromuscular targets. The importance of endogenous esterases in inactivation of poisonous esters has been recognized. Multiple cellular and plasma enzymes constitute barriers that play a role in natural defenses against toxicants. The presence of OP detoxifying enzymes in skin contributes to reduction of the amount of OP that penetrates into the body [116]. Tissue carboxylesterases react with OP, and participate in protection [58,117]. Certain secondary targets of OP also play a detoxifying role [118–120]. Finally, blood esterases significantly contribute to detoxication of OP molecules. Unlike plasma of most mammalians, there are no carboxylesterases in human plasma [121]. However, human plasma contains two enzymes capable of degrading poisonous esters: paraoxonase (PON1; EC 3.1.8.1) that displays arylesterase, lactonase and phosphotriesterase activities, and butyrylcholinesterase that hydrolyzes numerous poisonous esters and reacts stoichiometrically with OP. The concentration of paraoxonase in human serum is 500 nM, and its catalytic efficiency with OP is about 105 M−1 min−1. Albumin reacts with OP, but its reactivity is very slow compared to that of butyrylcholinesterase [122]. Though the concentration of butyrylcholinesterase in human serum is about 50 nM, its apparent second-order rate constant with OP is high ≈107 to 109 M−1 min−1. Thus, butyrylcholinesterase is the most significant stoichiometric OP scavenger in human plasma.
In the past decade, fast inactivation of OP in blood by pretreatment of animals with pure human butyrylcholinesterase has proven to be safe and efficient for protection against nerve agents [1]. In 2006, plasma-derived human butyrylcholinesterase was registered as an Investigational New Drug by the FDA [123,124]. In 2009 a phase I clinical trial on healthy volunteers (NCT00333528) was completed by Baxter Healthcare Corporation and butyrylcholinesterase became the first stoichiometric bioscavenger for prophylaxis of organophosphorus poisoning. It is estimated that protection from a dose of nerve agent lethal to 50% of the population requires 200–300 mg of butyrylcholinesterase per adult. Multiple 200–300 mg of butyrylcholinesterase are needed for human protection against higher doses of nerve agents [125]. Natural tetrameric butyrylcholinesterase purified from human plasma [21] and recombinant human butyrylcholinesterase (Protexia ™) from the milk of transgenic goat [126,127] are costly GMP-produced enzymes. Thus, it is important to develop new therapeutics that catalyze the destruction of OP. New mutants of human butyrylcholinesterase, paraoxonase, or phospholipase A2 [59] capable of degrading OP with a high turnover, would greatly reduce the cost of treatment and improve the bioscavenger approach against OP.
Conversion of butyrylcholinesterase into an OP-hydrolyzing enzyme
OP are hemi-substrates of butyrylcholinesterase and acetylcholinesterase. When butyrylcholinesterase reacts with carboxyl-esters, the acyl group is rapidly displaced from the planar acyl-enzyme intermediate by a water molecule. In contrast, the reaction with phosphyl-esters yields a stable tetrahedral adduct that restricts accessibility of water to the phosphorus atom and impairs proton transfer from the protonated catalytic histidine (H438) to the substrate [77]. Thus, spontaneous hydrolysis of phosphylated enzyme is very slow or even impossible. Jarv postulated that introduction of a second nucleophile in the active center could activate a water molecule. This water molecule could then attack the phosphorus atom on the back face, causing the P-serine bond to break [128]. Resolution of the three-dimensional structure of Torpedo californica acetylcholinesterase [129] opened the way to rational re-design of cholinesterases to make enzymes capable of hydrolyzing OP. Human butyrylcholinesterase was selected because its active center is larger (500 Å3) than that of acetylcholinesterase (300 Å3) and it is less stereospecific. A molecular model of human butyrylcholinesterase based on the crystal structure of Torpedo acetylcholinesterase was made for the design of butyrylcholinesterase mutants. The second nucleophile was created in the oxyanion hole where a glycine residue was replaced by a histidine. The first mutant, G117H, was capable of hydrolyzing paraoxon, DFP, sarin, echothiophate and VX at a slow rate [130,131]. Interestingly, the mutation G117H is in a position homologous to that of the carboxylesterase mutant, G137D, from a blowfly (Lucilia cuprina) resistant to OP. Though the OP hydrolase activity of the G137D carboxylesterase is very low, it is balanced by the abundance of the enzyme in insect organs [132]. The x-ray structure of the carboxylesterase has recently been solved to 2.5 A resolution [133]. The G117H mutant of butyrylcholinesterase, did not self-reactivate with soman because dealkylation of adduct (aging) was faster than dephosphonylation. Therefore, a second mutation was made on the glutamate residue important for the aging reaction: Glu197Gln. As expected, the double mutant G117H/E197Q was capable of hydrolyzing soman because the rate of aging was considerably reduced [134]. However, the catalytic activity of this double mutant was too slow to be of pharmacological interest.
More than 60 double and triple mutants of human butyrylcholinesterase with mutated Gly117 [135] and mutants of human acetylcholinesterase and Bungarus fasciatus acetylcholinesterase were made, using the same rationale [136]. Unfortunately, none of the mutated cholinesterases was more active than the first mutant, G117H. For a review see [106].
Strategies for designing new OP scavengers
Enzymes that hydrolyze or oxidize OP are of potential interest for detection, decontamination, protection and treatment of OP poisoning [137–139]. Among them, phosphotriesterases from bacteria (OP hydrolase), squid (DFPase) and human plasma (paraoxonase) are the most promising, but they are enantioselective, and pose immunological and pharmacotechnological problems. Because of the large variety of OP molecules and enantiomeric preference of cholinesterases and OP hydrolases, it will be impossible to make a universal mutant for protection against all OP. Rather, multiple mutated OP-degrading enzymes, differing in specificity, should be combined to make effective catalytic bioscavengers for prophylaxis and treatment of acute OP poisoning. Mixtures of effective enzymes against a large spectrum of OP could also be incorporated into active topical skin protectants, decontamination solutions and other protection equipment.
The considerable body of data obtained with human butyrylcholinesterase as the first bioscavenger, encourages further research for designing butyrylcholinesterase mutants capable of degrading nerve agents and pesticides [106]. However, new strategies for designing these enzymes have to be implemented. Computational design of mutants also called “intelligent” site-directed mutagenesis is the most promising strategy. Directed evolution of butyrylcholinesterase could be an alternative to computer-based methods. A recent study on the blowfly carboxylesterase mutant G137D showed that directed evolution of this enzyme led to 400-fold increase in OP hydrolase activity in 3 generations [133]. The drawback of directed evolution is that it requires expression in bacteria or yeast. However, expression of functional cholinesterases is difficult in yeast and has failed in bacteria so far. Such approaches in combination with chemical modifications and medium manipulations have been successfully used to improve properties of selected enzymes [140].
Basic requirements for operational butyrylcholinesterase-based catalytic bioscavengers
The concentration of toxic OP molecules in blood, [OP], even in the most severe cases of poisoning, is always very low (<11 nM), well below the Km of OP hydrolyzing enzymes [141]. Enzyme-catalyzed hydrolysis of OP in blood is a pseudo first-order process described by the equation v = kcat/Km·[E]·[OP]. The product of the bimolecular rate constant (kcat/Km) and enzyme active site concentration ([E]) is the pseudo first-order rate constant. Thus, the higher the catalytic efficiency (kcat/Km), the lower the dose of enzyme required to clean the blood of toxic molecules in a short time. Given that the molecular weight of butyrylcholinesterase per active site is 85 000, and taking the total volume of plasma as 3 liters per person, it can be calculated that an enzyme dose of 4.7 mg will reduce [OP] by 100-fold in 4.6 min if kcat/Km = 108 M−1 min−1. A dose of 47 mg enzyme will be needed if kcat/Km is 10 times lower. Therefore, the catalytic efficiency of operational mutants of butyrylcholinesterase needs to be increased by 3–4 orders of magnitude compared to the catalytic efficiency of G117H towards OP [130,136]. The success of strategies to increase the cocaine hydrolase activity of butyrylcholinesterase suggests that a butyrylcholinesterase mutant with improved OP hydrolase activity may be achieved.
