

Abstract
Background & Aims
Myofibroblast transdifferentiation generates hepatic myofibroblasts, which promote liver fibrogenesis. The peroxisome proliferator-activated receptor (PPAR)γ is a negative regulator of this process. We investigated epigenetic regulation of PPARγ and myofibroblast transdifferentiation.
Methods
Chromatin immunoprecipitation assays (ChIP) assessed binding of methyl-CpG binding protein 2 (MeCP2) to PPARγ and chromatin modifications that silence this gene. MeCP2-/y mice and an inhibitor (DZNep) of the epigenetic regulatory protein EZH2 were used in the CCl4 model of liver fibrosis. Liver tissues from mice were assessed by histological analysis; markers of fibrosis were measured by quantitative PCR (qPCR). Reverse-transcription PCR detected changes in expression of the microRNA miR132 and its target, elongated transcripts of MeCP2. Myofibroblasts were transfected with miR132; PPARγ and MeCP2 expression were analyzed by qPCR or immunoblotting.
Results
Myofibroblast transdifferentiation of hepatic stellate cells is controlled by a combination of MeCP2, EZH2, and miR132 in a relay pathway. The pathway is activated by downregulation of miR132, releasing the translational block on MeCP2. MeCP2 is recruited to the 5′ end of PPARγ, where it promotes methylation by H3K9 and recruits the transcription repressor HP1α. MeCP2 also stimulates expression of EZH2 and methylation of H3K27 to form a repressive chromatin structure in the 3′ exons of PPARγ. Genetic and pharmacological disruptions of MeCP2 or EZH2 reduced the fibrogenic characteristics of myofibroblasts and attenuated fibrogenesis.
Conclusions
Liver fibrosis is regulated by an epigenetic relay pathway that includes MeCP2, EZH2, and miR132. Reagents that interfere with this pathway might be developed to reduce fibrogenesis in chronic liver disease.
Keywords: Myofibroblast transdifferentiation, PPARγ, MeCP2, HP1, EZH2, histone methylation
Introduction
Myofibroblasts are sufficient and essential for wound repair; they appear in injured tissues at an early stage and stimulate both wound contraction and formation of a temporary scar (granulation tissue) which protects against infection and further damage 1. Persistence and proliferation of myofibroblasts in chronic injury is associated with progressive deposition of collagen-rich extracellular matrix (ECM) leading to tissue fibrosis. This fundamental role in tissue homeostasis necessitates an improved understanding of myofibroblast generation, function and fate. Myofibroblasts are absent in normal tissue but appear upon injury as a consequence of stimulation/transdifferentiation of resident fibroblasts, epithelia and pericytes 2. Despite the different cellular origins, myofibroblast transdifferentiation generates a similar phenotype including de novo expression of smooth muscle α-actin (α-SMA) and secretion of the profibrogenic growth factor TGFβ1. In addition the cell will secrete copious amounts of types I and III collagen and the collagenase inhibitor, tissue inhibitor of metalloproteinases-1 (TIMP-1) which in combination promotes the net deposition of cross-linking fibril-forming collagen 3. Myofibroblast transdifferentiation is therefore a highly conserved physiological process that must be tightly regulated to ensure limited and controlled scar formation.
Myofibroblast transdifferentiation is underpinned by changes in expression of hundreds of different genes that combine to generate the myofibroblast epigenome. Although numerous candidate transcription factors have been identified, the early regulatory events that trigger and orchestrate reprogramming of the myofibroblast epigenome are poorly understood. We recently described a role for DNA (CpG) methylation as a blue-print for the global alterations in the epigenome that drive transdifferentiation 4. Methylated CpGs act as a signal for transcriptional repression and silencing. Transcriptional silencing of the peroxisome proliferator-activated receptor-gamma (PPARγ) gene is required for conversion of hepatic stellate cells (HSC) into myofibroblasts 5, 6. PPARγ expression is associated with the adipogenic features of quiescent HSC (qHSC) and must be silenced for the cell to adopt its myofibroblastic characteristics. Forced over-expression of PPARγ in hepatic myofibroblasts results in reversion of transdifferentiation, with down-regulation of type I collagen, loss of proliferation and reacquisition of their adipogenic characteristics. Similar data are reported for lung fibroblasts in which expression of a constitutive active PPARγ protein inhibits the ability of TGFβ1 to induce myofibroblastic characteristics including expression of collagen I 7. TGFβ1 stimulation of myofibroblastic conversion of ocular fibroblasts is also prevented by adenoviral-mediated over-expression of PPARγ 8. PPARγ agonists can also partially reverse epithelial to mesenchymal transition (EMT) in anaplastic thyroid cancer cells 9. Taken together these studies suggest a pivotal regulatory role for PPARγ in transdifferentiation for cells of distinct origins and phenotypes. As such, we reasoned that investigation into how transcriptional silencing of PPARγ is regulated in HSC would lead to the discovery of novel and critical epigenetic regulators of fibrogenesis.
Gene silencing can be achieved by at least three epigenetic mechanisms; DNA methylation and the activities of the methyl-CpG binding proteins; remodelling of the histone code by enzymes that either add or remove acetyl and methyl groups to the lysine and arginine tails of the core histones; and the actions of microRNAs 10. Here we describe how all three mechanisms combine to generate a multi-component epigenetic relay pathway that regulates repression of PPARγ transcription in the hepatic myofibroblast. Furthermore, by disrupting the activities of two key components, the methyl-CpG binding protein 2 (MeCP2) and the histone methyltransferase EZH2 11, 12, we provide in vitro and in vivo evidence that the epigenetic relay has a wider function in controlling the myofibroblast phenotype and liver fibrosis.
Methods
Animals
Mecp2-/y mice were obtained from Jax labs (strain B6.129P2(C)-Mecp2tm1.1Bird/J) and are a cross of a constitutive CMVCre strain and a strain carrying an Mecp2 gene containing loxP sites around exons 3 and 4. The crossed line (pure C57BL/6) lack expression of Mecp2 mRNA and protein, appear normal at birth but develop mobility problems and have an expected lifespan of 50-60 days. Authors hold licences for work relating to all experiments carried out in animals issued/approved by local ethical committee and UK Home Office.
Chronic CCl4 liver injury model
Fibrogenesis was induced by 3-week CCl4 treatment of 6 week old Mecp2-/y or age matched Wt littermates. Mice were injected IP twice weekly with CCl4/olive oil in a 1:1[vol/vol] ratio at 1μl/g body weight. 24 hours after the final CCl4 administration, animals were sacrificed and liver samples prepared.
