

Abstract
β-Secretase is the rate limiting enzymatic activity in the production of the amyloid-β peptide (Aβ) and is thought to be involved in Alzheimer’s disease (AD) pathogenesis. Although BACE1 (β-site APP Cleaving Enzyme 1, EC 3.4.23.46) has received significant attention, the related BACE2 (EC 3.4.23.45) has not. Though BACE2 is also expressed in the brain, its potential role in AD has not been resolved. In this study, we compared the activities of both BACE1 and BACE2, which were isolated from the same samples of frontal cortex from both AD-affected individuals and age-matched controls. BACE1 activity showed a significant positive correlation with the amount of extractable Aβ, and BACE1 protein and activity were significantly increased in AD cases. Unexpectedly, there were substantial total amounts of BACE2 protein and enzymatic activity in the human brain. BACE2 activity did not change significantly in the AD brain, and was not related to Aβ concentration. These data indicate that BACE1 likely accounts for most of the Aβ produced in the human brain, and that BACE2 activity is not a likely contributor. However, since both forms of BACE compete for the same substrate pool, even small changes in BACE2 activity could have consequences for human disease.
Keywords: β-Secretase, BACE1, BACE2, β-amyloid precursor protein, amyloid-β peptide, Alzheimer’s Disease
Introduction
The development of Alzheimer’s Disease (AD) is marked by the progressive decline of cognitive function, including abnormalities of language, behavior, and memory (McKhann et al. 1984). As the amyloid precursor protein (APP) is trafficked through the secretory pathway, several key cleavage events occur, resulting in the release of products that are directed into the extracellular space and vesicle lumens. One of these cleavage products is the amyloid-β peptide (Aβ), which is derived from APP through sequential cleavage by two enzymatic activities: β- and γ-secretase. β-secretase first cleaves APP to release a large secreted derivative, sAPPβ. A membrane bound fragment, CTFβ, remains and is cleaved by γ-secretase to generate Aβ. The progressive fibrillization and deposition of Aβ within the brain is widely believed to be the primary causal factor in AD pathogenesis (Tanzi and Bertram 2005). β-secretase activity is believed to be the rate limiting step in the amyloidogenic pathway, processing ~10% of the total cellular APP. Due to their essential role in the generation of Aβ, both β- and γ-secretase are considered prime therapeutic targets (Selkoe 2001; Rochette and Murphy 2002).
The β-secretase enzyme (BACE, β-site APP cleaving enzyme) was independently discovered by five different laboratories approximately ten years ago (Vassar et al. 1999; Yan et al. 1999; Sinha et al. 1999; Hussain et al. 1999; Lin et al. 2000). There are two major forms of the enzyme, BACE1 (501 amino acids) and BACE2 (518 amino acids), which are ~75% homologous (45% identical) (Sun et al. 2005; Bennett et al. 2000). BACE is a type I integral membrane protein that contains two D(T/S)G(T/S) motifs within its extracellular domain, which are characteristic of an aspartyl protease. BACE1 primarily localizes to the Golgi apparatus and late endosomes, which is consistent with its optimal activity at low pH (Vassar et al. 1999). BACE1 is highly expressed in brain and pancreas, but is also found in other organs at much lower levels (Vassar et al. 1999; Marcinkiewicz and Seidah 2000). In contrast, BACE2 is present in most peripheral tissues at varying levels, with kidney being the highest (Bennett et al. 2000). Within the brain, BACE2 is believed to be mostly astrocytic, whereas BACE1 is largely neuronal (Basi et al. 2003; Irizarry et al. 2001). The knockout of BACE1 in the mouse leads to the abolishment of Aβ, sAPPβ and CTFβ production in brain (Luo et al. 2001). BACE2 is located on chromosome 21 in the obligate Down syndrome critical region and may contribute to amyloid pathology in these individuals (Acquati et al. 2000; Motonaga et al. 2002). BACE1 and BACE2 compete for substrate and can both cleave APP at the β-site (Farzan et al. 2000; Hussain et al. 2000; Basi et al. 2003); however, BACE2 displays a higher preference for cleavage within the Aβ peptide (Yan et al. 2001). Also, BACE1 and BACE2 combined knockout mice show increased morbidity over BACE1 knockouts alone, and BACE1 knockouts exhibit residual β-secretase activity that may be attributable to BACE2 (Cai et al. 2001; Luo et al. 2001; Dominguez et al. 2005). Thus, although there is good reason to believe that BACE1 is the principle β-secretase responsible for the production of Aβ in the brain, the role of both enzymes in the brain, and in disease, is not fully understood.
The activity and protein levels of BACE1 increase in brain in AD, with changes in BACE1 being significantly greater than those seen in normal aging (Holsinger et al. 2002; Fukumoto et al. 2002; Yang et al. 2003; Fukumoto et al. 2004; Li et al. 2004). The mechanism that underlies this process has yet to be explained. The strongest genetic link to late onset sporadic AD, the APOε4 allele, may explain only a fraction (<20%) of these cases (Roses 1994; Roses 1997), thus raising the intriguing possibility that changes in β-secretase expression may be directly connected to the development of the disease. BACE regulation has therefore become a major question in the AD field, and identification of the cellular factors that control BACE expression has become a significant endeavor. These factors are yet largely unknown but based on current data, are likely post-transcriptional. For example, the 5′ UTR of BACE1 is unusually long (~446 bases) and has significant secondary structure (77% GC), features typically indicative of a mRNA under tight translational regulation (Rossner et al. 2006). The 5′ UTR of BACE has been implicated in controlling BACE1 expression through an unknown inhibitory mechanism (Lammich et al. 2004). The 3′ UTR is also large (variable length: >500 to ~1800 bases) and may be responsible for stabilizing the mRNA and controlling half-life within the cell.
Though both forms of BACE compete for substrate, BACE2 activity, which is somewhat less abundant, is thought to be masked in brain (Hussain et al. 2000; Basi et al. 2003; Farzan et al. 2000; Yan et al. 2001; Sun et al. 2005). Little is known about the role BACE2 plays in development of AD, a short-coming in part due to the limitations of the currently available commercial assay systems. As β-secretase is the rate limiting enzymatic activity involved in Aβ generation, understanding both forms of the enzyme is fundamental to therapeutic target validation and future AD drug development. In the present study, we describe an approach that can independently measure both BACE1 and BACE2, and report the first simultaneous activity measures of both isoforms of β-secretase in the AD brain.