Lessons from mutagenesis of butyrylcholinesterase as an efficient cocaine esterase
Plasma butyrylcholinesterase is the major detoxifying enzyme of cocaine in humans [142]. However, wild-type butyrylcholinesterase slowly hydrolyzes (−)-cocaine with a kcat/Km of about 2.8 M min−1, so that under normal conditions, a large part of an administered dose of cocaine reaches its biological targets and triggers toxic effects. Efficient mutants of human butyrylcholinesterase have been made that hydrolyze cocaine at a high rate. A first mutation, A328Y, enhanced kcat/Km 4-fold [143]. See Table 2. Then, molecular dynamics simulation and computer-based ligand docking led to the A328W/Y332A double mutant that displays a higher kcat/Km=8.6 M min−1 [144]. Using the three-dimensional x-ray structure of human butyrylcholinesterase [81], molecular dynamic simulations of the deacylation transition state led to the design of highly active mutants against (−)-cocaine. A butyrylcholinesterase with 4 mutations A199S/S287G/A328W/Y332G had a catalytic efficiency 1500–5000 fold greater than wild-type butyrylcholinesterase [145,146]. Introduction of 5 mutations, A199S/F227A/S287G/A328W/Y332G, led to an enzyme 6500-fold more active than wild-type butyrylcholinesterase [147]. Thus, this strategy led to a progressive increase in kcat/Km for (−)cocaine of more than 3 orders of magnitude in 5 steps. The most active mutant is of therapeutic interest [148]. Computational (transition state simulations, free energy barrier perturbation simulations) and mutagenesis approaches are expected to lead to even more efficient mutants [149]. Application of this computational design approach on dephosphylation transition states should lead to new mutants of butyrylcholinesterase displaying high OP hydrolase activity.
Table 2.
Cocaine esterase activity of mutants of human butyrylcholinesterase
| enzyme | kcat/Km, M−1 min−1 | fold- increase | reference |
|---|---|---|---|
| wild-type BChE | 2.8 × 105 | 1 | [143] |
| A332Y | 11.3 × 105 | 4 | [143] |
| A328W/Y332A | 8.6 × 106 | 30 | [144] |
| F227A/S287G/A328W/Y332A | 3.1 × 107 | 100 | [150] |
| A199S/S287G/A328W/Y332G | 4.15 × 108 | 1500 | [145] |
| A199S/S287G/A328W/Y332G | 1.35 × 109 | 5000 | [148] |
| A199S/F227A/S287G/A328W/Y332G | 1.84 × 109 | 6500 | [147] |
Lessons from self-reactivating butyrylcholinesterase inhibited by OP
Though human butyrylcholinesterase is irreversibly inhibited by OP, mouse butyrylcholinesterase spontaneously self reactivates after inhibition by paraoxon, DFP or echothiophate [121]. The DFP-inhibited mouse butyrylcholinesterase reactivated to a lesser extent than the echothiophate-inhibited enzyme, suggesting that the fast aging of DFP-inhibited enzyme competed with self-reactivation. Other investigators have reported the peculiar behavior of phosphylated butyrylcholinesterase from some species (see [121]). An activity rebound of pig plasma butyrylcholinesterase after challenge of animals by VX was attributed to release of butyrylcholinesterase to the blood stream from organs [151]. However, study of highly purified pig butyrylcholinesterase inhibited by VX showed full spontaneous reactivation of the enzyme in less than 1 hour (Dorandeu et al., unpublished work). Comparison of butyrylcholinesterase sequences of mouse and pig with other species, as well as molecular modeling of structures of pig and mouse butyrylcholinesterase (Nachon, unpublished works) did not reveal any feature in the active center that could explain spontaneous reactivation of phosphylated enzymes. The amino acid differences responsible for spontaneous reactivation have not yet been identified, but it is clear that they are not within the active site gorge.
These observations, coming after the success of transition state simulations for the re-design of butyrylcholinesterase as a cocaine-esterase, indicate that the key is in the molecular dynamics of these enzymes. It may be hypothesized that the “breathing” of self-reactivatable butyrylcholinesterase causes transient adjustment of the active site geometry with unmasking of nucleophilic groups and polarization of a water molecule that acts to release the OP from serine. The functional architecture of the active center and the dynamics of butyrylcholinesterase in action result from a delicate balance between numerous interacting groups, suggesting that multiple mutations will be needed to convert human butyrylcholinesterase to an efficient OP hydrolase.
Lessons from the G117H mutant family
The crystal structure of the Gly117 His mutant of human butyrylcholinesterase [152,153] indicated that His117 is mobile enough to adopt a favorable conformation to activate a vicinal water molecule. However, the main conformation adopted by His117 in the phosphylated enzyme is not favorable for dephosphylation, so that an activated water molecule is produced infrequently. This explains the low turnover with OP substrates. In addition, evidence was provided that mutations at position Gly117 cause dislocation and loss of functionality of the oxyanion hole toward carboxyl-esters and carboxyl-thioesters [154]. Molecular modeling and empirical calculations on acetylation of the G117H mutant by acetylthiocholine are in agreement with these structural and kinetic data [155]. Therefore, the nucleophile needed for general base catalysis of future mutants of butyrylcholinesterase will have to be introduced in a location that does not impair enzyme functionality. Re-design of the active center pocket of butyrylcholinesterase with creation of a stable dyad as in Tyr124His/Tyr72Asp-based new mutants of human acetylcholinesterase [106] may be a starting point. However, “intelligent re-design” of multiple mutations in butyrylcholinesterase for high OP hydrolase activity will need implementation of an integrated computational-mutagenesis strategy as proposed above.
New OP binding motif to tyrosine and lysine in proteins that have no active site serine
There is general agreement that the acute toxicity of OP is due to inhibition of acetylcholinesterase. What is not understood is why low doses cause chronic illness in some people. Symptoms of low dose toxicity include headache, memory loss, inability to learn, difficulty sleeping, fatigue, and muscle weakness. Low dose is defined as a dose that causes no obvious signs of cholinergic toxicity and no significant acetylcholinesterase inhibition. A hypothesis to explain low dose effects is that noncholinesterase targets are modified by OP, and that modification damages nerve function.
A search for additional OP targets was begun by treating mice with a biotinylated OP [156] at a dose that did not inhibit acetylcholinesterase. The biotinylated OP made a covalent bond with albumin in plasma, muscle, and liver. The labeled peptide and the labeled amino acid were identified by mass spectrometry. In human albumin (accession # gi:122920512) the OP-labeled residue is Tyr411 [157,158]. When a homogenate of rat thymus was treated with radiolabeled DFP, the labeled protein was identified as albumin [159].
The question we asked next, was whether albumin was a special case or whether tyrosine in other proteins could also be modified by OP. Almost every pure protein when treated with a 20-fold molar excess of chlorpyrifos oxon, dichlorvos, DFP, soman, sarin, or FP-biotin had one or more OP-labeled tyrosines. In each case we isolated the labeled peptide and identified the labeled residue by mass spectrometry. Tyrosine was a common motif for OP binding in bovine tubulin alpha (Tyr 83), bovine tubulin beta (Tyr 59, Tyr 159, Tyr 281), mouse transferrin (Tyr 238, Tyr 319, Tyr 429, Tyr 491, Tyr 518), human transferrin (Tyr 238, Tyr 574), human alpha-2-glycoprotein 1 zinc-binding protein (Tyr 138, Tyr 174, Tyr 181), human kinesin 3C motor domain (Tyr 145), human keratin 1 (Tyr 230), bovine actin Tyr55, Tyr 200), murine ATP synthase beta (Tyr 431), murine adenine nucleotide translocase 1(Tyr 81), bovine chymotrypsinogen (Tyr 201), and porcine pepsin (Tyr 310) [160–162]. In addition we found OP covalently bound to lysine in keratin, albumin, bovine tubulin alpha, tubulin beta, bovine actin, and mouse transferrin [163]. A review of the 102 OP-labeled tyrosine peptides identified to date by mass spectrometry is in [164]. This review notes that certain ion masses always appear in the MS/MS spectrum of an OP-tyrosine labeled peptide and that these immonium ions can be used to help identify OP-labeled tyrosine.