Acute CCL4 liver injury
3 deazaneplanocin A was administered IP to two groups of five C57Bl6 mice (at 15mg/m2) 2 hours prior to a single dose of CCl4 (prepared as in chronic CCl4). Mice were sacrificed 24h after injury, bloods taken for assessment of liver enzyme levels and tissues harvested for histological/biochemical analysis.
Cell isolation
Rat and mouse HSC (from C57Bl6 Wt or Mecp2-/y livers) were isolated as previously described 29. The protocol for isolation of activated HSC from rat livers injured by bile duct ligation (BDL) for 10 days or acute or repeated injury with CCl4 for 3 weeks is provided in the supplementary material.
Immunohistochemistry
Mouse liver tissue was fixed in 10% formalin in phosphate-buffered saline, and liver sections stained with Sirius Red as previously described 30.
siRNA transfection
Cells were transfected with 2μgs siRNA targeting EZH2, MeCP2 or negative control using a square wave electroporator and allowed to grow for 48h prior to preparation of RNA and/or whole cell extracts.
Micro RNA detection, amplification and transfection
Micro RNAs were isolated from cells and reverse transcribed; rat miR132 was detected using miScript primer assay. To assess the effect of miR132 presence in MFBs, 2μg of miR132 mimic were transfected into myofibroblasts (as outlined for siRNA).
SDS-PAGE and Immunoblotting
SDS-PAGE and immunoblotting was done as previously described 30. Antibody recognizing MeCP2 (Abcam) was used at 1μg/ml; EZH2 (Active Motif) at 1/500 dilution and β actin (Sigma) at 1/1000 dilution.
Quantitative Reverse Transcriptase-Polymerase Chain Reaction (qRT-PCR)
Total RNA was obtained as in 30; cDNA was generated using a random hexamer primer and MMLV RT. Quantitative PCR program: 20sec at 94°C, 4 0 cycles of 20sec at 55°C, 30sec at 72°C and 5sec at 94°C. All reactions were normal ized to the internal control and relative level of transcriptional difference calculated using the following equation: [1/(2A)] ×100.
Long or short 3′UTR of MeCP2 analysis
Long or short 3′UTR of MeCP2 mRNA was detected by RT-PCR using primer pairs designed to specifically detect regions located at 2kb (short transcript) or 10kb (long transcript) of 3′UTR in MeCP2 mRNA.
Chromatin Immunoprecipitation Assay
ChIP for RNA pol II, MeCP2 and HP1α were carried out using 100μg cross-linked chromatin prepared as described previously4. ChIP grade antibodies were raised against MeCP2, RNA polymerase II CTD repeat YSPTSPS (phospho S2) and HP1α (Abcam). 10μg of antibody were used in each ChIP reaction. Native ChIP was carried out as described elsewhere 31, using 100μg native chromatin and antibodies raised against di/tri-methylated H3K4, H3K27 and H3K9 (Abcam). Primers used for qPCR analysis of ChIP reactions are listed in the supplementary data. Average values of eluates were normalized to average values of inputs.
Results
Culture-induced transdifferentiation of HSC was associated with a greater than 95% loss of PPARγ transcript (Fig 1a) and as determined by cross-linked ChIP, depletion of elongating RNA polymerase II (P-Ser2-RNAP) at the PPARγ gene (Fig 1b). These changes in transcription coincided with recruitment of the methyl-CpG binding protein MeCP2 to the promoter and exons A1 and A2 of the PPARγ gene (Fig 1c) which are spanned by a methylated CpG island (supplementary Figs 1 and 2). MeCP2 operates as a powerful epigenetic repressor of gene transcription 13. We therefore investigated the possibility that it controls silencing of PPARγ expression during myofibroblast transdifferentiation. siRNA knockdown of MeCP2 in myofibroblasts (Fig 1d) resulted in elevated PPARγ transcript expression (Fig 1e). Furthermore, 5-fold increased expression of PPARγ mRNA was observed in MeCP2 deficient mouse myofibroblasts (Mecp2-/y) compared with Wt (Fig 1f). We previously demonstrated that MeCP2 expression is barely detectable in normal liver and is induced selectively in myofibroblasts of the injured liver 4. To investigate a fibrogenic role for MeCP2 in vivo we compared hepatic PPARγ expression between Wt and Mecp2-/y mice repeatedly injured with CCl42. Absence of hepatic MeCP2 (Fig 2a) resulted in higher PPARγ mRNA expression in the injured liver (Fig 2b). PPARγ is a negative regulator of type I collagen expression and other phenotypic characteristics of the myofibroblast5, 6. Expression of type I collagen (Col1a1), TIMP-1 and αSMA transcripts were all reduced in injured Mecp2-/y livers compared to Wt (Fig 2b). Sirius red analysis revealed tracts of matrix that formed bridges of fibrotic tissue spanning hepatic vessels of injured Wt mice (Fig 2c, top panel). In contrast, bridging fibrosis was not evident in Mecp2-/y livers (Fig 2c, bottom panel). Blinded histopathology grading for fibrosis confirmed reduced fibrogenesis in Mecp2-/y livers with an average score grade of 1.8 (mild fibrosis) compared with grade 3 (severe bridging fibrosis) for Wt (Fig 2d). Quantification of area of Sirius red staining confirmed reduced fibrogenesis in Mecp2-/y livers (Fig 2e). ALT levels were similar between Wt and Mecp2-/y mice (supplementary Fig 3) and as hepatic MeCP2 expression is selective for myofibroblasts4, we conclude that deletion of MeCP2 protects against fibrosis due to the loss of influence on fibrogenesis rather than any effect on the mechanism of liver injury. Forced over-expression of PPARγ in myofibroblasts suppresses expression of type I collagen and protects against development of fibrosis 14. Analysis of type I collagen expression by HSC-derived myofibroblasts confirmed low levels of expression in MeCP2-depleted cells (Fig 2f). The attenuated fibrogenic response of the Mecp2-/y mouse is therefore explained, at least in part, by maintenance of PPARγ expression and suppression of the pro-fibrogenic characteristics of MeCP2-deficient myofibroblasts. However, MeCP2 is also required for repression of IκBα expression during transdifferentiation, this being critical for the expression of NF-κB-dependent genes4. MeCP2 therefore operates as a coordinator of the transcriptional silencing of multiple genes associated with the qHSC phenotype.
Figure 1. Diminution of PPARγ in myofibroblasts is regulated by MeCP2.