Materials and Methods
Tissue Collection and Preparation
Brain and kidney tissue samples used in assay development were isolated from female rats (Long Evans Hooded or Sprague Dawley; Charles River, MO) immediately following euthanatization by CO2 narcosis, and were stored frozen at −80°C for use in subsequent experiments. Some experiments also used tissue collected from C57/BL6 mice for comparison. Human brain samples were obtained from male and female control and AD brains from tissue repositories at the University of California, Irvine (UCI: Controls, N=5; AD, N=6) and the University of Kentucky (UK: Controls, N= 6; AD, N=3). Additional late stage AD samples from UK (N=3) were used for the development and validation of assay conditions. All samples used for experimental measures were derived from frontal cortex (Brodmann area 9). Control brains had no history of ante-mortem dementia (c.f. Table 1 for case details). Human and animal tissue collection and approval followed University of Kentucky IRB and IACUC guidelines.
Table 1.
Subjects Used in Present Study*
| Case** | Classification# | Sex | Braak Stage | Brain Weight (g) | Age (years) | PMI (hours) |
|---|---|---|---|---|---|---|
| UCI-02-99 | Control | F | II | N/A | 74.0 | 2.8 |
| UCI-19-01 | Control | F | II | 1320 | 74.5 | 2.3 |
| UCI-25-01 | Control | M | II | 1275 | 83.8 | 1.7 |
| UCI-07-03 | Control | F | III | 1119 | 84.8 | 4.3 |
| UCI-15-02 | Control | F | II | N/A | 88.1 | 5.0 |
| UK-1095 | Control | F | II | 1110 | 90.0 | 4.0 |
| UK-1142 | Control | M | - | 1200 | 92.0 | 3.3 |
| UK-1161 | Control | F | - | 1230 | 84.0 | 2.5 |
| UK-1170 | Control | F | I | 1100 | 84.0 | 2.5 |
| UK-1184 | Control | F | II | 1010 | 92.0 | 3.0 |
| UK-1187 | Control | F | - | 1210 | 85.0 | 2.5 |
| 1175 +/− 98 | 84.8 +/− 6.1 | 3.1 +/− 1.0 | ||||
| UCI-18-01 | Alzheimer’s Disease | F | VI | 907 | 74.3 | 4.5 |
| UCI-03-02 | Alzheimer’s Disease | F | VI | 1460 | 75.5 | 4.5 |
| UCI-01-01 | Alzheimer’s Disease | M | VI | 1071 | 81.4 | 2.5 |
| UCI-04-02 | Alzheimer’s Disease | M | VI | 1090 | 83.2 | 3.5 |
| UCI-08-02 | Alzheimer’s Disease | M | V | 1208 | 84.8 | 5.8 |
| UCI-15-05 | Alzheimer’s Disease | F | VI | 992 | 89.0 | 3.5 |
| UK-1036 | Alzheimer’s Disease | F | VI | 1055 | 93.0 | 3.0 |
| UK-1104 | Alzheimer’s Disease | M | VI | 1200 | 90.0 | 3.3 |
| UK-1196 | Alzheimer’s Disease | F | VI | 900 | 83.0 | 2.3 |
| 1099 +/− 174 | 83.8 +/− 6.3 | 3.7 +/− 1.1 |
Study measurements were made from frontal cortex (area 9); ELISAs for Aβ and BACE were done in UCI samples. Mean values +/− standard deviation.
Three additional AD cases from the UK ADC with longer PMIs (~7 hours) were used for the development of the assay, and were not used in the analysis.
MMSE scores: AD, 2.1 +/− 4.8; Control, 29.0 +/− 0.8.
Frozen samples were homogenized using a PowerMax VHS200 in five volumes (wet weight/volume) of tissue lysis buffer (TLB: 10 mM sodium acetate, 1.5 mM NaCl, 0.1% Triton X-100, 0.32 M sucrose, pH = 5.0). The buffer was supplemented with a complete protease inhibitor cocktail with EDTA (PIC; Amresco), with 100 nM pepstatin A added (Sigma). Whole tissue homogenate was centrifuged at 4,000 × g for 15 minutes to pellet insoluble material, followed by an additional spin at 14,000 × g for 30 minutes. Some samples were first separated on a 10 mL sucrose gradient (2.5 mL/layer TLB; weight/volume: 11%/22%/32%/43%; 100,000 × g for 2 hours) for comparison. Centrifugation was conducted in a refrigerated Eppendorf at 4°C. Collected fractions were used immediately for assay procedures.
Pelleted material was sequentially extracted in an equal volume of RIPA buffer (0.1% SDS, 0.5% deoxycholate, 1.0% Triton X-100, 50 mM Tris-Base, 150 mM NaCl, pH= 8.0, with PIC) followed by 70% formic acid (FA) for the determination of detergent soluble and insoluble Aβ, respectively (Murphy et al. 2007). In each case the pellet was extracted by brief sonication (10 × 0.5 s microtip pulses @ 20% power; Fisher Sonic Dismembrator, Model 500) followed by centrifugation to pellet insoluble material (RIPA fraction: 14,000 × g for 30 minutes; FA fraction: 100,000 × g for 1 hour; both spins were performed at 4°C). Protein content was determined by bicinchoninic acid (BCA) assay (Pierce Biotechnology) relative to bovine serum albumin (BSA) standards prepared in the same buffer.