Gaps in knowledge of proteins modified by low dose OP exposure
The studies with pure proteins suggest that the search for OP-labeled proteins should be broadened to include almost every protein in the body. The search is no longer restricted to enzymes with an active site serine. OP-labeled tubulin has been found in the brains of mice. Whether or not tubulin function is disrupted after low dose OP treatment is not yet known. Tubulin is an abundant protein in brain, but it is possible that a protein in low abundance could be responsible for symptoms of low dose OP toxicity. Likely candidates are enzymes that regulate axonal transport in nerve cells, because disruption of axonal transport is a common feature of neurodegenerative diseases [165].
Methods for detection of low dose exposure to organophosphorus agents
Why measure low dose exposure?
One reason for measuring low dose exposure is to understand why some people become chronically ill from a dose that has no ill effects on the majority. People with chemical sensitivity, chronic fatigue, toxic airline illness (www.toxicairlines.com), and Gulf War Illness suspect their illness is due to exposure to organophosphorus agents [166–168], but this belief is based on correlation rather than direct laboratory evidence. A method that detects low dose exposure could provide this missing evidence. This could lead to an understanding of the mechanism of illness from low doses of organophosphorus agents.
Forensic scientists could use a low dose exposure assay to identify persons who unlawfully synthesize and distribute nerve agents. Even though these people would have no symptoms of toxicity, nerve agent adducts in their blood would provide absolute evidence that they had been in contact with nerve agents. Workers involved in destruction and transportation of nerve agents could be tested for low dose exposure to provide assurance that their protective equipment is functioning properly. In the event of an incident where nerve agent is released, many people who report to emergency facilities would be the worried-well who need assurance that they have not been exposed. A highly sensitive method that can provide this assurance would calm the public. Pesticide applicators, including farmers and aerial sprayers, could be tested for low dose exposure to pesticides. This could protect them from Parkinson’s disease and depression, as these diseases have been linked to pesticide exposure [169,170].
Methods for measuring OP exposure
Laboratory tests for poisoning by nerve agents and pesticides measure the enzyme activity of acetylcholinesterase in red blood cells and the butyrylcholinesterase in plasma. Low levels indicate poisoning. The advantage of this method is that it is simple, inexpensive, and well established. However, activity assays are not useful for low dose exposures because normal levels of activity have a broad range, making it difficult to distinguish low endogenous levels from low dose exposure. Another disadvantage of this method is that it does not identify the inhibitor. Many chemicals inhibit cholinesterases including the carbamate pesticide carbaryl and the Alzheimer drug tacrine.
Intact nerve agent, nerve agent metabolites, pesticides, and pesticide metabolites in air, water, soil, fabric and urine can be extracted and identified by gas chromatography/mass spectrometry and liquid chromatography tandem mass spectrometry [171–173]. These methods identify the agent based on its mass and fragmentation pattern. The results leave no doubt as to the identity of the agent. Analysis of metabolites by gas chromatography has the drawback that several agents can yield identical metabolites, and most metabolites must be derivatized, thus adding an extra step to sample preparation. Identification of metabolites in urine does not allow one to know whether exposure was to the active agent or to breakdown products.
Organophosphorus agents bound to the active site serine of butyrylcholinesterase or acetylcholinesterase can be regenerated by treatment of blood with potassium fluoride. The freed agent is extracted and identified by gas chromatography/mass spectrometry [174,175]. The identification is highly reliable. The drawback of this method is that nerve agent or pesticide that has lost an alkyl group in the process called “aging”, cannot be regenerated. This means soman and other aged OP will give a negative result, indicating no exposure. Most pesticides yield identical dimethyl or diethylphosphate adducts of butyrylcholinesterase and acetylcholinesterase, so that identification of the pesticide is incomplete.
Electrospray mass spectrometry identifies adducts on the active site serine 198 of human butyrylcholinesterase. The butyrylcholinesterase is first purified from 0.5–1 ml plasma and then digested with trypsin or pepsin [68,176]. HPLC followed by mass spectrometry yields the mass of the poison. The advantage of this method is that it gives a positive result for “aged” adducts. The limitation is that most pesticides yield identical adducts on butyrylcholinesterase, so that one can confirm exposure but not identify the poison. For example, chlorpyrifos oxon and paraoxon both yield diethylphosphate adducts with an added mass of +136, while dichlorvos and malaoxon both yield dimethyl phosphate adducts with an added mass of +108.
Electrospray mass spectrometry can also be used to detect organophosphorus adducts on tyrosine 411 of human albumin [177]. The advantage of analyzing adducts on albumin is that organophosphorus labeled tyrosine does not age [178,179]. Therefore soman exposure can be distinguished from sarin exposure. The disadvantage of analyzing tyrosine adducts is that the percent labeling on albumin is low. Plasma from a patient poisoned with dichlorvos has been found to contain dichlorvos-labeled albumin by this method (unpublished). Guinea pigs poisoned with soman and tabun have OP-labeled albumin [178]. However, humans poisoned with OP other than dichlorvos have not yet been demonstrated to have OP-labeled albumin.
Mass spectrometers equipped with a source that requires no sample preparation are being evaluated but are not yet in routine use. The DESI source electrosprays boric acid solution onto a Teflon surface on which OP hydrolysate has been deposited [180]. The ions are analyzed in an LTQ mass spectrometer.
A drawback of mass spectrometry methods is that mass spectrometers are very expensive and require highly trained personnel to run them. Portable mass spectrometers are used by the military but they give a high rate of false positives [181].
An ELISA kit that uses a polyclonal antibody to detect chlorpyrifos in river and lake water is available from Strategic Diagnostics Inc, Newark, DE (catalog # 7250000). The limit of detection is in the nanomolar range. ELISA kits for detection of diazinon and carbofuran are also available. Analysis of environmental samples by commercial ELISA kits is less costly and labor intensive than analysis by gas or liquid chromatography. ELISA kit results tend to overestimate pesticide levels [182].
Gaps in knowledge on how to detect low dose exposure
Antibodies that recognize OP-modified proteins in human fluids (plasma, saliva, and urine) are not yet available. Several laboratories are developing antibody-based detection devices using Quantum dots [183], an array biosensor [184], and a fluorogenic sensor (Ateris Technologies, Missoula, MT) but no antibody has as yet been made with adequate binding affinity and specificity to serve in these devices. It is anticipated that a hand held device, and possibly even a dip-stick type of assay, will become available in the future after suitable antibodies have been created.
Conclusion
Human butyrylcholinesterase is highly effective for preventing the toxicity of nerve agents and OP pesticides. Its limitation is that one molecule of butyrylcholinesterase can inactivate only one molecule of OP. A more cost effective therapeutic would rapidly destroy many OP molecules. Such a therapeutic has not yet been made, though several laboratories are designing mutant proteins with OP hydrolase activity. The low dose toxicity of OP is not due to inhibition of cholinesterases, but may involve modification of proteins that have no active site serine.
Acknowledgments
Supported by NIH grant U01 NS058056-03, NIH Cancer Center Support grant CA036727, US Army Medical Research & Materiel Command W81XWH-07-2-0034, French Procurement Agency DGA/PEA 08co501 and Agence Nationale pour la Recherche ANR-06-BLAN-0163.
Abbreviations
- AChE
acetylcholinesterase
- BChE
butyrylcholinesterase
- DFP
diisopropyl fluorophosphates
- G117H
mutation of Glycine 117 to Histidine
- OP
organophosphorus compound
- 2-PAM
pralidoxime
- VX
nerve agent; O-ethyl S-[2-(diisopropylamino)ethyl] methylphosphonothioate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Patrick Masson, Email: pmasson@unmc.edu.
Oksana Lockridge, Email: olockrid@unmc.edu.