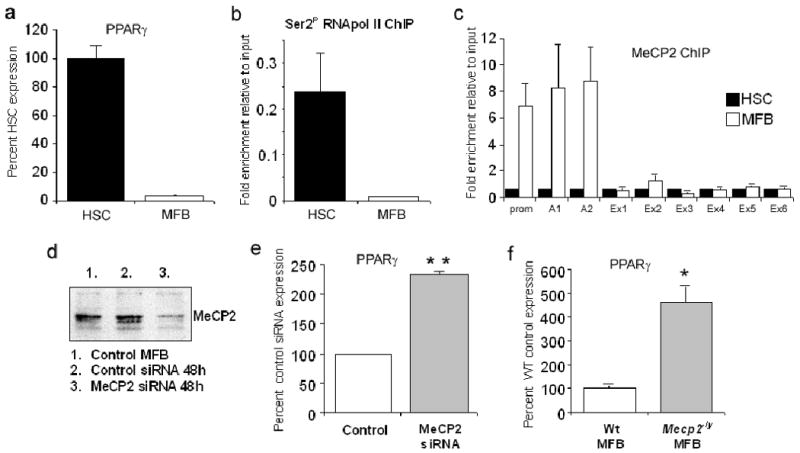
(a) PPARγ mRNA levels were quantified by qRT-PCR in rat qHSCs and day 10 transdifferentiated myofibroblasts. (b) 100μg of crosslinked chromatin obtained from rat HSC and myofibroblasts was incubated with 10μg of anti RNAPolII phosphoSer2; ChIP assay was carried out and exon A2 of rat PPARγ amplified. (c) ChIP assay was carried as in (1b) using 10μg anti MeCP2 Ab; PPARγ exons and promoter were amplified by qPCR. (d) 5×106 rat myofibrobasts were electroporated with 2μgs total siRNA designed to target rat MeCP2 and knockdown confirmed by a western blot. (e) Total RNA was isolated from control or rat MeCP2 siRNA transfected cells and used used as template in qRT-PCR reactions with primers specific for rat PPARγ. p=0.0019 (f) Quiescent HSCs were isolated from Wt C57Bl6 or Mecp2-/y mice and transdifferentiated in vitro for 14 days. Total RNA was prepared from both cell populations and PPARγ mRNA quantified by qRT-PCR p=0.031.
Figure 2. MeCP2 knockouts are attenuated for fibrogenesis.
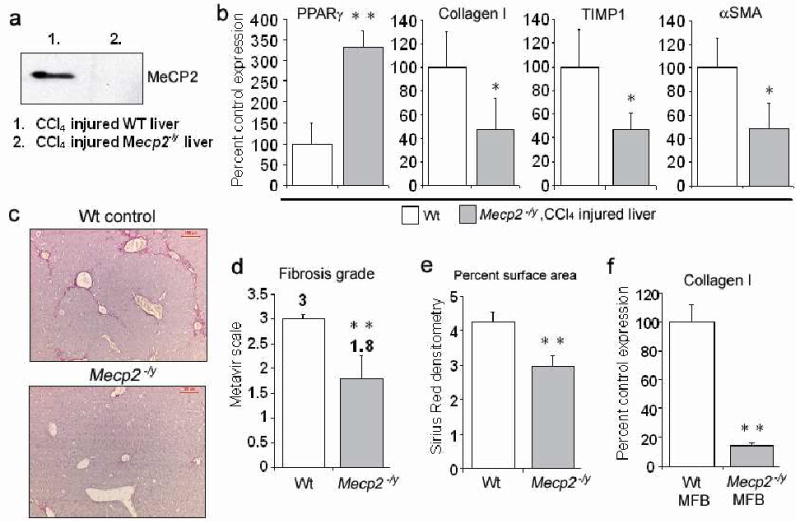
(a) 200μg whole cell protein extract isolated from frozen CCl4 injured Wt or Mecp2-/y liver was separated by SDS PAGE, proteins transferred onto membrane and immunoblotted for MeCP2. (b) RNA was isolated from frozen CCl4 injured Wt or Mecp2-/y liver, and used in qRT-PCR reactions with primers specific for mouse PPARγ (p=0.0025), collagen I (p=0.036), TIMP-1 (p=0.036) and α-SMA (p=0.036). (c) Sirius Red immunostaining on sections cut from CCl4 injured Wt or Mecp2-/y liver (×5 magnification). (d) Sirius red immunostained sections from 5 Wt and Mecp2-/y CCl4 injured livers were blinded and scored for the grade of fibrosis based on Metavir scale (p=0.0015) and (e) quantified by morphometry (p=0.0029). Averaged results for 5 animals. (f) qHSC were isolated from Wt or Mecp2-/y livers and transdifferentiated in vitro for 14 days. Total RNA was prepared from both cell populations and used for qRT-PCR with primers specific for mouse collagen1 (p=0.032).
We next investigated how MeCP2 expression is controlled during myofibroblast transdifferentiation. MeCP2 protein is undetectable in qHSC (both in vitro and in vivo (see supplemental figure 4 for in vivo data)) and is induced with transdifferentiation 4. Here (Fig 3a) we show that MeCP2 protein expression is induced during the early transitionary phase (culture day 1) of transdifferentiation and increases in expression with each subsequent day of culture, reaching high levels when the cells are fully transdifferentiated (day 7). However, MeCP2 transcript was detected in freshly isolated qHSC (Fig 3b) and similar levels of MeCP2 mRNA were measured in HSC isolated from control livers and HSC isolated from livers injured by either BDL or CCl4 that are undergoing transdifferentiation (supplemental data 5a). These data suggest a post-transcriptional mechanism of regulation for MeCP2 during transdifferentiation. Klein et al previously described how the microRNA miR132 exerts a repressive influence on MeCP215. The Mecp2 gene contains multiple polyadenylation sites and generates several transcripts which differ in their length of 3′ UTR, ranging from 1.8kb to 10kb 15. The 10kb transcript predominates in the brain and unlike the shorter transcripts includes recognition elements for several microRNAs including miR132 which is also selectively enriched in the brain 15. Primers located between 1 and 8.5kb from the translational stop codon of the MeCP2 transcript were employed for RT-PCR detection of short and long transcripts respectively. This analysis showed that qHSC and myofibroblasts express the elongated form of MeCP2 transcript inclusive of the miR132 binding site (Fig 3c). qRT-PCR analysis confirmed similar levels of the elongated transcript expressed in HSC isolated from control, BDL and CCl4 injured livers (supplemental Fig5b). We next showed that MFB transdifferentiation is accompanied by a greater than 90% diminution of miR132 (Fig 4a). Loss of miR132 expression in myofibroblasts during liver injury was confirmed by quantification of the microRNA in HSC isolated from BDL- and CCl4-injured rat livers (Fig 4b). Transfection of miR132 increased expression of the microRNA in myofibroblasts by 300-fold (Fig 4c) which was associated with diminished expression of MeCP2 protein (Fig 4d). This treatment was also accompanied by increased PPARγ mRNA expression (Fig 4e). We conclude that MeCP2, and in turn the myofibroblast phenotype are under the negative regulation of miR132 in qHSC.
Figure 3. MeCP2 expression in HSC.
