

Abstract
BACKGROUND & AIMS
The mechanisms by which reflux of bile acids into the pancreas induces pancreatitis are unknown. We reasoned that key events responsible for this phenomenon might be mediated by Gpbar1, a recently identified and widely expressed G protein-coupled, cell surface, bile acid receptor.
METHODS
Acute pancreatitis was induced in wild type and Gpbar1−/− mice by either retrograde ductal infusion of taurolithocholic acid-3-sulfate (TLCS) or supramaximal secretagogue stimulation with caerulein. In vitro experiments were performed in which acini obtained from wild type and Gpbar1−/− mice were exposed to either sub-micellar concentrations of TLCS (200–500 μM) or a supramaximally stimulating concentration of caerulein (10 nM).
RESULTS
Gpbar1 is expressed at the apical pole of acinar cells and its genetic deletion is associated with reduced hyperamylasemia, edema, inflammation, and acinar cell injury in TLCS- but not in caerulein-induced pancreatitis. In vitro, genetic deletion of Gpbar1 is associated with markedly reduced generation of pathological calcium transients, intracellular activation of digestive zymogens, and cell injury when these responses are induced by exposure to TLCS but not when they are induced by exposure to caerulein.
CONCLUSIONS
Gpbar1 may play a critical role in the evolution of bile acid-induced pancreatitis by coupling exposure to bile acids with generation of pathological intracellular calcium transients, intra acinar cell zymogen activation, and acinar cell injury. Acute biliary pancreatitis may be a “receptor-mediated” disease and interventions that interfere with Gpbar1 function might prove beneficial in the treatment and/or prevention of biliary acute pancreatitis.
INTRODUCTION
Acute pancreatitis is a frequently severe and sometimes lethal disease that is most commonly triggered by the passage of biliary stones or sludge into or through the terminal biliopancreatic duct. Events responsible for induction of this so-called “biliary” or “gallstone” form of acute pancreatitis are poorly understood but two mechanisms have been proposed: (a) the “common channel” theory which argues that stones or sludge cause pancreatitis by promoting reflux of bile into the pancreatic duct through a common biliopancreatic channel1, and (b) the “duct obstruction” theory which argues that the stones or sludge promote ductal hypertension by interfering with juice out-flow and that hypertension triggers pancreatitis2.
To date, experimental evidence that duct obstruction/hypertension, without bile reflux, can cause pancreatic cell injury and pancreatitis is scant and limited, exclusively, to studies involving the American opossum3. On the other hand, a large number of studies employing many other species have shown that simple pancreatic duct outflow obstruction leads, almost exclusively, to pancreatic atrophy with little evidence of acute pancreatitis. These observations, along with the fact that pancreatitis can be induced in virtually all of those species if transient ductal obstruction is accompanied by retrograde, intraductal infusion of bile or bile acids4, 5, clearly support the bile reflux theory as an explanation for the pathogenesis of pancreatitis and even further support comes from the unfortunate, but frequent, finding that acute pancreatitis can be triggered in patients by retrograde injection of contrast agents into the pancreatic duct during performance of endoscopic retrograde cholangiopancreatography (ERCP)6.
The “common channel” theory assumes that bile acids. or some other constituent of bile, can injure pancreatic parenchymal cells and it was initially believed that that injury resulted from detergent- or ionophore-like properties of bile acids7–10. Recently, however, several studies have suggested that bile acids may adversely effect pancreatic acinar cells by more specific mechanisms. For example, exposure of acinar cells to bile acid concentrations below their critical micellar concentration has been shown to trigger PI3K-mediated inhibition of the sarco(endoplasmic) Ca-ATPase (SERCA), thus leading to pathological increases in intra-acinar cell calcium levels, intracellular activation of digestive zymogens, cell injury/death, and activation of inflammatory pathways11–13. Sub-micellar concentrations of bile acids can also trigger both IP3- and ryanodine-receptor mediated intra-acinar cell calcium release from stores within the endoplasmic reticulum as well as from stores within acidic organelles at the apical pole of acinar cells which, presumably, are zymogen granules14. This calcium release can result in pathological as well as physiological intracellular calcium transients.