References
- 1.Saxena A, Sun W, Luo C, Myers TM, Koplovitz I, Lenz DE, Doctor BP. J Mol Neurosci. 2006;30:145–148. doi: 10.1385/jmn:30:1:145. [DOI] [PubMed] [Google Scholar]
- 2.Manoharan I, Boopathy R, Darvesh S, Lockridge O. Clin Chim Acta. 2007;378:128–135. doi: 10.1016/j.cca.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 3.Darvesh S, Hopkins DA. J Comp Neurol. 2003;463:25–43. doi: 10.1002/cne.10751. [DOI] [PubMed] [Google Scholar]
- 4.Wetherell JR, French MC. Biochem Pharmacol. 1986;35:939–945. doi: 10.1016/0006-2952(86)90080-8. [DOI] [PubMed] [Google Scholar]
- 5.Duysen EG, Li B, Xie W, Schopfer LM, Anderson RS, Broomfield CA, Lockridge O. J Pharmacol Exp Ther. 2001;299:528–535. [PubMed] [Google Scholar]
- 6.Chatonnet F, Boudinot E, Chatonnet A, Taysse L, Daulon S, Champagnat J, Foutz AS. Eur J Neurosci. 2003;18:1419–1427. doi: 10.1046/j.1460-9568.2003.02867.x. [DOI] [PubMed] [Google Scholar]
- 7.Wang Y, Boeck AT, Duysen EG, Van Keuren M, Saunders TL, Lockridge O. Toxicol Appl Pharmacol. 2004;196:356–366. doi: 10.1016/j.taap.2003.12.018. [DOI] [PubMed] [Google Scholar]
- 8.Hartmann J, Kiewert C, Duysen EG, Lockridge O, Greig NH, Klein J. J Neurochem. 2007;100:1421–1429. doi: 10.1111/j.1471-4159.2006.04347.x. [DOI] [PubMed] [Google Scholar]
- 9.Adler M, Filbert MG. FEBS Lett. 1990;267:107–110. doi: 10.1016/0014-5793(90)80300-8. [DOI] [PubMed] [Google Scholar]
- 10.Norel X, Angrisani M, Labat C, Gorenne I, Dulmet E, Rossi F, Brink C. Br J Pharmacol. 1993;108:914–919. doi: 10.1111/j.1476-5381.1993.tb13486.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reubsaet JL, Ringvold A. J Chromatogr Sci. 2005;43:401–405. doi: 10.1093/chromsci/43.8.401. [DOI] [PubMed] [Google Scholar]
- 12.Henschler D. Hoppe Seylers Z Physiol Chem. 1956;305:97–104. [PubMed] [Google Scholar]
- 13.Darvesh S, MacDonald SE, Losier AM, Martin E, Hopkins DA, Armour JA. J Auton Nerv Syst. 1998;71:75–84. doi: 10.1016/s0165-1838(98)00064-2. [DOI] [PubMed] [Google Scholar]
- 14.Duysen EG, Li B, Darvesh S, Lockridge O. Toxicology. 2007;233:60–69. doi: 10.1016/j.tox.2006.11.069. [DOI] [PubMed] [Google Scholar]
- 15.Casida JE, Quistad GB. Chem Res Toxicol. 2004;17:983–998. doi: 10.1021/tx0499259. [DOI] [PubMed] [Google Scholar]
- 16.Mahmood NA, Carmichael WW. Toxicon. 1987;25:1221–1227. doi: 10.1016/0041-0101(87)90140-1. [DOI] [PubMed] [Google Scholar]
- 17.Whittaker M. Anaesthesia. 1980;35:174–197. doi: 10.1111/j.1365-2044.1980.tb03800.x. [DOI] [PubMed] [Google Scholar]
- 18.Li B, Duysen EG, Carlson M, Lockridge O. J Pharmacol Exp Ther. 2008;324:1146–1154. doi: 10.1124/jpet.107.133330. [DOI] [PubMed] [Google Scholar]
- 19.Lockridge O, Schopfer LM, Winger G, Woods JH. J Med CBR Def. 2005;3 doi: 10.1901/jaba.2005.3-nihms5095. online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grunwald J, Marcus D, Papier Y, Raveh L, Pittel Z, Ashani Y. J Biochem Biophys Methods. 1997;34:123–135. doi: 10.1016/s0165-022x(97)01208-6. [DOI] [PubMed] [Google Scholar]
- 21.Saxena A, Luo C, Doctor BP. Protein Expr Purif. 2008;61:191–196. doi: 10.1016/j.pep.2008.05.021. [DOI] [PubMed] [Google Scholar]
- 22.Kronman C, Velan B, Gozes Y, Leitner M, Flashner Y, Lazar A, Marcus D, Sery T, Papier Y, Grosfeld H, et al. Gene. 1992;121:295–304. doi: 10.1016/0378-1119(92)90134-b. [DOI] [PubMed] [Google Scholar]
- 23.Masson P, Legrand P, Bartels CF, Froment MT, Schopfer LM, Lockridge O. Biochemistry. 1997;36:2266–2277. doi: 10.1021/bi962484a. [DOI] [PubMed] [Google Scholar]
- 24.Barak D, Ordentlich A, Bromberg A, Kronman C, Marcus D, Lazar A, Ariel N, Velan B, Shafferman A. Biochemistry. 1995;34:15444–15452. doi: 10.1021/bi00047a008. [DOI] [PubMed] [Google Scholar]
- 25.Rush RS, Main AR, Kilpatrick BF, Faulkner GD. J Pharmacol Exp Ther. 1981;216:586–591. [PubMed] [Google Scholar]
- 26.Masson P, Marnot B, Lombard JY, Morelis P. Biochimie. 1984;66:235–249. doi: 10.1016/0300-9084(84)90067-1. [DOI] [PubMed] [Google Scholar]
- 27.Li H, Schopfer LM, Masson P, Lockridge O. Biochem J. 2008;411:425–432. doi: 10.1042/BJ20071551. [DOI] [PubMed] [Google Scholar]
- 28.Radic Z, Pickering NA, Vellom DC, Camp S, Taylor P. Biochemistry. 1993;32:12074–12084. doi: 10.1021/bi00096a018. [DOI] [PubMed] [Google Scholar]
- 29.Saxena A, Redman AM, Jiang X, Lockridge O, Doctor BP. Chem Biol Interact. 1999;119–120:61–69. doi: 10.1016/s0009-2797(99)00014-9. [DOI] [PubMed] [Google Scholar]
- 30.Cashman JR, Berkman CE, Underiner G, Kolly CA, Hunter AD. Chem Res Toxicol. 1998;11:895–901. doi: 10.1021/tx9800203. [DOI] [PubMed] [Google Scholar]
- 31.Zhan CG, Zheng F, Landry DW. J Am Chem Soc. 2003;125:2462–2474. doi: 10.1021/ja020850+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Balasubramanian AS, Bhanumathy CD. Faseb J. 1993;7:1354–1358. doi: 10.1096/fasebj.7.14.8224608. [DOI] [PubMed] [Google Scholar]
- 33.Quinn D. Chem Rev. 1987;87:955–979. [Google Scholar]
- 34.Masson P, Froment MT, Gillon E, Nachon F, Darvesh S, Schopfer LM. Biochim Biophys Acta. 2007;1774:1139–1147. doi: 10.1016/j.bbapap.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 35.Masson P, Froment MT, Gillon E, Nachon F, Lockridge O, Schopfer LM. Febs J. 2008;275:2617–2631. doi: 10.1111/j.1742-4658.2008.06409.x. [DOI] [PubMed] [Google Scholar]
- 36.Montenegro MF, Maria TM, de la Cadena MP, Campoy FJ, Munoz-Delgado E, Vidal CJ. Biol Chem. 2008;389:425–432. doi: 10.1515/BC.2008.041. [DOI] [PubMed] [Google Scholar]
- 37.Montenegro MF, Moral-Naranjo MT, Munoz-Delgado E, Campoy FJ, Vidal CJ. Febs J. 2009;276:2074–2083. doi: 10.1111/j.1742-4658.2009.06942.x. [DOI] [PubMed] [Google Scholar]
- 38.Imai T. Drug Metab Pharmacokinet. 2006;21:173–185. doi: 10.2133/dmpk.21.173. [DOI] [PubMed] [Google Scholar]
- 39.Sanghani SP, Sanghani PC, Schiel MA, Bosron WF. Protein Pept Lett. 2009;16 doi: 10.2174/092986609789071324. in press. [DOI] [PubMed] [Google Scholar]