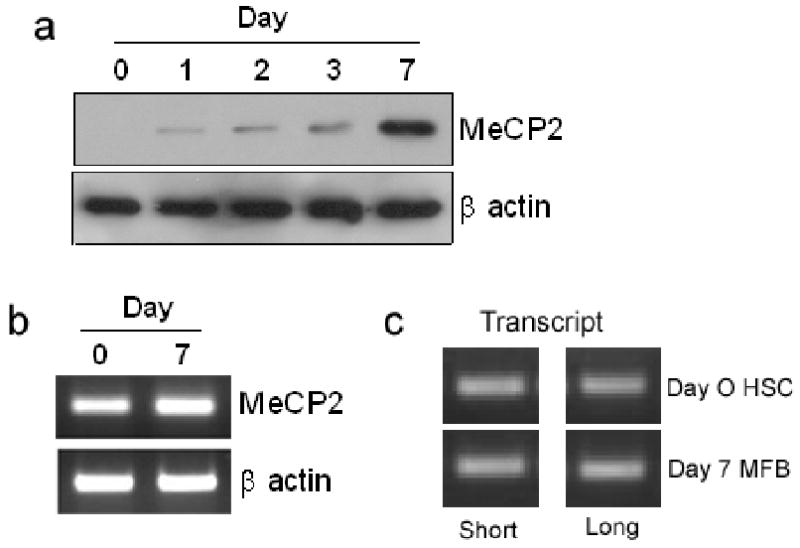
(a) 30μg whole cell extract from freshly isolated HSC harvested at day 1, 2, 3 and 7 of culture were separated by SDS-PAGE and immunoblotted for MeCP2 and β actin. (b) RNA isolated from qHSC (culture day 0) and myofibroblasts (culture day 7) was used for semi-quantitiative RT-PCR analysis of MeCP2 and β actin or (c) or interrogated with primers specific for short/long MeCP2 transcript.
Figure 4. MeCP2 expression is regulated by miR132.
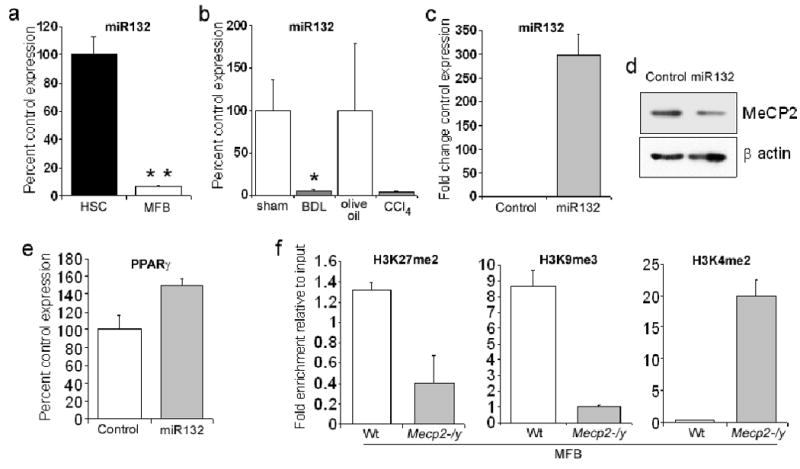
(a) Micro RNAs were isolated from rat qHSC and myofibroblasts using miRNeasy mini kit and reverse transcribed using miScript Reverse Transcription Kit. miR132 was detected with miScript primer assay 218300. (b) Micro RNA was purified from HSC isolated from livers of mice following BDL (or sham control) and CCl4 (or olive control) injuries and miR132 detected as described above. (c to e) 5×106 myofibroblasts were mixed with 2μg miR132 mimic or control siRNA and electroporated. Cells were harvested 48 hours later for RNA and whole cell protein extract. (c) mIR132 was quantified by miScript primer assay 218300 (d) Whole cell protein extracts were separated by SDS-PAGE and immunoblotted for MeCP2 and β–actin. (e) cDNA obtained in (b) was further used as template for qRT-PCR analysis of PPARγ. (f) Native chromatin was prepared from in vitro transdifferentiated Wt or Mecp2-/y myofibroblasts. 100μg of native chromatin was incubated with 10μg of anti-dimethyl H3K27, H3K9 or H3K4 and ChIP assays carried out. Immunoprecipitated DNA was used as template in qPCR reactions using mouse PPARγ promoter specific primers.
We next determined downstream regulatory events through which MeCP2 controls PPARγ transcription. Native ChIP detected MeCP2-dependent histone methylation signatures at the 5′ end (promoter region) of the PPARγ gene (Fig 4f). Methylated H3K9 16 and H3K27 11, 16 are signatures of repressive chromatin that were enriched at the PPARγ gene in Wt myofibroblasts; however both of these modifications were depleted in Mecp2-/y myofibroblasts. By contrast the transcriptional active signature H3K4me2 16 was almost absent in Wt myofibroblasts, but was highly enriched in Mecp2-/y cells. MeCP2 must therefore orchestrate multiple epigenetic events at the PPARγ chromatin. It has been previously reported that MeCP2 facilitates H3K9 methylation and that this repressive mark recruits the transcriptional repressor HP1α 17. ChIP analysis across the PPARγ gene confirmed that HP1α is recruited selectively to the promoter and exons A1 and A2 (Fig 5a) which coincides with the binding pattern for MeCP2 shown in Fig 1c. Since HP1α is selectively recruited to the 5′ region of the PPARγ gene in myofibroblasts and is absent from the gene in qHSC, we conclude that MeCP2-dependent methylation of H3K9 recruits HP1α. HP1α is predominantly associated with heterochromatic DNA but is also found at euchromatic sites in association with transcriptionally repressed genes where it promotes a repressive chromatin structure 17, 18. Hence one mechanism by which MeCP2 regulates transcriptional silencing of PPARγ is the recruitment of HP1α to methylated H3K9 at the 5′ region of the PPARγ gene.
Figure 5. MeCP2 regulates HP1α recruitment, expression of EZH2 and H3K27 methylation.