Immunoprecipitation and Western Blotting
For immunoprecipitation, a monoclonal antibody raised against the BACE1 ectodomain (R&D Systems, MAB931) or a polyclonal antibody raised against amino acids 496-511 of BACE2 (EMD Biosciences, Ab1) were added to the appropriate tissue samples (0.5 μg/1 mL of sample). Protein A/G sepharose beads (20 μL) were added to each sample and samples were incubated overnight at 4°C on a Rotoshake Genie. Beads were washed 3 times with TLB. Activity of the immunoprecipitated BACE1 or BACE2 were assayed directly on the beads (see below). For western blotting, samples were prepared in Laemmli buffer (2% SDS, 10% glycerol, 62.5 mM Tris-HCl, 0.002% bromophenol blue) with 10% (v/v) β-mercaptoethanol, and heated to 95°C for 5 minutes prior to loading. Proteins were separated using 10–20% SDS-PAGE Tris-Glycine Criterion gels (Bio-Rad), and electrically transferred to 0.45 μm nitrocellulose membranes. Membranes were blocked overnight with 2% BSA/1% BlockAce (Serotec) in 1X PBS, pH= 7.4 (Fisher). Blots were incubated in primary antibody (1 μg/mL) for 1–2 hours at room temperature (RT), and in secondary antibody (100 pg/mL) for 30 minutes to 1 hour (at RT). Between antibody applications, blots were washed 3 × 10 minutes in TBST buffer (1X TBS + 0.1% Tween-20; Fisher). Primary antibodies used were: MAB931 (BACE1 ectodomain, R&D Systems), MAB5308 (BACE1 C-terminus, Millipore), Ab1 (BACE2 C-terminus, EMD Biosciences), Ab5670 (BACE2, amino acids 441-457; Abcam), Ab8025 (BACE2, amino acids 44-59; Abcam), AF4097 (BACE2, amino acids 63-466; R&D Systems), and monoclonal antibody AC15 (against β-Actin; Sigma). Horseradish peroxidase (HRP) conjugated secondary antibodies and enhanced chemiluminescent detection reagents were obtained from Pierce Biotechnology.
BACE1 and BACE2 Activity Assay
Samples for assay were precleared with 20 μL of protein A/G beads for 30 minutes, then loaded (200 μL) onto black 96-well protein A/G plates (Pierce Biotechnology), along with 0.5 μg of antibody (MAB931 or MAB5308 for BACE1; Ab1 or Ab5670 for BACE2)/well. All samples were run at least in duplicate. A second set of samples, loaded in absence of antibody, served as controls for background activity. After overnight capture at 4°C, plates were washed extensively (5x) with TLB, and then loaded with 200 μL of TLB containing a BACE fluorogenic peptide substrate (1–10 μM) conforming to either human APPΔNL or APP wild type β-secretase cleavage site (EMD Biosciences). The Swedish familial mutation in APP (ΔNL), which results in early-onset, autosomal dominant AD, enhances β-secretase cleavage (Mullan et al. 1992; Citron et al. 1992; Cai et al. 1993); the APPΔNL sequence is a preferred substrate for both BACE1 and BACE2 (Andrau et al. 2003). For determination of substrate specificity, fluorogenic substrates VII (APPWT) or VIII (APPΔNL) were used (Abz/EDDnp; excitation/emission: 320 nm/420 nm). Fluorescence was measured at 10 minute intervals for up to 8 hours at 37°C on a BioTek plate reader. For the study of recombinant enzymes (BACE1, BACE2 or cathepsin D; R&D systems, 0.2 – 0.5 nM/well), we used APPWT (Calbiochem) or APPΔNL (R&D Systems) with MCA/DNP (7-methoxycoumarin-4-acetic acid/2,4-dinitrophenol; excitation/emission: 320 nm/400 nm; 0 – 10 μM/well) and read fluorescence relative to known concentrations of MCA. For measurement of activity in disease cases, we used an APPΔNL substrate with an EDANS/DABCYL combination (excitation/emission: 350 nm/490 nm; Peptides International, Louisville, KY), as this gave improved sensitivity in preliminary tests on tissue samples. The peptide used was a octamer corresponding to the P4′ to P4 amino acids flanking the β-secretase site in the human APP sequence. Based on the results of earlier experiments, readings for disease cases were endpoint measures taken after an incubation of 2 hours at 37°C. Assays performed on beads were similar, except 10 μM of a specific BACE inhibitor was added to some assays (EMD Biosciences BACE inhibitor IV).
Enzyme Linked Immunosorbent Assays (ELISA)
Measurement of Aβ is a straightforward technique that we routinely perform in our lab (Murphy et al. 1999; Murphy et al. 2000; Murphy et al. 2003; Murphy et al. 2007). Detergent soluble and formic acid soluble pools of Aβ were measured in tissue samples using a standard, well characterized sandwich ELISA. Details of this procedure, and the antibodies used, have been published (Das et al. 2003; Kukar et al. 2005; Levites et al. 2006; McGowan et al. 2005; Murphy et al. 2007). Briefly, plates (Immulon 4HBX) were coated with 1.0 μg/well of antibody, and blocked with a solution of Synblock (Serotec). Aβ capture was performed using monoclonal antibody Ab9, and detection was performed using biotinylated 4G8 (Covance) followed by HRP conjugated neutravidin (Pierce Biotechnology). Formic acid extracted material was initially neutralized by a 1:20 dilution in TP buffer (1 M Tris base, 0.5 M Na2HPO4), followed by a further dilution as needed (1:100 to 1:400) in AC buffer (0.02 M sodium phosphate buffer [pH = 7], 0.4 M NaCl, 2 mM EDTA, 0.4% Block Ace, 0.2% BSA, 0.05% CHAPS, and 0.05% NaN3). RIPA soluble fractions were diluted (1:5 to 1:20) in AC buffer alone. A peptide standard curve of Aβ40 was run on the same plate for comparison, and standards and samples were run at least in duplicate. Plates were washed between steps with standard PBS (2-4x) followed by PBS containing 0.05% Tween-20 (2-4x). Plates were developed with TMB reagent (Kirkegaard & Perry Laboratories), stopped (after approximately 30 minutes) with 6% o-phosphoric acid, and read at 450 nm using a BioTek multiwell plate reader. Aβ values were determined by interpolation relative to the standard curve. For the BACE1 and BACE2 ELISAs, recombinant BACE1 and BACE2 (R&D Systems) adsorbed directly to the plate were used as standards, and MAB5308/biotinylated-MAB931 (for BACE1) and Ab1/biotinylated-AF4097 (for BACE2) were used for capture and detection, respectively. ELISAs were conducted on the same samples from which activity data were collected.