Kim et al11 suggested that bile acids exert their potentially injurious calcium-related effects on acinar cells by acting from within the cell subsequent to their uptake via bile acid transporters located on the apical (lumenal) and basolateral acinar cell membrane. In the current communication, we evaluate the role of the recently discovered15, 16 cell surface G protein-coupled bile acid receptor-1 (Gpbar1). We show that the bile acid taurolithocholic acid 3-sulfate sodium salt (TLCS) triggers pathological changes in pancreatic acinar cells by interacting with Gpbar1 at the lumenal cell surface and that genetic deletion17, 18 of Gpbar1 protects mice from bile acid-induced acute pancreatitis. These studies suggest that biliary acute pancreatitis may be a “receptor-mediated” disease.
MATERIALS AND METHODS
All experiments conformed to protocols approved by the Tufts Medical Center Animal Care and Use Committee. Most reagents, including tetrodotoxin, caerulein TLCS, and Na-taurocholate were purchased from Sigma (St. Louis, MO). Fura 2/AM and 1,2-bis(2-aminophenoxy)ethane-N,N,N,N′-tetraacetic acid (BAPTA), were from Molecular Probes (Eugene, OR). Substrates for measuring trypsin and chymotrypsin activity were from Peptides International, (Louisville, KY) and substrates for LDH and amylase were from Genzyme (Framingham, MA). Concanamycin A was purchased from Axxora (San Diego, CA).
Animals and expression of Gpbar1
C57Bl/6 Gpbar1−/− mice and their C57Bl/6 wild type littermates, generated as described by Vassileva et al17, were used to found our colonies of Gpbar1−/− and wild type control animals. Gpbar1−/− mice develop normally, are fertile, and appear normal on gross examination for at least 20 months after birth17. Gpbar1 expression in the mouse pancreas was evaluated by rt-PCR. Immunofluorescence microscopy on cryostat acetone fixed sections was performed with antibodies raised against the C-terminal domain of mouse Gpbar1 at the Tufts Center for Neuroscience Research, P30 NS047423) (Supplemental Methods).
Induction of pancreatitis and evaluation of pancreatitis severity
We employed two dissimilar models of acute experimental pancreatitis. One model involved retrograde infusion of 3 mM TLCS or Na-taurocholate into the pancreatic duct as previously described by our group5. It was used to test the effects of bile acids on the pancreas. The second model, induced by supramaximal stimulation with caerulein19, was used for control studies. Maximal severity in both models is reached 12–24 hours after induction and the pancreatitis resolves over the following week5, 21. Mice were sacrificed 24 hours after induction of pancreatitis and blood samples were taken for measurement of serum amylase activity. The pancreas was removed and used for other measurements. In bile acid-induced pancreatitis, those measurements were made using the head region of the gland (defined as the portion of the pancreas lying within 5 mm of the lesser duodenal curvature) since previously reported studies by our group have shown that the changes in this model are confined to that area. In the caerulein-induced model, randomly selected portions of the pancreas were used because, in that model, changes are uniformly distributed throughout the pancreas. Pancreatic edema was evaluated by measuring water content and expressing that as a percent of tissue wet weight22. Inflammation was evaluated by quantitating tissue myeloperoxidase activity22 using a commercially available ELISA (Hycult Biotechnology, Uden, Netherlands) according to the manufacturer’s instructions). Acinar cell injury/death was morphometrically determined22 in hematoxylin/eosin-stained sections by an observer unfamiliar with sample identity. Apoptosis was quantitated using the TUNEL technique22 and changes related to Gpbar1 deletion were evaluated by comparing the ratio of TUNEL-positive cells to the area of acinar cell injury/death in samples from Gpbar1−/− and wild type mice.