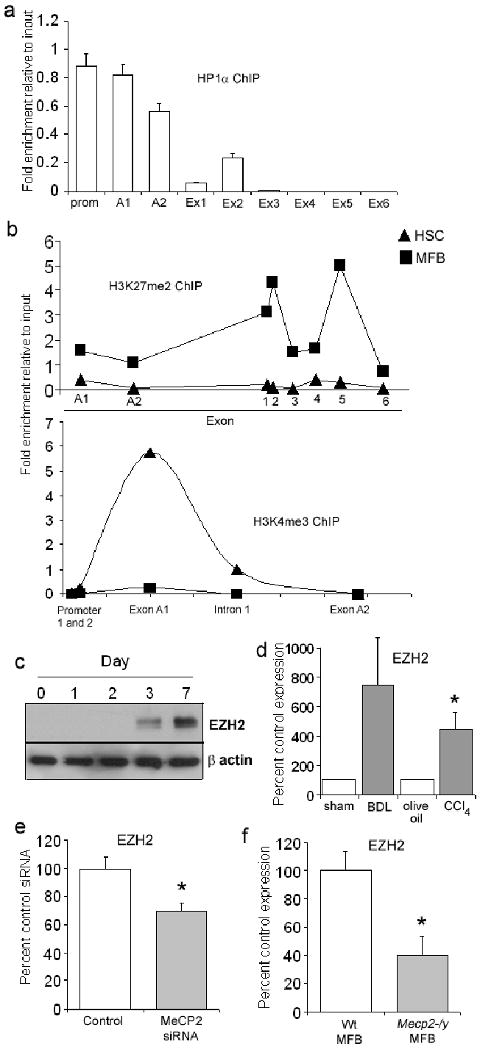
(a) 100μg of crosslinked chromatin from rat myofibroblasts was incubated with 10μg of anti HP1α and ChIP assay carried out. Immunoprecipitated DNA was used as template in qPCR reactions using primers specific for PPARγ promoter/exons in the rat gene. (b) 100μg of native chromatin prepared from rat HSC or myofibroblasts was incubated with 10μg of anti-dimethyl H3K27 (top) or anti-trimethyl H3K4 (bottom) and ChIP assay carried out. Immunoprecipitated DNA was used as template in qPCR reactions using primers specific for rat PPARγ promoter/exons (c) 30μg whole cell extract from freshly isolated HSC or myofibroblasts harvested at day 1, 2, 3 and 7 culture were separated by SDS-PAGE and immunoblotted for EZH2 and β actin. (d) qRT-PCR analysis of EZH2 mRNA was performed using RNA extracted from HSC isolated from BDL (or sham operated) and CCl4 (or olive oil) injured rat liver (n=5)(p=0.0184). (e) RNA isolated from myofibroblasts transfected with 2μgs control or MeCP2 siRNA (from figure 1e) was for qRT-PCR analysis of EZH2 (p=0.0152)(f) RNA from Wt or Mecp2-/y myofibroblasts (from figure 1f) was utilized for qRT-PCR analysis of EZH2 expression (p=0.0478).
The observation that methylation of H3K27 was depleted in Mecp2-/y myofibroblasts was of interest as to date there is no known association between MeCP2 and this histone modification. To investigate the relationship between MeCP2 and H3K27 methylation in more detail we employed high resolution ChIP to determine if methylation of H3K27 occurs at regions of the PPARγ gene associated with chromatin bound MeCP2. This involved digestion of chromatin with monococcal nuclease to generate mononucleosomes (covering roughly 146 nucleotides) prior to IP followed by qPCR using primers specific for the A1, A2 and downstream exons. H3K27 methylation (H3K27me2) was barely detected at the PPARγ gene in qHSC. In myofibroblasts, low levels of H3K27me2 were detected at exons A1 and A2, however the modification was particularly enriched in myofibroblasts at exons 1, 2 and 5 (Fig 5b). Of note, H3K4 trimethylation (H3K4me3) a marker of transcriptional active chromatin, was enriched at exon 1 and intron 1 in qHSC but was absent in myofibroblasts confirming that transdifferentiation involves coordinated modulation of active and repressive epigenetic signatures (Fig 5b). As MeCP2 does not associate with exons downstream of exon A2 at the 5′ end of the PPARγ gene (Fig 1c) it is unlikely to be directly responsible for H3K27 methylation at exons 1, 2 or 5. Methylation of H3K27 is specifically mediated by the evolutionarily conserved Polycomb repressor complex 2 (PRC2) and its constituent H3K27 methyl-transferase EZH2 11, 19. EZH2 protein expression was absent in qHSC and was induced with transdifferentiation, although later (culture day 3) than observed for MeCP2 (Fig 5c). EZH2 mRNA was also induced in HSC transdifferentiating in vivo in response to BDL and CCl4 injury (Fig 5d). Cells depleted of MeCP2 by siRNA expressed lower levels of EZH2 compared with control cultures (Fig 5e). We also measured 60% lower EZH2 transcript expression in Mecp2-/y myofibroblasts compared with Wt cells. These results reveal an unexpected role for MeCP2 as a positive regulator of EZH2 expression and provide an explanation of how MeCP2 stimulates H3K27 methylation. It has recently emerged from studies with neurons that MeCP2 can function as an activator of transcription for a subset of genes including somatostatin, opioid receptor kappa 1, guanidinoacetate methyltransferase and G protein–regulated inducer of neurite outgrowth 1 20.
To provide direct evidence that EZH2 is a regulator of PPARγ gene transcription we employed siRNA-mediated knockdown to achieve 80% depletion of EZH2 in myofibroblasts (Fig 6a), this resulted in elevated expression of PPARγ transcript (Fig 6b) and a modest decrease in expression of type I collagen (supplemental Fig 6). A similar effect was observed when myofibroblasts were treated with 3 deazaneplanocin A (Fig 6c) which depletes cells of EZH2 21. Furthermore, treatment of freshly isolated quiescent hepatic stellate cells with 3 deazaneplanocin A completely prevented morphological signs of transdifferentiation (Fig 6d) and suppressed the induction of α1 (I) collagen and tissue inhibitor of metalloproteinase-1 (TIMP-1) (Fig 6e). Administration of the EZH2 inhibitor to mice during acute CCl4 injury also suppressed the induction of type I collagen and TIMP-1 which provide surrogate markers for in vivo transdifferentiation of HSC and the hepatic fibrogenic response (Fig 6f). EZH2 is therefore a negative regulator of PPARγ transcription in myofibroblasts, but also appears to play a wider regulatory role in promoting myofibroblast transdifferentiation and fibrosis. Furthermore, the stimulation of EZH2 expression and subsequent methylation of H3K27 in the downstream exons of the PPARγ gene is identified as a second mechanism through which MeCP2 achieves transcriptional silencing of PPARγ. The combined effect of H3K9 methylation and HP1α binding at the 5′ end of the PPARγ gene with H3K27 methylation and PRC1 recruitment in the downstream exons would be to prevent both transcriptional initiation and elongation which reflects the biological importance of suppressing PPARγ expression for MTD and fibrosis.
Figure 6. EZH2 regulates PPARγ, myofibroblast transdifferentiation and fibrogenesis.