Data Analyses
Data were analyzed using SPSS®. Group comparisons were made using Student’s t-test. A single tailed t-test was used for BACE1 comparisons based on the a priori hypothesis, derived from multiple published reports, that BACE1 is elevated in AD; as little is known about BACE2, a two tailed test was used instead. For comparison between experiments, data were analyzed by a general linear model ANOVA. Basic enzymology parameters were determined graphically in Sigmaplot®, from either Lineweaver-Burk or Hanes-Woolf plots. Significance of correlations (Pearson’s r) were always determined based on 2-tailed hypotheses.
Results
We first determined how precise standard conditions are for measuring BACE1 activity in tissue homogenate. Using conditions similar to those outlined in a commercial β-secretase assay kit (R&D Systems), we homogenized mouse brain samples in a low salt sodium acetate buffer. The BACE activity assay was performed using Substrate VIII APPΔNL, a substrate containing the Swedish APP mutation. Samples were first separated on a sucrose gradient and evaluated for activity, and a BACE1 immunoassay performed to locate the protein. There was a dissociation between where BACE1 protein was found on the gradient and where enzymatic activity was located (Fig 1). Though 84% of the measured activity is in the highest density fraction (43% sucrose), only 16% of the BACE1 protein is found in this fraction. In contrast, although only 20% of the measured activity is in the lower density fractions, 80% of the BACE1 protein can be found here. Therefore, most of the activity measured following the addition of a suitable substrate to a permissive buffer does not necessarily correspond to cleavage by BACE1, since most of the enzyme is located elsewhere. This indicates that, at best, the common procedure of adding substrate to homogenized tissue likely only yields a very rough approximation of BACE1 activity, even though the APPΔNL substrate is not preferably cleaved by other common aspartyl proteases. Although some portion of the activity measured under these conditions may be BACE2, it is probable that most of the residual activity can be attributed to other enzymes. In any case, since APPΔNL based substrates can be cleaved by both BACE1 and BACE2, this approach is unable to separate the contributions of both forms of the enzyme to the total activity. In order to independently measure each enzyme in the absence of other contaminating activities, we chose to refine an immunocapture method previously reported for BACE1 (Fukumoto et al. 2002) for use with both enzymes.
Figure 1.

The majority of BACE1 protein (as determined by ELISA) does not track with the majority of enzymatic cleavage activity against the APPΔNL substrate when brain homogenate is separated on a discontinuous sucrose gradient. The bulk of BACE1 protein is located in the third (32% sucrose) fraction, whereas the bulk of activity is found in last (43% sucrose) fraction.
We performed extensive assay validation experiments to verify that this methodology was correctly measuring BACE1 and BACE2. First, isolated β-secretase activity was insensitive to inhibition by the prototypical aspartyl protease inhibitor pepstatin A at concentrations up to 100 nM. This indicates that the activity is not a typical aspartyl protease, like cathepsin D, which is pepstatin sensitive; BACE cannot be inhibited by pepstatin except at moderate to high μM concentrations (Vassar et al. 1999). Broad spectrum inhibitors of metallo (EDTA), serine (aprotinin), and cysteine (leupeptin, E64) proteases failed to inhibit the activity. In contrast, an inhibitor of BACE1 and BACE2 (β-secretase inhibitor IV; EMD biosciences) was able to completely abolish activity at low μM concentrations (Fig. 2). Finally, the immobilized activity showed a many fold preference for the APPΔNL sequence over the wild type APP sequence (Fig. 3). These results are what would be expected if the assay is measuring the activity of BACE1 or BACE2, and not some other proteolytic activity. Results were highly comparable when using antibodies directed against either the N- or C-terminus of the enzymes, or with similar antibodies against the same or slightly different epitope from different suppliers, suggesting that this approach is versatile and robust. We noted no substantial difference between BACE1 and BACE2 isolated from mouse, rat or human tissue.
Figure 2.

Specificity of the BACE immunocapture method for measuring enzymatic activity. Isolated activity can be blocked by the addition of a commercially available specific inhibitor (β-secretase inhibitor IV; EMD biosciences) selective against both forms of BACE (BACE1 IC50, 15 nM; BACE2 IC50, 230 nM). For example, this compound will not effectively inhibit the highly abundant kidney aspartyl protease renin (IC50 >50 μM), and still blocks all of the activity isolated by either BACE1 or BACE2 antibody pull down. Data are expressed as base fluorescence units over two hours, corrected to background fluorescence.
Figure 3.

Immunocaptured BACE1 and BACE2 prefer the APPΔNL sequence over the corresponding hexameric sequence (P3′ to P3) from wild type human APP, as shown by time dependent cleavage of both substrates at 37°C. Data are expressed as base fluorescence units corrected to background (control wells, identical except with antibody omitted) at each time point. Capture at either the N- or C-terminus gave similar results, although N-terminal capture usually resulted in greater total activity. Antibodies: MAB931 (N-terminal) or MAB5308 (C-terminal) for BACE1; Ab1 (N-terminal) or Ab5670 (C-terminal) for BACE2.
We examined the ability of recombinant enzymes (BACE1, BACE2, and cathepsin D) to cleave the APPWT or APPΔNL sequence in the buffer conditions chosen for our assay. As expected, Kcat values for both BACE1 and BACE2 were higher for the APPΔNL substrate (169 h−1 and 185 h−1, respectively) as compared to the APPWT substrate (26 h−1 and 42 h−1, respectively). Interestingly, cathepsin D followed a similar pattern, preferring the APPΔNL substrate (50 h−1) over the APPWT substrate (3 h−1). Overall, the APPΔNL substrate was processed more efficiently (kcat/km) by BACE1 (0.12 h−1nM−1) and BACE2 (0.14 h−1nM−1) as compared to cathepsin D (0.03 h−1nM−1). These results differ from those observed in other studies, where Cathepsin D exhibited similar enzymatic properties to both BACE1 and BACE2 (Gruninger-Leitch et al. 2002; Schechter and Ziv 2008). The most likely reason for this discrepancy are substantial differences in the buffer conditions (e.g., presence or absence of detergents, salt concentrations, etc.) under which the assay is performed. Also, recombinant renin (R&D systems) was unable to appreciably cleave the APPΔNL substrate under these conditions (not shown). These data indicate that the APPΔNL substrate is preferred by both BACE1 and BACE2 under the conditions selected for our assay.