- 40.Masson P, Froment MT, Darvesh S, Schopfer LM, Lockridge O. J Enzyme Inhib Med Chem. 2007;22:463–469. doi: 10.1080/14756360701383932. [DOI] [PubMed] [Google Scholar]
- 41.Darvesh S, Hopkins DA, Geula C. Nat Rev Neurosci. 2003;4:131–138. doi: 10.1038/nrn1035. [DOI] [PubMed] [Google Scholar]
- 42.Rajesh RV, Chitra L, Layer PG, Boopathy R. Biochimie. 2009;91:1087–1094. doi: 10.1016/j.biochi.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 43.Masson P, Froment MT, Fort S, Ribes F, Bec N, Balny C, Schopfer LM. Biochim Biophys Acta. 2002;1597:229–243. doi: 10.1016/s0167-4838(02)00265-0. [DOI] [PubMed] [Google Scholar]
- 44.Masson P, Goldstein BN, Debouzy JC, Froment MT, Lockridge O, Schopfer LM. Eur J Biochem. 2004;271:220–234. doi: 10.1046/j.1432-1033.2003.03924.x. [DOI] [PubMed] [Google Scholar]
- 45.Frieden C. J Biol Chem. 1970;245:5788–5799. [PubMed] [Google Scholar]
- 46.Masson P, Schopfer LM, Froment MT, Debouzy JC, Nachon F, Gillon E, Lockridge O, Hrabovska A, Goldstein BN. Chem Biol Interact. 2005;157–158:143–152. doi: 10.1016/j.cbi.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 47.Hrabovska A, Debouzy JC, Froment MT, Devinsky F, Paulikova I, Masson P. Febs J. 2006;273:1185–1197. doi: 10.1111/j.1742-4658.2006.05144.x. [DOI] [PubMed] [Google Scholar]
- 48.Sadar MH, Laidler KJ. Can J Biochem. 1975;53:380–387. doi: 10.1139/o75-052. [DOI] [PubMed] [Google Scholar]
- 49.Badiou A, Froment MT, Fournier D, Masson P, Belzunces LP. Chem Biol Interact. 2008;175:410–412. doi: 10.1016/j.cbi.2008.05.039. [DOI] [PubMed] [Google Scholar]
- 50.Pattison S, Bernhard S. Proc Natl Acad Sci U S A. 1978;75:3613–3617. doi: 10.1073/pnas.75.8.3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bolger MB, Taylor P. Biochemistry. 1979;18:3622–3629. doi: 10.1021/bi00583a029. [DOI] [PubMed] [Google Scholar]
- 52.Loudwig S, Nicolet Y, Masson P, Fontecilla-Camps JC, Bon S, Nachon F, Goeldner M. Chembiochem. 2003;4:762–767. doi: 10.1002/cbic.200300571. [DOI] [PubMed] [Google Scholar]
- 53.Morrison JF, Stone SR. Comments Mol Cell Biophys. 1985;2:347–368. [Google Scholar]
- 54.Maxwell DM, Brecht KM, Koplovitz I, Sweeney RE. Arch Toxicol. 2006;80:756–760. doi: 10.1007/s00204-006-0120-2. [DOI] [PubMed] [Google Scholar]
- 55.Jokanovic M, Prostran M. Curr Med Chem. 2009;16:2177–2188. doi: 10.2174/092986709788612729. [DOI] [PubMed] [Google Scholar]
- 56.Polhuijs M, Langenberg JP, Benschop HP. Toxicol Appl Pharmacol. 1997;146:156–161. doi: 10.1006/taap.1997.8243. [DOI] [PubMed] [Google Scholar]
- 57.Harel M, Su CT, Frolow F, Ashani Y, Silman I, Sussman JL. J Mol Biol. 1991;221:909–918. doi: 10.1016/0022-2836(91)80183-u. [DOI] [PubMed] [Google Scholar]
- 58.Fleming CD, Edwards CC, Kirby SD, Maxwell DM, Potter PM, Cerasoli DM, Redinbo MR. Biochemistry. 2007;46:5063–5071. doi: 10.1021/bi700246n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Epstein TM, Samanta U, Kirby SD, Cerasoli DM, Bahnson BJ. Biochemistry. 2009;48:3425–3435. doi: 10.1021/bi8023527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yanagisawa N, Morita H, Nakajima T. J Neurol Sci. 2006;249:76–85. doi: 10.1016/j.jns.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 61.Shafferman A, Ordentlich A, Barak D, Stein D, Ariel N, Velan B. Biochem J. 1996;318(Pt 3):833–840. doi: 10.1042/bj3180833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Davies DR, Green AL. Biochem J. 1956;63:529–535. doi: 10.1042/bj0630529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nachon F, Asojo OA, Borgstahl GE, Masson P, Lockridge O. Biochemistry. 2005;44:1154–1162. doi: 10.1021/bi048238d. [DOI] [PubMed] [Google Scholar]
- 64.Heilbronn E. Biochem Pharmacol. 1963;12:25–36. doi: 10.1016/0006-2952(63)90006-6. [DOI] [PubMed] [Google Scholar]
- 65.Hobbiger F. Br J Pharmacol Chemother. 1956;11:295–303. doi: 10.1111/j.1476-5381.1956.tb01069.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Worek F, Diepold C, Eyer P. Arch Toxicol. 1999;73:7–14. doi: 10.1007/s002040050580. [DOI] [PubMed] [Google Scholar]
- 67.Masson P, Fortier PL, Albaret C, Froment MT, Bartels CF, Lockridge O. Biochem J. 1997;327(Pt 2):601–607. doi: 10.1042/bj3270601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fidder A, Hulst AG, Noort D, de Ruiter R, van der Schans MJ, Benschop HP, Langenberg JP. Chem Res Toxicol. 2002;15:582–590. doi: 10.1021/tx0101806. [DOI] [PubMed] [Google Scholar]
- 69.Li H, Schopfer LM, Nachon F, Froment MT, Masson P, Lockridge O. Toxicol Sci. 2007;100:136–145. doi: 10.1093/toxsci/kfm215. [DOI] [PubMed] [Google Scholar]
- 70.Carletti E, Li H, Li B, Ekstrom F, Nicolet Y, Loiodice M, Gillon E, Froment MT, Lockridge O, Schopfer LM, Masson P, Nachon F. J Am Chem Soc. 2008;130:16011–16020. doi: 10.1021/ja804941z. [DOI] [PubMed] [Google Scholar]
- 71.Beauregard G, Lum J, Roufogalis BD. Biochem Pharmacol. 1981;30:2915–2920. doi: 10.1016/0006-2952(81)90252-5. [DOI] [PubMed] [Google Scholar]
- 72.Saxena A, Doctor BP, Maxwell DM, Lenz DE, Radic Z, Taylor P. Biochem Biophys Res Commun. 1993;197:343–349. doi: 10.1006/bbrc.1993.2481. [DOI] [PubMed] [Google Scholar]
- 73.Smith TE, Usdin E. Biochemistry. 1966;5:2914–2918. doi: 10.1021/bi00873a021. [DOI] [PubMed] [Google Scholar]
- 74.Michel HO, Hackley BE, Jr, Berkowitz L, List G, Hackley EB, Gillilan W, Pankau M. Arch Biochem Biophys. 1967;121:29–34. doi: 10.1016/0003-9861(67)90006-9. [DOI] [PubMed] [Google Scholar]
- 75.Viragh C, Kovach IM, Pannell L. Biochemistry. 1999;38:9557–9561. doi: 10.1021/bi991112+. [DOI] [PubMed] [Google Scholar]
- 76.Viragh C, Akhmetshin R, Kovach IM, Broomfield C. Biochemistry. 1997;36:8243–8252. doi: 10.1021/bi962764q. [DOI] [PubMed] [Google Scholar]
- 77.Kovach IM. J Phys Org Chem. 2004;17:602–614. [Google Scholar]
- 78.Sanson B, Nachon F, Colletier JP, Froment MT, Toker L, Greenblatt HM, Sussman JL, Ashani Y, Masson P, Silman I, Weik M. J Med Chem. 2009 doi: 10.1021/jm900433t. [DOI] [PubMed] [Google Scholar]