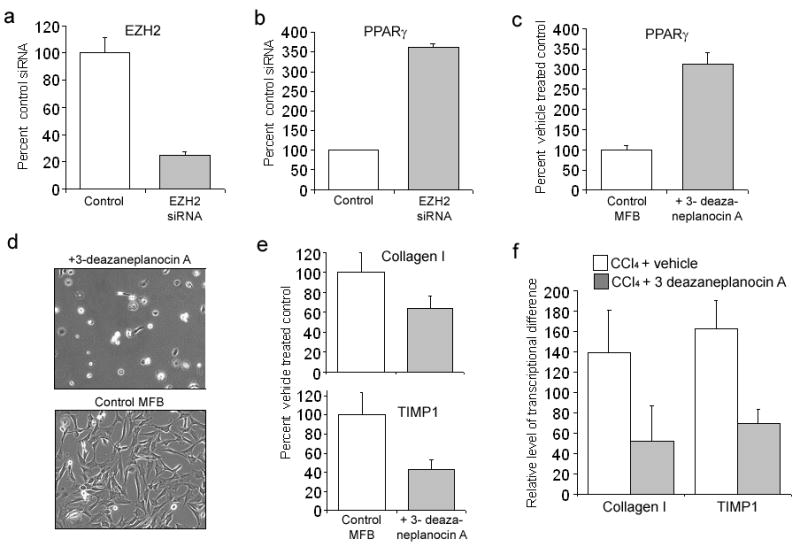
(a) and (b) 5×106 rat myofibroblasts electroporated with 2μgs of control or EZH2 siRNA. Total RNA was isolated and used for qRT-PCR detection of EZH2 (a) or PPARγ (p=0.0012) (b). (c) Myofibroblasts were treated with 1μM 3-deazaneplanocin A or vehicle for 72h; total RNA prepared and used for qRT-PCR detection of PPARγ. (d) Freshly isolated rat HSC were cultured for 12 hrs in standard media (bottom photomicrograph) or media containing 1μM 3-deazaneplanocin A (top photomicrograph), cultures were washed and replaced with standard media and cells incubated for a further 10 days. (e) RNA obtained in 5c was used for qRT-PCR detection of collagen I and TIMP-1. (f) qRT-PCR analysis of type 1 collagen and TIMP-1 transcripts from RNA isolated from CCl4 injured livers from control or mice pre-treated for 1hrs with 3-deazaneplanocin A (n=4).
Discussion
The liver is frequently exposed to a wide variety of toxins and damaging microbes, it is therefore critical that it is able to respond to these insults by mounting a rapid and effective wound-healing response. Similar to other vital organs (e.g. kidney, pancreas, heart and lung) scar formation or fibrogenesis is an important protective response of the liver to cellular damage. The major fibrogenic cell in the liver (and other organs) is the myofibroblast, which can be generated locally at the site of injury by transdifferentiation of resident differentiated cells 1-3. Selective depletion of liver myofibroblasts attenuates fibrosis in rodent models of chronic and progressive liver disease, moreover the clearance of myofibroblasts by apoptosis is associated with regression of fibrosis 2, 22, 23. Hence, both the production and the life-span of the myofibroblast must be subject to exquisite control mechanisms to ensure a measured wound-healing response. Regulatory events that determine the balance between survival and apoptosis of myofibroblasts have received a great deal of recent attention 23. By contrast, relatively little is understood about the mechanisms that regulate myofibroblast transdifferentiation. Here, we describe a novel multi-step epigenetic relay network that operates to control transdifferentiaion and fibrogenesis in the injured liver (Fig 7). To discover this pathway we used a candidate gene approach focusing on mechanisms responsible for transcriptional suppression of PPARγ. The rationale for this approach is based on the fact that PPARγ is a suppressor of MTD which must be silenced for the cell to acquire myofibroblastic characteristics 5. By first discovering the epigenetic regulators (MeCP2 and EZH2) that are responsible for PPARγ silencing during transdifferentiation and then investigating their contribution to fibrogenesis in rodent models of liver injury, we have identified important epigenetic regulators of fibrosis and new targets for therapeutic application in chronic liver disease.
Figure 7. mIR132, MeCP2 and EZH2 operate in a regulatory epigenetic relay pathway that culminates in transcriptional repression of PPARγ.
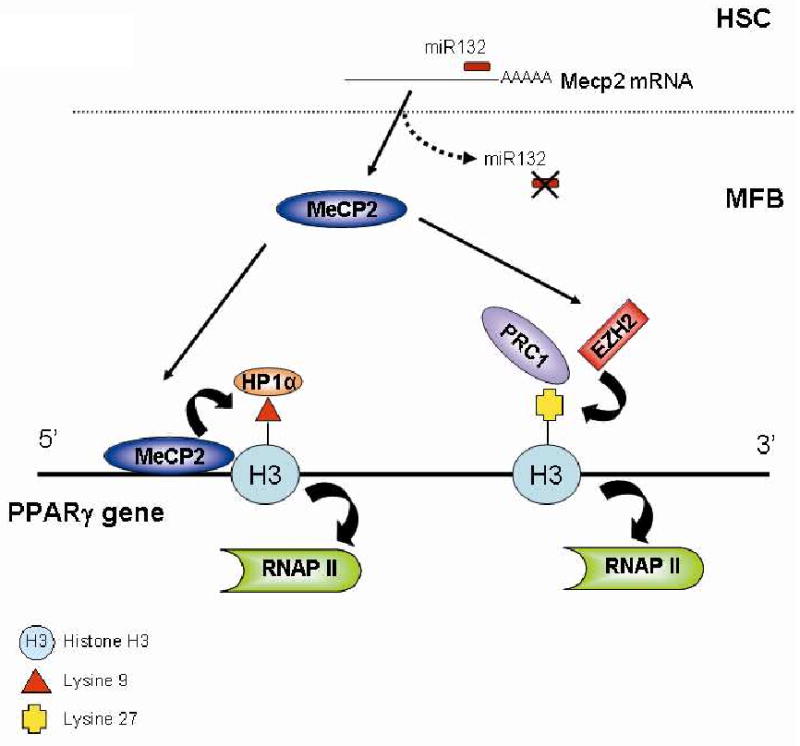
In qHSC miR132 exerts a translational block on MeCP2 transcript. Upon transdifferentiation mIR132 expression is down regulated enabling translation of MeCP2 protein and binding to the 5′ end of the PPARγ gene where it promotes H3K9 methylation and recruitment of the transcription repressor HP1α. Additionally, MeCP2 stimulates expression of EZH2 which methylates H3K27 to form a binding site for the polycomb repressor 1 complex which promotes chromatin condensation in the 3′ PPARγ exons. The culmination of the pathway is transcriptional repression of the master anti-fibrogenic gene PPARγ.