We next wanted to determine how well our procedure could measure BACE1 and BACE2 activities in both cases of AD and in cognitively normal, age-matched controls. Surprisingly, there was a substantial amount of BACE2 in human brain. This was contrary to our expectations based on mRNA expression data published for the rodent brain (Bennett et al. 2000). BACE1 and BACE2 were approximately equal in expression (as seen by western blot, relative to recombinant protein; Fig. 4). Based on observations of its apparent molecular weight, and its pattern of epitope reactivity, the predominant form of BACE2 in human brain appears to be splice form C, in which exon 7 is omitted. In this case series, it was difficult to detect a difference between AD and control cases by immunoblot alone, although BACE1 was slightly higher (p<0.1). This observation was born out by ELISA, in which BACE1 was clearly elevated in the AD cases (p<0.04). The amount of BACE2 protein did not differ between AD and control cases, as determined by either immunoblot or ELISA.
Figure 4.

(A) BACE1 (MAB931) and (B) BACE2 (Ab1) are both readily detected in human brain (recombinant protein in ng/lane). Comparison between multiple immunoblots with either recombinant BACE1 or BACE2 protein indicated that the amount of each enzyme in human brain, at least in the frontal cortex, is approximately equal. The predominant form of BACE2 in brain (consistent with that of rodent kidney), based on its apparent molecular weight and pattern of epitope reactivity, is likely splice form C (exon 7 omitted). (C) Differences between control cases and AD were difficult to detect by immunoblot alone, although BACE1 was slightly higher (p<0.1). Additional evaluation by ELISA confirmed that BACE1 was in fact elevated in the frontal cortex in AD brain (* = p<0.05) whereas BACE2 was unchanged.
BACE1 activity was substantially increased (p<0.04) in the superior frontal gyrus in AD cases, whereas BACE2 showed no significant change. These findings were similar for both specific activity (activity standardized to the total amount of protein) and when activity was standardized to the amount of BACE1 or BACE2 protein determined by ELISA (Fig. 5). As expected, Aβ extracted in either RIPA buffer or in 70% FA was significantly (p<0.01) elevated in the AD cases (Fig 6.). The amount of Aβ was significantly correlated with the amount of BACE1 (RIPA: R2 = 0.43, p<0.03; FA: R2 = 0.49, p<0.02), but not with BACE2 (RIPA: R2 = 0.04, p<0.6; FA: R2 = 0.04, p<0.6), in this brain region.
Figure 5.

BACE1 activity is significantly elevated in AD cases as compared to controls (* = p<0.05) when specific activity is determined either by standardization to total protein (A) or to BACE1 protein as measured by ELISA (B). BACE2 did not change significantly.
Figure 6.
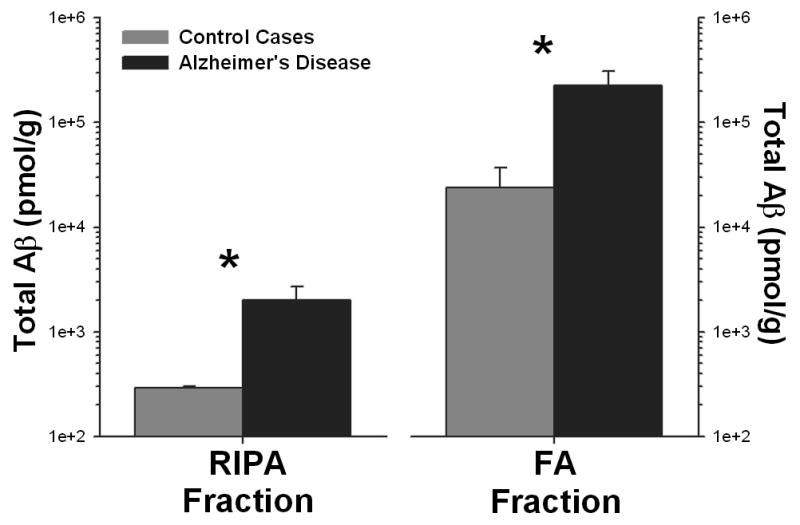
The total amount of Aβ increases in AD (* = p<0.01), as measured in both the RIPA and FA extracted fractions from human brain (note the exponential scale). The amount of Aβ in each fraction was correlated (p<0.05) with BACE1, but not BACE2, activity.
Discussion
β-Secretase is known to increase with age and is involved in human disease. Commercially available BACE assay kits provide limited insight into β-secretase activity, as non-β-secretase enzymes are capable, albeit inefficiently, of cleaving the BACE substrate. Other techniques looking at protein expression, such as western blotting, are only semi-quantitative. Although the preferences for substrate site are slightly different, BACE2 is also able to generate the same cleavage products as BACE1 (Marcinkiewicz and Seidah 2000; Hussain et al. 2000; Yan et al. 2001; Farzan et al. 2000; Bennett et al. 2000; Dominguez et al. 2005; Sun et al. 2005). It is therefore possible that BACE2 may be responsible for some component of the measured activity noted in current literature.
We observed a substantial increase in BACE1 protein and activity in the frontal cortex (Brodmann area 9) of AD brain. This is consistent with several other groups (Holsinger et al. 2002; Fukumoto et al. 2002; Yang et al. 2003; Li et al. 2004). We observed a positive correlation between the amount of BACE1 and the amount of Aβ in the brain, in both the detergent soluble (RIPA) and insoluble (FA) fractions, and no correlation with BACE2 activity. This is consistent with a role of BACE1 as the primary β-secretase enzyme in the brain, in spite of substantial quantities of both forms of the enzyme. However, since the ELISA that we used can only detect Aβ cleaved at the normal β-site, we cannot rule out the presence of peptides derived through internal cleavage (e.g., F19-F20) by BACE2 (Yan et al. 2001). BACE1 has been shown to be elevated around plaques in the brains of transgenic mice (Zhao et al. 2007), and β-secretase activity has been reported to be correlated with plaque number in the brains of sporadic AD cases (Li et al. 2004). However, other studies have failed to find a correlation between the amount of extractable Aβ and β-secretase activity (Fukumoto et al. 2002). It is likely that the difficulty in detecting robust correlations between Aβ and β-secretase enzymatic activity in the brain is because this activity is one step removed from the final step in Aβ production, cleavage of CTFβ by γ-secretase.