- 79.Segall Y, Waysbort D, Barak D, Ariel N, Doctor BP, Grunwald J, Ashani Y. Biochemistry. 1993;32:13441–13450. doi: 10.1021/bi00212a009. [DOI] [PubMed] [Google Scholar]
- 80.Grunwald J, Segall Y, Shirin E, Waysbort D, Steinberg N, Silman I, Ashani Y. Biochem Pharmacol. 1989;38:3157–3168. doi: 10.1016/0006-2952(89)90608-4. [DOI] [PubMed] [Google Scholar]
- 81.Nicolet Y, Lockridge O, Masson P, Fontecilla-Camps JC, Nachon F. J Biol Chem. 2003;278:41141–41147. doi: 10.1074/jbc.M210241200. [DOI] [PubMed] [Google Scholar]
- 82.Amitai G, Ashani Y, Gafni A, Silman I. Biochemistry. 1982;21:2060–2069. doi: 10.1021/bi00538a013. [DOI] [PubMed] [Google Scholar]
- 83.Berman HA, Decker MM. J Biol Chem. 1986;261:10646–10652. [PubMed] [Google Scholar]
- 84.Masson P, Goasdoue JL. Biochim Biophys Acta. 1986;869:304–313. doi: 10.1016/0167-4838(86)90070-1. [DOI] [PubMed] [Google Scholar]
- 85.Masson P, Gouet P, Clery C. J Mol Biol. 1994;238:466–478. doi: 10.1006/jmbi.1994.1305. [DOI] [PubMed] [Google Scholar]
- 86.Masson P, Clery C, Guerra P, Redslob A, Albaret C, Fortier PL. Biochem J. 1999;343(Pt 2):361–369. [PMC free article] [PubMed] [Google Scholar]
- 87.Ashani Y, Gentry MK, Doctor BP. Biochemistry. 1990;29:2456–2463. doi: 10.1021/bi00462a004. [DOI] [PubMed] [Google Scholar]
- 88.Gabel F, Masson P, Froment MT, Doctor BP, Saxena A, Silman I, Zaccai G, Weik M. Biophys J. 2009;96:1489–1494. doi: 10.1016/j.bpj.2008.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hobbiger F. In: Cholinesterases and anticholinesterase agents. Koelle GB, editor. Springer-Verlag; Berlin: 1963. pp. 941–943. [Google Scholar]
- 90.Harris LW, Fleisher JH, Clark J, Cliff WJ. Science. 1966;154:404–407. doi: 10.1126/science.154.3747.404. [DOI] [PubMed] [Google Scholar]
- 91.Kirby AJ, Younas M. J Chem Soc B. 1970:1165–1172. [Google Scholar]
- 92.Behrman EJ, Biallas MJ, Brass HJ, Edwards JO, Isaks M. J Org Chem. 1970;35:3063–3069. [Google Scholar]
- 93.Behrman EJ, Biallas MJ, Brass HJ, Edwards JO, Isaks M. J Org Chem. 1970;35:3069–3075. [Google Scholar]
- 94.Carletti E, Aurbek N, Gillon E, Loiodice M, Nicolet Y, Fontecilla-Camps JC, Masson P, Thiermann H, Nachon F, Worek F. Biochem J. 2009;421:97–106. doi: 10.1042/BJ20090091. [DOI] [PubMed] [Google Scholar]
- 95.Masson P, Froment MT, Bartels CF, Lockridge O. Biochem J. 1997;325(Pt 1):53–61. doi: 10.1042/bj3250053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Schoene K. Biochim Biophys Acta. 1978;525:468–471. doi: 10.1016/0005-2744(78)90243-7. [DOI] [PubMed] [Google Scholar]
- 97.Schoene K, Steinhanses J, Wertmann A. Biochim Biophys Acta. 1980;616:384–388. doi: 10.1016/0005-2744(80)90156-4. [DOI] [PubMed] [Google Scholar]
- 98.Stalc A, Sentjurc M. Biochem Pharmacol. 1990;40:2511–2517. doi: 10.1016/0006-2952(90)90093-z. [DOI] [PubMed] [Google Scholar]
- 99.Dawson RM, Dowling MH, Poretski M. Neurochem Int. 1991;19:125–133. [Google Scholar]
- 100.Puu G. Biochem Pharmacol. 1988;37:969–970. doi: 10.1016/0006-2952(88)90191-8. [DOI] [PubMed] [Google Scholar]
- 101.Dawson RM. Neurosci Lett. 1989;100:227–230. doi: 10.1016/0304-3940(89)90689-7. [DOI] [PubMed] [Google Scholar]
- 102.Johnson JL, Cusack B, Hughes TF, McCullough EH, Fauq A, Romanovskis P, Spatola AF, Rosenberry TL. J Biol Chem. 2003;278:38948–38955. doi: 10.1074/jbc.M304797200. [DOI] [PubMed] [Google Scholar]
- 103.Taylor P, Kovarik Z, Reiner E, Radic Z. Toxicology. 2007;233:70–78. doi: 10.1016/j.tox.2006.11.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mizutani MY, Itai A. J Med Chem. 2004;47:4818–4828. doi: 10.1021/jm030605g. [DOI] [PubMed] [Google Scholar]
- 105.Ekstrom FJ, Astot C, Pang YP. Clin Pharmacol Ther. 2007;82:282–293. doi: 10.1038/sj.clpt.6100151. [DOI] [PubMed] [Google Scholar]
- 106.Masson P, Nachon F, Broomfield CA, Lenz DE, Verdier L, Schopfer LM, Lockridge O. Chem Biol Interact. 2008;175:273–280. doi: 10.1016/j.cbi.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 107.Saxena A, Maxwell DM, Quinn DM, Radic Z, Taylor P, Doctor BP. Biochem Pharmacol. 1997;54:269–274. doi: 10.1016/s0006-2952(97)00180-9. [DOI] [PubMed] [Google Scholar]
- 108.Kovarik Z, Radic Z, Berman HA, Taylor P. Toxicology. 2007;233:79–84. doi: 10.1016/j.tox.2006.08.032. [DOI] [PubMed] [Google Scholar]
- 109.Mazor O, Cohen O, Kronman C, Raveh L, Stein D, Ordentlich A, Shafferman A. Mol Pharmacol. 2008;74:755–763. doi: 10.1124/mol.108.047449. [DOI] [PubMed] [Google Scholar]
- 110.Eddleston M, Buckley NA, Eyer P, Dawson AH. Lancet. 2008;371:597–607. doi: 10.1016/S0140-6736(07)61202-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Aas P. Prehosp Disaster Med. 2003;18:208–216. doi: 10.1017/s1049023x00001072. [DOI] [PubMed] [Google Scholar]
- 112.Albuquerque EX, Pereira EF, Aracava Y, Fawcett WP, Oliveira M, Randall WR, Hamilton TA, Kan RK, Romano JA, Jr, Adler M. Proc Natl Acad Sci U S A. 2006;103:13220–13225. doi: 10.1073/pnas.0605370103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wetherell J, Price M, Mumford H, Armstrong S, Scott L. Toxicology. 2007;233:120–127. doi: 10.1016/j.tox.2006.07.028. [DOI] [PubMed] [Google Scholar]
- 114.Eyer P, Szinicz L, Thiermann H, Worek F, Zilker T. Toxicology. 2007;233:108–119. doi: 10.1016/j.tox.2006.08.033. [DOI] [PubMed] [Google Scholar]
- 115.Thiermann H, Szinicz L, Eyer P, Felgenhauer N, Zilker T, Worek F. Toxicology. 2007;233:145–154. doi: 10.1016/j.tox.2006.11.056. [DOI] [PubMed] [Google Scholar]
- 116.Schallreuter KU, Gibbons NC, Elwary SM, Parkin SM, Wood JM. Biochem Biophys Res Commun. 2007;355:1069–1074. doi: 10.1016/j.bbrc.2007.02.078. [DOI] [PubMed] [Google Scholar]
- 117.Redinbo MR, Potter PM. Drug Discov Today. 2005;10:313–325. doi: 10.1016/S1359-6446(05)03383-0. [DOI] [PubMed] [Google Scholar]