MeCP2 is best known for its role as a transcriptional repressor that operates in the normal development of the nervous system; mutations in the Mecp2 gene are associated with Rett syndrome and other forms of X-linked mental retardation 12. Functions for MeCP2 outside of the nervous system, other than in cancers, are unexplored. Our data suggest a new function for MeCP2 as a pivotal regulator of hepatic fibrogenesis. Two key observations support this concept. First, mice that are deficient for MeCP2 are attenuated for their fibrogenic response to toxic injury of the liver. This observation probably reflects the inability of MeCP2-deficient hepatic stellate cells to undergo normal transdifferentiation, retaining expression of PPARγ and expressing reduced levels of type 1 collagen. Second, hepatic MeCP2 expression is restricted to myofibroblasts and is subject to a tight mechanism of regulation involving the suppression of protein translation by miR132. miR132 only operates as a translation repressor for MeCP2 transcripts that carry an extended 3′UTR that includes the miR132 recognition motif and which were thought to be selectively expressed in neuronal cells. However, our data suggest that HSC also express the extended miR132-regulated MeCP2 transcripts. Furthermore, miR132 expression is lost with transdifferentiation and when experimentally reconstituted leads to diminished MeCP2 protein expression and elevated levels of PPARγ transcript. There is a growing literature describing a variety of neuronal-like features of HSC and the contributions of these characteristics to hepatic wound-repair24. Our discovery that a “neuron-specific” mechanism controls MeCP2 in HSC extends the similarities and explains the restricted expression of MeCP2 protein to myofibroblasts in the liver. It will be of interest to determine if other stellate-like cells of the kidney, pancreas and respiratory system 1, 2 also utilise a miR132/MeCP2 control mechanism to regulate transdifferention. Klein et al reported that miR132 is induced via a CREB-dependent pathway involving CREB phosphorylation 15. Of note, induction of CREB phosphorylation has previously been described to suppress the fibrogenic characteristics of HSC-derived myofibroblasts 25. It therefore seems likely that CREB phosphorylation suppresses transdifferentiation via induction of miR132. Although we delineate function for miR132 in HSC transdifferentiation, it is possible that this process may be additionally controlled by other micro RNA species.
Interrogation of the mechanism by which MeCP2 represses PPARγ transcription revealed at least two alterations in the histone code associated with gene silencing, H3K9 and H3K27 methylation. Both modifications were diminished in MeCP2-deficient myofibroblasts. Association of MeCP2 with methylation of H3K9 is previously reported and is a requirement for recruitment of the chromatin silencer HP1α 17 which is selectively bound to the same 5′ regions of the PPARγ gene as MeCP2 and is induced with transdifferentiation. H3K9 methylation and recruitment of HP1α would therefore generate transcriptionally repressed chromatin structure in the PPARγ promoter and exons A1 and A2 to suppress transcription initiation. An unexpected discovery was that H3K27 methylation was influenced by MeCP2. Since H3K27 methylation was enriched in the 3′ exons of PPARγ which did not correspond with binding of MeCP2 suggesting an indirect regulatory mechanism. EZH2 is the only known H3K27 methyltransferase and we confirmed by both siRNA and pharmacological inhibition that EZH2 activity is required for transcriptional repression of PPARγ. Just as for MeCP2, EZH2 is not expressed in HSC and is induced during transdifferentiation. However, expression of EZH2 follows the earlier induction of MeCP2. This latter observation combined with our finding of reduced levels of EZH2 in MeCP2-depleted myofibroblasts suggests that MeCP2 relays its signal for H3K27 methylation at least in part by stimulating high level expression of EZH2. H3K27 methylation leads to recruitment of the PRC1 Polycomb complex which promotes chromatin condensation and when found in downstream exons would suppress elongation of RNApol II transcripts 26. MeCP2 therefore functions as a regulator of PPARγ transcription (and transdifferentiation) by suppressing both transcriptional initiation (H3K9 methylation) and elongation (H3K27 methylation).
The discovery of new functions for three epigenetic regulators (miR132, MeCP2 and EZH2) in myofibroblast transdifferentiation has important implications for our understanding of the control of wound-healing and fibrosis in the diseased liver. There is still no clear explanation from genetic studies as to why fibrosis develops only in a minority of patients with chronic liver disease 27. Genetic or environmental influences on the expression or activity of mIR132, MeCP2 or EZH2 would attenuate transdifferentiation and impact on the numbers of myofibroblasts and their production of fibrogenic molecules. As demonstrated here, there is a clear inverse correlation between expression of MeCP2 or EZH2 and the expression of type I collagen and TIMP-1. Further studies on miR132 in human liver disease would be particularly interesting since its expression is sensitive to environmental cues that influence fibrosis 25. Furthermore, microRNAs may be transmitted through both the male and female germ lines and can influence non-mendelian inheritance of epigenetic traits from one generation to the next 28.
Finally, the therapeutic potential for targeting the miR132-MeCP2-EZH2 relay should be explored in the context of chronic liver disease, especially as this is a regulatory network that is selective to the myofibroblast.
Supplementary Material
Acknowledgments
Supported by NIAAA/NIH grants, R21AA016682, R24AA012885, P50AA11999; and Medical Research Service of Veterans Affairs. DAM and JM are supported by grants from the Medical Research Council, Wellcome Trust and British Liver Trust.
Grant support: Supported by grants from NIH NIAAA (to H.T and D.A.M) and MRC (to D.A.M) and Newcastle Health Care Charity and the Newcastle upon Tyne Hospitals NHS Charity (to J.M)
Abbreviations
- EMT
epithelial to mesenchymal transition
- HSC
hepatic stellate cell
- PPARγ
peroxisome proliferator-activated receptor-gamma
- MeCP2
methyl-CpG binding protein 2
- TIMP-1
tissue inhibitor of metalloproteinases
Footnotes
Conflict of interest: Authors report no conflict of interest
Author contribution to the manuscript:
Jelena Mann- Study concept and design, analysis and interpretation of data, carried out 90% of experiments, had a major part in writing the manuscript
David CK Chu- Material provision and support
Aidan Maxwell- Acquisition of data
Fiona Oakley- Acquisition of data, technical support
Nian-Ling Zhu- generated mRNA from hepatic stellate cells isolated from in vivo models of liver injury.
Hidekazu Tsukamoto- Obtained funding and generated mRNA from hepatic stellate cells isolated from in vivo models of liver injury.
Derek Mann- Study concept and design, analysis and interpretation of data, writing of the manuscript.