In-vitro studies
Acini for in vitro measurements were prepared as described previously22 (Supplemental Methods). Calcium transients within single acinar cells were measured and analyzed using small pancreatic acini22 (Supplemental Methods). Intracellular activation of digestive enzyme zymogens (trypsinogen and chymotrypsinogen) was quantitated, fluorometrically, in homogenized samples of standard-sized acini using Boc-Gln-Ala-Arg-MCA and Suc-Ala-Ala-Pro-Phe-MCA, respectively, as the substrate as previously described25 and net stimulated activation was calculated after subtracting enzyme activation noted in the absence of either bile acid (TLCS or Na-taurocholate) or caerulein. Cell injury/lysis was quantitated by measuring LDH leakage from standard-sized acini as previously described25. For this purpose, acini were suspended in DMEM/F12 buffer A containing caerulein, TLCS, or Na-taurocholate at 37 °C for varying times, centrifuged, and cells in the pellet were lysed with 0.5% Triton X-100. LDH activity was separately measured in the supernatant and pellet. LDH leakage was expressed as the percent of total LDH activity that was present in the supernatant. Amylase secretion, over 30 min, was quantitated using standard-sized acini suspended in buffer A as previously described and expressed as a percent of total amylase content that was discharged into the suspending medium25. Net stimulated secretion was calculated after subtracting amylase discharge noted in the absence of either TLCS or caerulein. Selected acini were pre-loaded with BAPTA before exposure to either TLCS or caerulein in order to evaluate the effect of intra-cellular calcium chelation on bile acid- or secretagogue-induced secretion. Pre-loading with BAPTA was achieved by incubating acini in buffer A containing 20 μM BAPTA and 0.2% DMSO for 30 min at 37 °C followed by extensive washing in buffer A to remove residual BAPTA and DMSO from the suspending medium.
Analysis of data
The results reported in this study are expressed as mean +/− SD values obtained from 3 or more independent experiments. In all figures, the vertical bars denote SD values. Statistical evaluation was accomplished using student’s t test when two groups were compared and analysis of variance when multiple groups were compared. A p value of <0.05 was considered to indicate a significant difference.
RESULTS
The bile acid receptor Gpbar1 is expressed in the pancreas
Gpbar1 expression in the pancreas of wild type mice can be demonstrated by rt-PCR (Fig. 1) and it is lost following genetic deletion of Gpbar1. Immunofluorescence microscopy with a Leica TCS SP2 confocal microscope (Fig. 1) indicates that most of the anti-Gpbar1 staining is localized to the region very near, but not identical, to the location of f-actin at the acinar cell apical pole. Positive Gpbar1 stain was also seen in gallbladder of wild type mice but not in that of Gpbar1−/− mice (Suppl. Fig 1).
Figure 1. Gpbar1 is expressed in the mouse pancreas.

Gpbar1 expression in the pancreas of wild type and Gpbar1−/− mice was evaluated by rt-PCR (Panel A), and immunofluorescence microscopy (Panel B) as described in the text. Note intense anti-Gpbar1 antibody fluorescence at the apical pole of wild type acinar cells which is adjacent but not identically localized to the sites occupied by f-actin (Panel B, overlay) and absence of Gpbar1 expression in Gpbar1−/− pancreas (Panels A, B).
Genetic deletion of Gpbar1 reduces the severity of bile acid-induced pancreatitis
Retrograde intraductal infusion of TLCS (50 μl, 3 mM) and repeated administration of caerulein (50 μg/injection, 12 injections) each induce acute pancreatitis that is characterized by hyperamylasemia, pancreatic edema, inflammation, and acinar cell injury/necrosis (Fig. 2A and B). In TLCS-induced pancreatitis, hyperamylasemia, pancreatic edema, pancreatic inflammation and acinar cell injury/death are all significantly reduced (p<0.05) when Gpbar1 has been deleted (Fig. 2A) but, when pancreatitis is induced by caerulein administration, none of these parameters of pancreatitis severity are altered by genetic deletion of Gpbar1 (Fig. 1B). None of the features of TLCS-induced pancreatitis (i.e. hyperamylasemia, edema, and acinar cell injury/death) are observed when 50 μl of 3 mM Na-taurocholate, rather than TLCS, is infused into the pancreatic duct (Suppl. Fig. 2A).
FIGURE 2. Genetic deletion of Gpbar1 reduces the severity of TLCS- but not caerulein-induced pancreatitis.