Although it is widely believed that BACE2 is expressed in the brain at much lower levels than BACE1, these data are primarily based on expression data from rodents (Bennett et al. 2000); BACE2 has not been studied at the same level of detail in the human brain. Even though BACE2 lies in the Down syndrome obligate region of chromosome 21 (Acquati et al. 2000), its role in the development of amyloid pathology in DS has remained controversial. Although there has been some suggestion of a role for BACE2 in the development of AD pathology in the DS brain (Motonaga et al. 2002), other reports have not shown consistent changes in the amount of BACE2 protein, even though transcription of BACE2 mRNA may be increased (Sun et al. 2006; Cheon et al. 2008). The role of BACE2 in AD is, at this time, unknown. However, an association was reported between one haplotype of BACE2 and AD in two independent data sets, (Myllykangas et al. 2005) suggesting that a link to disease may exist. In AD brain, BACE2 protein has been reported to be inversely correlated with Braak stage (Stockley et al. 2006), suggesting that it may decrease with increasing severity of neurodegeneration. Although this study is far from exhaustive, we did not detect significant changes in BACE2 activity in the AD brain. Given the limited number of studies on BACE2 in the human brain, further examination is warranted, especially since we detected a relatively large quantity of BACE2 protein and activity. BACE2 can act as an alternative α-secretase-like enzyme, cleaving within the normal Aβ sequence, and hence precluding the formation of the full length Aβ peptide (Yan et al. 2001; Sun et al. 2006). Since both forms of BACE can compete for the same substrate pool (Basi et al. 2003), even small changes in BACE2 in the human brain may indirectly contribute to the development of AD, simply by altering the available substrate pool for BACE1. It is possible that a much larger data set, including other forms of neurodegenerative disease and additional brain regions, may answer this question in greater detail.
The involvement of β-secretase as a key player in AD is by now a well-established matter. Although it seems likely that BACE1 is the major player in the disease process in the brain, there is still much we do not know about BACE2. For instance, if BACE2 is primarily expressed in glial cells (Basi et al. 2003), it may be selectively increased in areas of the AD brain that harbor substantial gliosis. We still do not know whether BACE1, BACE2, or both forms of the enzyme are altered in early stages of the disease, such as mild cognitive impairment (MCI) (Petersen 2004; Price et al. 2009). Changes in BACE1 (or 2) activity early on in the disease process would imply that β-secretase is more likely a cause, rather than a consequence, of AD.
Acknowledgments
This work was supported by grants awarded to M.P.M. by NIH (NS058382, AG005119, RR020171) and the state of Kentucky (KSEF-07-RDE-010). Tissue samples were provided by the Alzheimer’s Disease Centers of the Institute for Brain Aging and Dementia (P50 AG16573) and the Sanders-Brown Center on Aging (P50 AG028383). The authors thank Harry LeVine, Louis B. Hersh, William R. Markesbery, Christa M. Studzinski and Dana Niedowicz for helpful advice with these studies. The authors declare no financial conflict of interest.
Abbreviations
- Aβ
amyloid-β peptide
- AD
Alzheimer’s disease
- APP
β-amyloid precursor protein
- BACE
β-site amyloid precursor protein cleaving enzyme
- DS
Down Syndrome
- FA
formic acid
- RIPA
radio-immunoprecipitation assay
- SDS
sodium dodecyl sulfate
References
- Acquati F, Accarino M, Nucci C, Fumagalli P, Jovine L, Ottolenghi S, Taramelli R. The gene encoding DRAP (BACE2), a glycosylated transmembrane protein of the aspartic protease family, maps to the down critical region. FEBS Lett. 2000;468:59–64. doi: 10.1016/s0014-5793(00)01192-3. [DOI] [PubMed] [Google Scholar]
- Andrau D, Dumanchin-Njock C, Ayral E, et al. BACE1- and BACE2-expressing human cells: characterization of beta-amyloid precursor protein-derived catabolites, design of a novel fluorimetric assay, and identification of new in vitro inhibitors. J Biol Chem. 2003;278:25859–25866. doi: 10.1074/jbc.M302622200. [DOI] [PubMed] [Google Scholar]
- Basi G, Frigon N, Barbour R, Doan T, Gordon G, McConlogue L, Sinha S, Zeller M. Antagonistic effects of beta-site amyloid precursor protein-cleaving enzymes 1 and 2 on beta-amyloid peptide production in cells. J Biol Chem. 2003;278:31512–31520. doi: 10.1074/jbc.M300169200. [DOI] [PubMed] [Google Scholar]
- Bennett BD, Babu-Khan S, Loeloff R, Louis JC, Curran E, Citron M, Vassar R. Expression analysis of BACE2 in brain and peripheral tissues. J Biol Chem. 2000;275:20647–20651. doi: 10.1074/jbc.M002688200. [DOI] [PubMed] [Google Scholar]
- Cai H, Wang Y, McCarthy D, Wen H, Borchelt DR, Price DL, Wong PC. BACE1 is the major beta-secretase for generation of Abeta peptides by neurons. Nat Neurosci. 2001;4:233–234. doi: 10.1038/85064. [DOI] [PubMed] [Google Scholar]
- Cai XD, Golde TE, Younkin SG. Release of excess amyloid s protein from a mutant amyloid A protein precursor. Science. 1993;259:514–517. doi: 10.1126/science.8424174. [DOI] [PubMed] [Google Scholar]