References
- 1.Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iredale JP. Models of liver fibrosis: exploring the dynamic nature of inflammation and repair in a solid organ. J Clin Invest. 2007;117:539–48. doi: 10.1172/JCI30542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Desmouliere A, Chaponnier C, Gabbiani G. Tissue repair, contraction, and the myofibroblast. Wound Repair Regen. 2005;13:7–12. doi: 10.1111/j.1067-1927.2005.130102.x. [DOI] [PubMed] [Google Scholar]
- 4.Mann J, Oakley F, Akiboye F, et al. Regulation of myofibroblast transdifferentiation by DNA methylation and MeCP2: implications for wound healing and fibrogenesis. Cell Death Differ. 2007;14:275–85. doi: 10.1038/sj.cdd.4401979. [DOI] [PubMed] [Google Scholar]
- 5.Hazra S, Xiong S, Wang J, et al. J Biol Chem. 2004;279:11392–401. doi: 10.1074/jbc.M310284200. [DOI] [PubMed] [Google Scholar]
- 6.Miyahara T, Schrum L, Rippe R, et al. Peroxisome proliferator-activated receptors and hepatic stellate cell activation. J Biol Chem. 2000;275:35715–22. doi: 10.1074/jbc.M006577200. [DOI] [PubMed] [Google Scholar]
- 7.Milam JE, Keshamouni VG, Phan SH, et al. PPAR-gamma agonists inhibit profibrotic phenotypes in human lung fibroblasts and bleomycin-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2008;294:L891–901. doi: 10.1152/ajplung.00333.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saika S, Yamanaka O, Okada Y, et al. Effect of overexpression of PPARgamma on the healing process of corneal alkali burn in mice. Am J Physiol Cell Physiol. 2007;293:C75–86. doi: 10.1152/ajpcell.00332.2006. [DOI] [PubMed] [Google Scholar]
- 9.Aiello A, Pandini G, Frasca F, et al. Peroxisomal proliferator-activated receptor-gamma agonists induce partial reversion of epithelial-mesenchymal transition in anaplastic thyroid cancer cells. Endocrinology. 2006;147:4463–75. doi: 10.1210/en.2005-1610. [DOI] [PubMed] [Google Scholar]
- 10.Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell. 2007;128:635–8. doi: 10.1016/j.cell.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 11.Cao R, Zhang Y. The functions of E(Z)/EZH2-mediated methylation of lysine 27 in histone H3. Curr Opin Genet Dev. 2004;14:155–64. doi: 10.1016/j.gde.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 12.Hite KC, Adams VH, Hansen JC. Recent advances in MeCP2 structure and function. Biochem Cell Biol. 2009;87:219–27. doi: 10.1139/o08-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chadwick LH, Wade PA. MeCP2 in Rett syndrome: transcriptional repressor or chromatin architectural protein? Curr Opin Genet Dev. 2007;17:121–5. doi: 10.1016/j.gde.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 14.Yang L, Chan CC, Kwon OS, et al. Regulation of peroxisome proliferator-activated receptor-gamma in liver fibrosis. Am J Physiol Gastrointest Liver Physiol. 2006;291:G902–11. doi: 10.1152/ajpgi.00124.2006. [DOI] [PubMed] [Google Scholar]
- 15.Klein ME, Lioy DT, Ma L, et al. Homeostatic regulation of MeCP2 expression by a CREB-induced microRNA. Nat Neurosci. 2007;10:1513–4. doi: 10.1038/nn2010. [DOI] [PubMed] [Google Scholar]
- 16.Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol. 2005;6:838–49. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- 17.Fuks F, Hurd PJ, Deplus R, et al. The DNA methyltransferases associate with HP1 and the SUV39H1 histone methyltransferase. Nucleic Acids Res. 2003;31:2305–12. doi: 10.1093/nar/gkg332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheutin T, McNairn AJ, Jenuwein T, Gilbert DM, Singh PB, Misteli T. Maintenance of stable heterochromatin domains by dynamic HP1 binding. Science. 2003;299:721–5. doi: 10.1126/science.1078572. [DOI] [PubMed] [Google Scholar]
- 19.Kirmizis A, Bartley SM, Kuzmichev A, et al. Silencing of human polycomb target genes is associated with methylation of histone H3 Lys 27. Genes Dev. 2004;18:1592–605. doi: 10.1101/gad.1200204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chahrour M, Jung SY, Shaw C, et al. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–9. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tan J, Yang X, Zhuang L, et al. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 2007;21:1050–63. doi: 10.1101/gad.1524107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Douglass A, Wallace K, Parr R, et al. Antibody-targeted myofibroblast apoptosis reduces fibrosis during sustained liver injury. J Hepatol. 2008;49:88–98. doi: 10.1016/j.jhep.2008.01.032. [DOI] [PubMed] [Google Scholar]
- 23.Gieling RG, Burt AD, Mann DA. Fibrosis and cirrhosis reversibility - molecular mechanisms. Clin Liver Dis. 2008;12:915–37. xi. doi: 10.1016/j.cld.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 24.Ebrahimkhani MR, Elsharkawy AM, Mann DA. Wound healing and local neuroendocrine regulation in the injured liver. Expert Rev Mol Med. 2008;10:e11. doi: 10.1017/S146239940800063X. [DOI] [PubMed] [Google Scholar]
- 25.Houglum K, Lee KS, Chojkier M. Proliferation of hepatic stellate cells is inhibited by phosphorylation of CREB on serine 133. J Clin Invest. 1997;99:1322–8. doi: 10.1172/JCI119291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schwartz YB, Pirrotta V. Polycomb silencing mechanisms and the management of genomic programmes. Nat Rev Genet. 2007;8:9–22. doi: 10.1038/nrg1981. [DOI] [PubMed] [Google Scholar]
- 27.Weber S, Gressner OA, Hall R, et al. Genetic determinants in hepatic fibrosis: from experimental models to fibrogenic gene signatures in humans. Clin Liver Dis. 2008;12:747, vii. doi: 10.1016/j.cld.2008.07.012. [DOI] [PubMed] [Google Scholar]
- 28.Rassoulzadegan M, Grandjean V, Gounon P, et al. RNA-mediated non-mendelian inheritance of an epigenetic change in the mouse. Nature. 2006;441:469–74. doi: 10.1038/nature04674. [DOI] [PubMed] [Google Scholar]
- 29.Wright MC, Issa R, Smart DE, et al. Gliotoxin stimulates the apoptosis of human and rat hepatic stellate cells and enhances the resolution of liver fibrosis in rats. Gastroenterology. 2001;121:685–98. doi: 10.1053/gast.2001.27188. [DOI] [PubMed] [Google Scholar]
- 30.Oakley F, Mann J, Nailard S, et al. Nuclear factor-kappaB1 (p50) limits the inflammatory and fibrogenic responses to chronic injury. Am J Pathol. 2005;166:695–708. doi: 10.1016/s0002-9440(10)62291-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O'Neill LP, Turner BM. Immunoprecipitation of native chromatin: NChIP. Methods. 2003;31:76–82. doi: 10.1016/s1046-2023(03)00090-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.