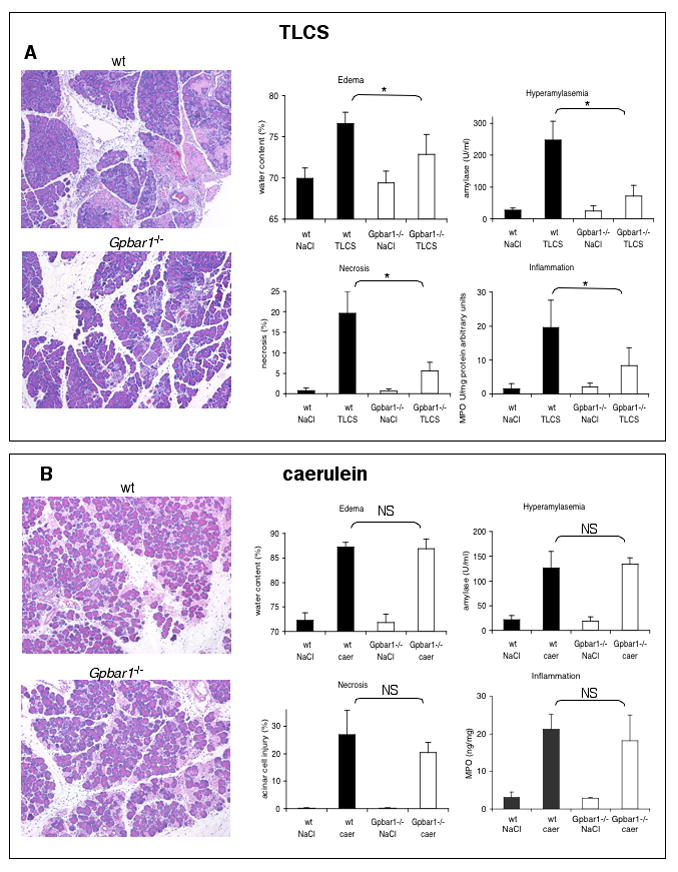
Acute pancreatitis was induced, in wild type and Gpbar1−/− mice either by retrograde ductal infusion with 3 mM TLCS (Panel A) or supramaximal stimulation with repeated administration of 50 μg/kg/injection of caerulein (Panel B) as described in the text. Twenty-four hours after the start of pancreatitis-induction, the animals were sacrificed and pancreatitis severity was evaluated as described in the text. Solid bars report results obtained using wild type mice while open bars report results from Gpbar1−/− animals. Asterisks denote significant differences in bracketed groups in which results from wild type and Gpbar1−/− mice are compared. NS = not significant.
In wild type mice, TUNEL-positive acinar cells account for 31.7% of total injured/dead acinar cells in TLCS-induced pancreatitis (Suppl. Fig 3). Genetic deletion of Gpbar1 markedly reduces the extent of acinar cell injury/death during TLCS-induced pancreatitis but the % of injured/dead acinar cells that is TUNEL-positive is not significantly altered (Suppl. Fig. 3). These findings indicate that (a) both apoptosis and necrosis account for acinar cell death during TLCS-induced pancreatitis but (b) genetic deletion of Gpbar1 does not reduce the severity of TLCS-induced pancreatitis by altering the mode of TLCS-induced acinar cell death.
Genetic deletion of Gpbar1 reduces TLCS-induced generation of intra-acinar cell pathological calcium transients
The four types of intracellular calcium responses to TLCS and caerulein stimulation are shown in Fig. 3A. It is widely agreed that calcium transients consisting of pure oscillations are physiological responses, those consisting of only a peak/plateau are pathological responses, and those consisting of oscillations + peak plateau represent a transitional response. Therefore, we will use that terminology in this presentation.
Figure 3. Genetic deletion of Gpbar1 alters frequency with which pathological calcium transients are observed after exposure of cells to TLCS but not after exposure of cells to a supramaximally stimulating concentration of caerulein.