- 118.Wang Q, Sun M, Zhang H, Huang C. J Biochem Mol Toxicol. 1998;12:213–217. doi: 10.1002/(sici)1099-0461(1998)12:4<213::aid-jbt3>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 119.Nomura DK, Fujioka K, Issa RS, Ward AM, Cravatt BF, Casida JE. Toxicol Appl Pharmacol. 2008;228:42–48. doi: 10.1016/j.taap.2007.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Nomura DK, Leung D, Chiang KP, Quistad GB, Cravatt BF, Casida JE. Proc Natl Acad Sci U S A. 2005;102:6195–6200. doi: 10.1073/pnas.0501915102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Li B, Sedlacek M, Manoharan I, Boopathy R, Duysen EG, Masson P, Lockridge O. Biochem Pharmacol. 2005;70:1673–1684. doi: 10.1016/j.bcp.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 122.Li B, Nachon F, Froment MT, Verdier L, Debouzy JC, Brasme B, Gillon E, Schopfer LM, Lockridge O, Masson P. Chem Res Toxicol. 2008;21:421–431. doi: 10.1021/tx700339m. [DOI] [PubMed] [Google Scholar]
- 123.Saxena A, Luo C, Chilukuri N, Maxwell DM, Doctor BP. In: Chemical Warfare Agents Chemistry, Pharmacology, Toxicology and Therapeutics. Romano JA, Lukey BJ, Salem H, editors. CRC Press; Boca Raton, FL: 2007. pp. 145–173. [Google Scholar]
- 124.Lenz DE, Broomfield CA, Yeung DT, Masson P, Maxwell DM, Cerasoli DM. In: Chemical Warfare Agents Chemistry, Pharmacology, Toxicology and Therapeutics. Romano JA, Lukey BJ, Salem H, editors. CRC Press; Boca Raton, FL: 2007. pp. 175–202. [Google Scholar]
- 125.Ashani Y, Pistinner S. Toxicol Sci. 2004;77:358–367. doi: 10.1093/toxsci/kfh012. [DOI] [PubMed] [Google Scholar]
- 126.Huang YJ, Huang Y, Baldassarre H, Wang B, Lazaris A, Leduc M, Bilodeau AS, Bellemare A, Cote M, Herskovits P, Touati M, Turcotte C, Valeanu L, Lemee N, Wilgus H, Begin I, Bhatia B, Rao K, Neveu N, Brochu E, Pierson J, Hockley DK, Cerasoli DM, Lenz DE, Karatzas CN, Langermann S. Proc Natl Acad Sci U S A. 2007;104:13603–13608. doi: 10.1073/pnas.0702756104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Huang YJ, Lundy PM, Lazaris A, Huang Y, Baldassarre H, Wang B, Turcotte C, Cote M, Bellemare A, Bilodeau AS, Brouillard S, Touati M, Herskovits P, Begin I, Neveu N, Brochu E, Pierson J, Hockley DK, Cerasoli DM, Lenz DE, Wilgus H, Karatzas CN, Langermann S. BMC Biotechnol. 2008;8:50. doi: 10.1186/1472-6750-8-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Jarv J. In: Enzymes hydrolysing organophosphorus compounds. Reiner E, Aldridge WN, GHoskin FC, editors. Ellis Horwood Ltd; Chichester, UK: 1989. pp. 221–225. [Google Scholar]
- 129.Sussman JL, Harel M, Frolow F, Oefner C, Goldman A, Toker L, Silman I. Science. 1991;253:872–879. doi: 10.1126/science.1678899. [DOI] [PubMed] [Google Scholar]
- 130.Lockridge O, Blong RM, Masson P, Froment MT, Millard CB, Broomfield CA. Biochemistry. 1997;36:786–795. doi: 10.1021/bi961412g. [DOI] [PubMed] [Google Scholar]
- 131.Millard CB, Lockridge O, Broomfield CA. Biochemistry. 1995;34:15925–15933. doi: 10.1021/bi00049a007. [DOI] [PubMed] [Google Scholar]
- 132.Newcomb RD, Campbell PM, Ollis DL, Cheah E, Russell RJ, Oakeshott JG. Proc Natl Acad Sci U S A. 1997;94:7464–7468. doi: 10.1073/pnas.94.14.7464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Jackson CJ, Liu J, Russell R, Weik M, Tawfik DS, Oakeshott JG. 10th International Meeting on Cholinesterases; Sibenik, Croatia. 2009. p. 61. [Google Scholar]
- 134.Millard CB, Lockridge O, Broomfield CA. Biochemistry. 1998;37:237–247. doi: 10.1021/bi972057c. [DOI] [PubMed] [Google Scholar]
- 135.Schopfer LM, Ticu-Boeck A, Broomfield CA, Lockridge O. J Med Chem Def online journal. 2004;2:1–21. [Google Scholar]
- 136.Poyot T, Nachon F, Froment MT, Loiodice M, Wieseler S, Schopfer LM, Lockridge O, Masson P. Biochim Biophys Acta. 2006;1764:1470–1478. doi: 10.1016/j.bbapap.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 137.Costa LG, Furlong C. In: Handbook of Toxicology of Chemical Warfare Agents. Gupta RC, editor. Elsevier-Academic Press; Amsterdam: 2009. pp. 1023–1031. [Google Scholar]
- 138.Kern RJ, Wales ME, Tiffany-Castiglioni E, Wild JR. In: Handbook of Toxicology of Chemical Warfare Agents. Gupta RC, editor. Elsevier-Academic Press; Amsterdam: 2009. pp. 1041–1051. [Google Scholar]
- 139.Masson P, Rochu D. In: Handbook of Toxicology of Chemical Warfare Agents. Gupta RC, editor. Elsevier-Academic Press; Amsterdam: 2009. pp. 1053–1065. [Google Scholar]
- 140.Bershtein S, Tawfik DS. Curr Opin Chem Biol. 2008;12:151–158. doi: 10.1016/j.cbpa.2008.01.027. [DOI] [PubMed] [Google Scholar]
- 141.Sweeney RE, Maxwell DM. Math Biosci. 2003;181:133–143. doi: 10.1016/s0025-5564(02)00154-2. [DOI] [PubMed] [Google Scholar]
- 142.Inaba T, Stewart DJ, Kalow W. Clin Pharmacol Ther. 1978;23:547–552. doi: 10.1002/cpt1978235547. [DOI] [PubMed] [Google Scholar]
- 143.Xie W, Altamirano CV, Bartels CF, Speirs RJ, Cashman JR, Lockridge O. Mol Pharmacol. 1999;55:83–91. doi: 10.1124/mol.55.1.83. [DOI] [PubMed] [Google Scholar]
- 144.Sun H, Pang YP, Lockridge O, Brimijoin S. Mol Pharmacol. 2002;62:220–224. doi: 10.1124/mol.62.2.220. [DOI] [PubMed] [Google Scholar]
- 145.Pan Y, Gao D, Yang W, Cho H, Yang G, Tai HH, Zhan CG. Proc Natl Acad Sci U S A. 2005;102:16656–16661. doi: 10.1073/pnas.0507332102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zheng F, Zhan CG. Org Biomol Chem. 2008;6:836–843. doi: 10.1039/b716268e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zheng F, Yang W, Ko MC, Liu J, Cho H, Gao D, Tong M, Tai HH, Woods JH, Zhan CG. J Am Chem Soc. 2008;130:12148–12155. doi: 10.1021/ja803646t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Brimijoin S, Gao Y, Anker JJ, Gliddon LA, Lafleur D, Shah R, Zhao Q, Singh M, Carroll ME. Neuropsychopharmacology. 2008;33:2715–2725. doi: 10.1038/sj.npp.1301666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Yang W, Pan Y, Zheng F, Cho H, Tai HH, Zhan CG. Biophys J. 2009;96:1931–1938. doi: 10.1016/j.bpj.2008.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Pancook JD, Pecht G, Ader M, Lockridge O, Watkins JD. Faseb J. 2003;17:A565. [Google Scholar]