- Cheon MS, Dierssen M, Kim SH, Lubec G. Protein expression of BACE1, BACE2 and APP in Down syndrome brains. Amino Acids. 2008;35:339–343. doi: 10.1007/s00726-007-0618-9. [DOI] [PubMed] [Google Scholar]
- Citron M, Oltersdorf T, Haass C, McConlogue L, Hung AY, Seubert P, Vigo-Pelfrey C, Lieberburg I, Selkoe DJ. Mutation of the beta-amyloid precursor protein in familial Alzheimer’s disease increases beta-protein production. Nature. 1992;360:672–674. doi: 10.1038/360672a0. [DOI] [PubMed] [Google Scholar]
- Das P, Howard V, Loosbrock N, Dickson D, Murphy MP, Golde TE. Amyloid-beta immunization effectively reduces amyloid deposition in FcRgamma−/− knock-out mice. J Neurosci. 2003;23:8532–8538. doi: 10.1523/JNEUROSCI.23-24-08532.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez D, Tournoy J, Hartmann D, et al. Phenotypic and biochemical analyses of BACE1- and BACE2-deficient mice. J Biol Chem. 2005;280:30797–30806. doi: 10.1074/jbc.M505249200. [DOI] [PubMed] [Google Scholar]
- Farzan M, Schnitzler CE, Vasilieva N, Leung D, Choe H. BACE2, a beta-secretase homolog, cleaves at the beta-site and within the amyloid-beta region of the amyloid-beta precursor protein. Proc Natl Acad Sci USA. 2000;97:9712–9717. doi: 10.1073/pnas.160115697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumoto H, Cheung BS, Hyman BT, Irizarry MC. beta-Secretase Protein and Activity Are Increased in the Neocortex in Alzheimer Disease. Arch Neurol. 2002;59:1381–1389. doi: 10.1001/archneur.59.9.1381. [DOI] [PubMed] [Google Scholar]
- Fukumoto H, Rosene DL, Moss MB, Raju S, Hyman BT, Irizarry MC. Beta-secretase activity increases with aging in human, monkey, and mouse brain. Am J Pathol. 2004;164:719–725. doi: 10.1016/s0002-9440(10)63159-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruninger-Leitch F, Schlatter D, Kung E, Nelbock P, Dobeli H. Substrate and inhibitor profile of BACE (beta-secretase) and comparison with other mammalian aspartic proteases. J Biol Chem. 2002;277:4687–4693. doi: 10.1074/jbc.M109266200. [DOI] [PubMed] [Google Scholar]
- Holsinger RM, McLean CA, Beyreuther K, Masters CL, Evin G. Increased expression of the amyloid precursor beta-secretase in Alzheimer’s disease. Ann Neurol. 2002;51:783–786. doi: 10.1002/ana.10208. [DOI] [PubMed] [Google Scholar]
- Hussain I, Powell D, Howlett DR, et al. Identification of a Novel Aspartic Protease (Asp 2) as beta-Secretase. Mol Cell Neurosci. 1999;14:419–427. doi: 10.1006/mcne.1999.0811. [DOI] [PubMed] [Google Scholar]
- Hussain I, Powell DJ, Howlett DR, et al. ASP1 (BACE2) cleaves the amyloid precursor protein at the beta-secretase site. Mol Cell Neurosci. 2000;16:609–619. doi: 10.1006/mcne.2000.0884. [DOI] [PubMed] [Google Scholar]
- Irizarry MC, Locascio JJ, Hyman BT. beta-site APP cleaving enzyme mRNA expression in APP transgenic mice: anatomical overlap with transgene expression and static levels with aging. Am J Pathol. 2001;158:173–177. doi: 10.1016/s0002-9440(10)63955-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukar T, Murphy MP, Eriksen JL, et al. Diverse compounds mimic Alzheimer disease-causing mutations by augmenting Abeta42 production. Nat Med. 2005;11:545–550. doi: 10.1038/nm1235. [DOI] [PubMed] [Google Scholar]
- Lammich S, Schobel S, Zimmer AK, Lichtenthaler SF, Haass C. Expression of the Alzheimer protease BACE1 is suppressed via its 5′-untranslated region. EMBO Rep. 2004;5:620–625. doi: 10.1038/sj.embor.7400166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levites Y, Das P, Price RW, Rochette MJ, Kostura LA, McGowan EM, Murphy MP, Golde TE. Anti-Abeta(42)- and anti-Abeta(40)-specific mAbs attenuate amyloid deposition in an Alzheimer disease mouse model. J Clin Invest. 2006;116:193–201. doi: 10.1172/JCI25410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Lindholm K, Yang LB, et al. Amyloid beta peptide load is correlated with increased beta-secretase activity in sporadic Alzheimer’s disease patients. Proc Natl Acad Sci U S A. 2004;101:3632–3637. doi: 10.1073/pnas.0205689101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Koelsch G, Wu S, Downs D, Dashti A, Tang J. Human aspartic protease memapsin 2 cleaves the beta-secretase site of beta-amyloid precursor protein. Proc Natl Acad Sci U S A. 2000;97:1456–1460. doi: 10.1073/pnas.97.4.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Bolon B, Kahn S, et al. Mice deficient in BACE1, the Alzheimer’s beta-secretase, have normal phenotype and abolished beta-amyloid generation. Nat Neurosci. 2001;4:231–232. doi: 10.1038/85059. [DOI] [PubMed] [Google Scholar]
- Marcinkiewicz M, Seidah NG. Coordinated expression of beta-amyloid precursor protein and the putative beta-secretase BACE and alpha-secretase ADAM10 in mouse and human brain. J Neurochem. 2000;75:2133–2143. doi: 10.1046/j.1471-4159.2000.0752133.x. [DOI] [PubMed] [Google Scholar]
- McGowan E, Pickford F, Kim J, et al. Abeta42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron. 2005;47:191–199. doi: 10.1016/j.neuron.2005.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan E. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Task Force on Alzheimer’s Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- Motonaga K, Itoh M, Becker LE, Goto Y, Takashima S. Elevated expression of beta-site amyloid precursor protein cleaving enzyme 2 in brains of patients with Down syndrome. Neurosci Lett. 2002;326:64–66. doi: 10.1016/s0304-3940(02)00287-2. [DOI] [PubMed] [Google Scholar]