- 151.Dorandeu F, Foquin A, Briot R, Delacour C, Denis J, Alonso A, Froment MT, Renault F, Lallement G, Masson P. Toxicology. 2008;248:151–157. doi: 10.1016/j.tox.2008.03.013. [DOI] [PubMed] [Google Scholar]
- 152.Masson P, Carletti E, Nachon F. Protein Pept Lett. 2009 doi: 10.2174/092986609789071207. [DOI] [PubMed] [Google Scholar]
- 153.Nachon F, Nicolet Y, Ticu-Boeck A, Masson P, Lockridge O. IXth International Meeting on Cholinesterases, Poster P-IV-2; Suzhou, China. 2007. [Google Scholar]
- 154.Masson P, Froment MT, Gillon E, Nachon F, Lockridge O, Schopfer LM. Biochim Biophys Acta. 2007;1774:16–34. doi: 10.1016/j.bbapap.2006.10.012. [DOI] [PubMed] [Google Scholar]
- 155.Amitay M, Shurki A. Proteins. 2009;77:370–377. doi: 10.1002/prot.22442. [DOI] [PubMed] [Google Scholar]
- 156.Peeples ES, Schopfer LM, Duysen EG, Spaulding R, Voelker T, Thompson CM, Lockridge O. Toxicol Sci. 2005;83:303–312. doi: 10.1093/toxsci/kfi023. [DOI] [PubMed] [Google Scholar]
- 157.Williams NH, Harrison JM, Read RW, Black RM. Arch Toxicol. 2007;81:627–639. doi: 10.1007/s00204-007-0191-8. [DOI] [PubMed] [Google Scholar]
- 158.Li B, Schopfer LM, Hinrichs SH, Masson P, Lockridge O. Anal Biochem. 2007;361:263–272. doi: 10.1016/j.ab.2006.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Carter WG, Tarhoni M, Rathbone AJ, Ray DE. Hum Exp Toxicol. 2007;26:347–353. doi: 10.1177/0960327107074617. [DOI] [PubMed] [Google Scholar]
- 160.Grigoryan H, Schopfer LM, Thompson CM, Terry AV, Masson P, Lockridge O. Chem Biol Interact. 2008;175:180–186. doi: 10.1016/j.cbi.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Li B, Schopfer LM, Grigoryan H, Thompson CM, Hinrichs SH, Masson P, Lockridge O. Toxicol Sci. 2009;107:144–155. doi: 10.1093/toxsci/kfn211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Grigoryan H, Li B, Anderson EK, Xue W, Nachon F, Lockridge O, Schopfer LM. Chem Biol Interact. 2009;180:492–498. doi: 10.1016/j.cbi.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Grigoryan H, Li B, Xue W, Grigoryan M, Schopfer LM, Lockridge O. Anal Biochem. 2009;394:92–100. doi: 10.1016/j.ab.2009.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Schopfer LM, Grigoryan H, Li B, Masson P, Lockridge O. J Chromatography B. 2009. in press. [Google Scholar]
- 165.Morfini GA, Burns M, Binder LI, Kanaan NM, LaPointe N, Bosco DA, Brown RH, Jr, Brown H, Tiwari A, Hayward L, Edgar J, Nave KA, Garberrn J, Atagi Y, Song Y, Pigino G, Brady ST. J Neurosci. 2009;29:12776–12786. doi: 10.1523/JNEUROSCI.3463-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Miller CS, Mitzel HC. Arch Environ Health. 1995;50:119–129. doi: 10.1080/00039896.1995.9940889. [DOI] [PubMed] [Google Scholar]
- 167.Tahmaz N, Soutar A, Cherrie JW. Ann Occup Hyg. 2003;47:261–267. doi: 10.1093/annhyg/meg042. [DOI] [PubMed] [Google Scholar]
- 168.Golomb BA. Proc Natl Acad Sci U S A. 2008;105:4295–4300. doi: 10.1073/pnas.0711986105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Hancock DB, Martin ER, Mayhew GM, Stajich JM, Jewett R, Stacy MA, Scott BL, Vance JM, Scott WK. BMC Neurol. 2008;8:6. doi: 10.1186/1471-2377-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Beseler C, Stallones L, Hoppin JA, Alavanja MC, Blair A, Keefe T, Kamel F. J Occup Environ Med. 2006;48:1005–1013. doi: 10.1097/01.jom.0000235938.70212.dd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Barr DB, Allen R, Olsson AO, Bravo R, Caltabiano LM, Montesano A, Nguyen J, Udunka S, Walden D, Walker RD, Weerasekera G, Whitehead RD, Jr, Schober SE, Needham LL. Environ Res. 2005;99:314–326. doi: 10.1016/j.envres.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 172.Shih ML, Smith JR, McMonagle JD, Dolzine TW, Gresham VC. Biol Mass Spectrom. 1991;20:717–723. doi: 10.1002/bms.1200201111. [DOI] [PubMed] [Google Scholar]
- 173.D’Agostino PA, Hancock JR, Chenier CL, Lepage CR. J Chromatogr A. 2006;1110:86–94. doi: 10.1016/j.chroma.2006.01.083. [DOI] [PubMed] [Google Scholar]
- 174.Van Der Schans MJ, Polhuijs M, Van Dijk C, Degenhardt CE, Pleijsier K, Langenberg JP, Benschop HP. Arch Toxicol. 2004;78:508–524. doi: 10.1007/s00204-004-0568-x. [DOI] [PubMed] [Google Scholar]
- 175.Jakubowski EM, McGuire JM, Evans RA, Edwards JL, Hulet SW, Benton BJ, Forster JS, Burnett DC, Muse WT, Matson K, Crouse CL, Mioduszewski RJ, Thomson SA. J Anal Toxicol. 2004;28:357–363. doi: 10.1093/jat/28.5.357. [DOI] [PubMed] [Google Scholar]
- 176.Li H, Ricordel I, Tong L, Schopfer LM, Baud F, Megarbane B, Maury E, Masson P, Lockridge O. J Appl Toxicol. 2009;29:149–155. doi: 10.1002/jat.1392. [DOI] [PubMed] [Google Scholar]
- 177.Li B, Schopfer LM, Hinrichs SH, Masson P, Lockridge O. Anal Biochem. 2007;361:263–272. doi: 10.1016/j.ab.2006.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Williams NH, Harrison JM, Read RW, Black RM. Arch Toxicol. 2007;81:627–639. doi: 10.1007/s00204-007-0191-8. [DOI] [PubMed] [Google Scholar]
- 179.Li B, Nachon F, Froment MT, Verdier L, Debouzy JC, Brasme B, Gillon E, Schopfer LM, Lockridge O, Masson P. Chem Res Toxicol. 2008;21:421–431. doi: 10.1021/tx700339m. [DOI] [PubMed] [Google Scholar]
- 180.Song Y, Cooks RG. J Mass Spectrom. 2007;42:1086–1092. doi: 10.1002/jms.1244. [DOI] [PubMed] [Google Scholar]
- 181.Seto Y. In: Handbook of Toxicology of Chemical Warfare Agents. Gupta RC, editor. Chapter 53. Academic Press; 2009. pp. 813–825. [Google Scholar]
- 182.Sullivan JJ, Chen YG, Goh KS. J Agric Food Chem. 2007;55:6407–6416. doi: 10.1021/jf070700o. [DOI] [PubMed] [Google Scholar]
- 183.Wang H, Wang J, Timchalk C, Lin Y. Anal Chem. 2008;80:8477–8484. doi: 10.1021/ac801211s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Ligler FS, Sapsford KE, Golden JP, Shriver-Lake LC, Taitt CR, Dyer MA, Barone S, Myatt CJ. Anal Sci. 2007;23:5–10. doi: 10.2116/analsci.23.5. [DOI] [PubMed] [Google Scholar]
- 185.Pan Y, Muzyka JL, Zhan CG. J Phys Chem B. 2009;113:6543–6552. doi: 10.1021/jp8114995. [DOI] [PMC free article] [PubMed] [Google Scholar]