- Mullan M, Crawford F, Axelman K, Houlden H, Lilius L, Winblad B, Lannfelt L. A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N-terminus of s-amyloid. Nat Genet. 1992;1:345–347. doi: 10.1038/ng0892-345. [DOI] [PubMed] [Google Scholar]
- Murphy MP, Beckett TL, Ding Q, Patel E, Markesbery WR, St Clair DK, LeVine H, 3rd, Keller JN. Abeta solubility and deposition during AD progression and in APPxPS-1 knock-in mice. Neurobiol Dis. 2007;27:301–311. doi: 10.1016/j.nbd.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Murphy MP, Das P, Nyborg AC, et al. Overexpression of nicastrin increases Abeta production. Faseb J. 2003;17:1138–1140. doi: 10.1096/fj.02-1050fje. [DOI] [PubMed] [Google Scholar]
- Murphy MP, Hickman LJ, Eckman CB, Uljon SN, Wang R, Golde TE. gamma-Secretase, evidence for multiple proteolytic activities and influence of membrane positioning of substrate on generation of amyloid beta peptides of varying length. J Biol Chem. 1999;274:11914–11923. doi: 10.1074/jbc.274.17.11914. [DOI] [PubMed] [Google Scholar]
- Murphy MP, Uljon SN, Fraser PE, et al. Presenilin 1 regulates pharmacologically distinct gamma-secretase activities. Implications for the role of presenilin in gamma-secretase cleavage. J Biol Chem. 2000;275:26277–26284. doi: 10.1074/jbc.M002812200. [DOI] [PubMed] [Google Scholar]
- Myllykangas L, Wavrant-De Vrieze F, Polvikoski T, et al. Chromosome 21 BACE2 haplotype associates with Alzheimer’s disease: a two-stage study. J Neurol Sci. 2005;236:17–24. doi: 10.1016/j.jns.2005.04.008. [DOI] [PubMed] [Google Scholar]
- Petersen RC. Mild cognitive impairment as a diagnostic entity. J Intern Med. 2004;256:183–194. doi: 10.1111/j.1365-2796.2004.01388.x. [DOI] [PubMed] [Google Scholar]
- Price JL, McKeel DW, Jr, Buckles VD, et al. Neuropathology of nondemented aging: presumptive evidence for preclinical Alzheimer disease. Neurobiol Aging. 2009;30:1026–1036. doi: 10.1016/j.neurobiolaging.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochette MJ, Murphy MP. Gamma-Secretase: substrates and inhibitors. Mol Neurobiol. 2002;26:81–95. doi: 10.1385/MN:26:1:081. [DOI] [PubMed] [Google Scholar]
- Roses AD. Apolipoprotein E affects the rate of Alzheimer disease expression: beta-amyloid burden is a secondary consequence dependent on APOE genotype and duration of disease [see comments]. [Review] Journal of Neuropathology & Experimental Neurology. 1994;53:429–437. doi: 10.1097/00005072-199409000-00002. [DOI] [PubMed] [Google Scholar]
- Roses AD. Apolipoprotein E, a gene with complex biological interactions in the aging brain. Neurobiol Dis. 1997;4:170–185. doi: 10.1006/nbdi.1997.0161. [DOI] [PubMed] [Google Scholar]
- Rossner S, Sastre M, Bourne K, Lichtenthaler SF. Transcriptional and translational regulation of BACE1 expression--implications for Alzheimer’s disease. Prog Neurobiol. 2006;79:95–111. doi: 10.1016/j.pneurobio.2006.06.001. [DOI] [PubMed] [Google Scholar]
- Schechter I, Ziv E. Kinetic properties of cathepsin D and BACE 1 indicate the need to search for additional beta-secretase candidate(s) Biol Chem. 2008;389:313–320. doi: 10.1515/BC.2008.025. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Sinha S, Anderson JP, Barbour R, et al. Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature. 1999;402:537–540. doi: 10.1038/990114. [DOI] [PubMed] [Google Scholar]
- Stockley JH, Ravid R, O’Neill C. Altered beta-secretase enzyme kinetics and levels of both BACE1 and BACE2 in the Alzheimer’s disease brain. FEBS Lett. 2006;580:6550–6560. doi: 10.1016/j.febslet.2006.10.076. [DOI] [PubMed] [Google Scholar]
- Sun X, He G, Song W. BACE2, as a novel APP theta-secretase, is not responsible for the pathogenesis of Alzheimer’s disease in Down syndrome. FASEB J. 2006;20:1369–1376. doi: 10.1096/fj.05-5632com. [DOI] [PubMed] [Google Scholar]
- Sun X, Wang Y, Qing H, et al. Distinct transcriptional regulation and function of the human BACE2 and BACE1 genes. Faseb J. 2005;19:739–749. doi: 10.1096/fj.04-3426com. [DOI] [PubMed] [Google Scholar]
- Tanzi RE, Bertram L. Twenty years of the Alzheimer’s disease amyloid hypothesis: a genetic perspective. Cell. 2005;120:545–555. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Vassar R, Bennett BD, Babu-Khan S, et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- Yan R, Bienkowski MJ, Shuck ME, et al. Membrane-anchored aspartyl protease with Alzheimer’s disease beta-secretase activity. Nature. 1999;402:533–537. doi: 10.1038/990107. [DOI] [PubMed] [Google Scholar]
- Yan R, Munzner JB, Shuck ME, Bienkowski MJ. BACE2 functions as an alternative alpha-secretase in cells. J Biol Chem. 2001;276:34019–34027. doi: 10.1074/jbc.M105583200. [DOI] [PubMed] [Google Scholar]
- Yang LB, Lindholm K, Yan R, et al. Elevated beta-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat Med. 2003;9:3–4. doi: 10.1038/nm0103-3. [DOI] [PubMed] [Google Scholar]
- Zhao J, Fu Y, Yasvoina M, et al. Beta-site amyloid precursor protein cleaving enzyme 1 levels become elevated in neurons around amyloid plaques: implications for Alzheimer’s disease pathogenesis. J Neurosci. 2007;27:3639–3649. doi: 10.1523/JNEUROSCI.4396-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]