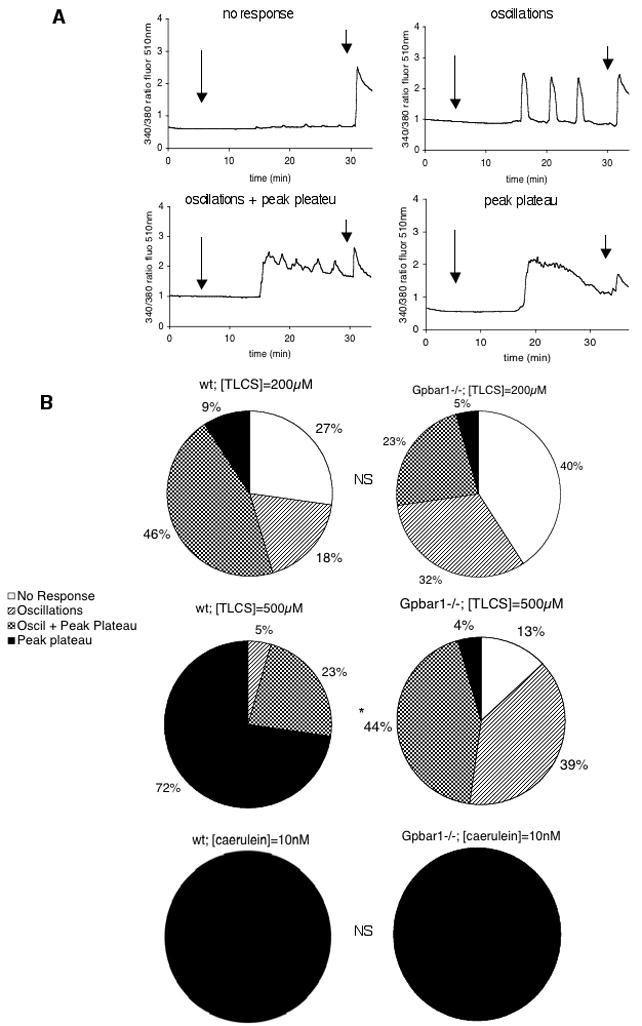
Small pancreatic acini from wild type and Gpbar1−/− mice were exposed to 200 μM and 500 μM TLCS or to 10 nM caerulein and calcium transients were monitored in individual acinar cells as described in the text. Panel A shows tracings that are representative of the 4 patterns as described in the text. Long arrows denote time of TLCS addition while short arrows denote time of caerulein addition. This latter addition was used to confirm maintenance of cell viability and functionality throughout the experiment. Panel B reports frequency with which each response pattern was observed after exposure of wild type or Gpbar1 −/− acini to either TLCS or caerulein. Asterisks denote significant differences (p<0.01) when Gpbar1−/− acini are compared to wild type acini. NS = not significant.
Most wild type or Gpbar1−/− acinar cells display physiological calcium transients, or simply fail to respond, when exposed to 200 μM TLCS and most wild type acinar cells exposed to 500 μM TLCS display pathological or transitional calcium transients (Fig. 2B). Physiological transients are infrequent when wild type cells are exposed to 500 μM TLCS. In contrast, most Gpbar1−/− acinar cells exposed to 500 μM TLCS display either physiological or transitional calcium transients and pathological transients are infrequent (Fig. 2B). This differing response of wild type and Gpbar1−/− cells to 500 μM TLCS is significant (p < 0.01). In contrast, the effect of Gpbar1 deletion on calcium transients is not observed when acinar cells are exposed to caerulein. When either wild type or Gpbar1−/− cells are exposed to 10 pM caerulein, all cells of either type display physiological calcium transients characterized by an oscillatory response (not shown) while, in the presence of 10 nM caerulein, all cells of either type display pathological calcium transients (Fig. 3B). These findings suggest that genetic deletion of Gpbar1 reduces the generation of pathological calcium transients that occurs when acinar cells are exposed to 500 μM TLCS but it does not effect the generation of pathological calcium transients in response to a supramaximally stimulating concentration of caerulein.
Genetic deletion of Gpbar1 reduces TLCS-induced activation of digestive zymogens within pancreatic acini
Both trypsinogen and chymotrypsinogen activation are observed when wild type acini are exposed to 500 μM TLCS (Fig. 4) but no activation of these zymogens is observed when acini are exposed to either 100 μM or 200 μM TLCS (not shown). In Gpbar1−/− acini, 500 μM TLCS triggers little, if any, zymogen activation. Zymogen activation is also not observed when wild type acini are exposed to 500 μM Na-taurocholate (Suppl. Fig. 2B).
Figure 4. Genetic deletion of Gpbar1 prevents intra-acinar cell activation of trypsinogen and chymotrypsinogen after exposure to TLCS but not after exposure to caerulein.
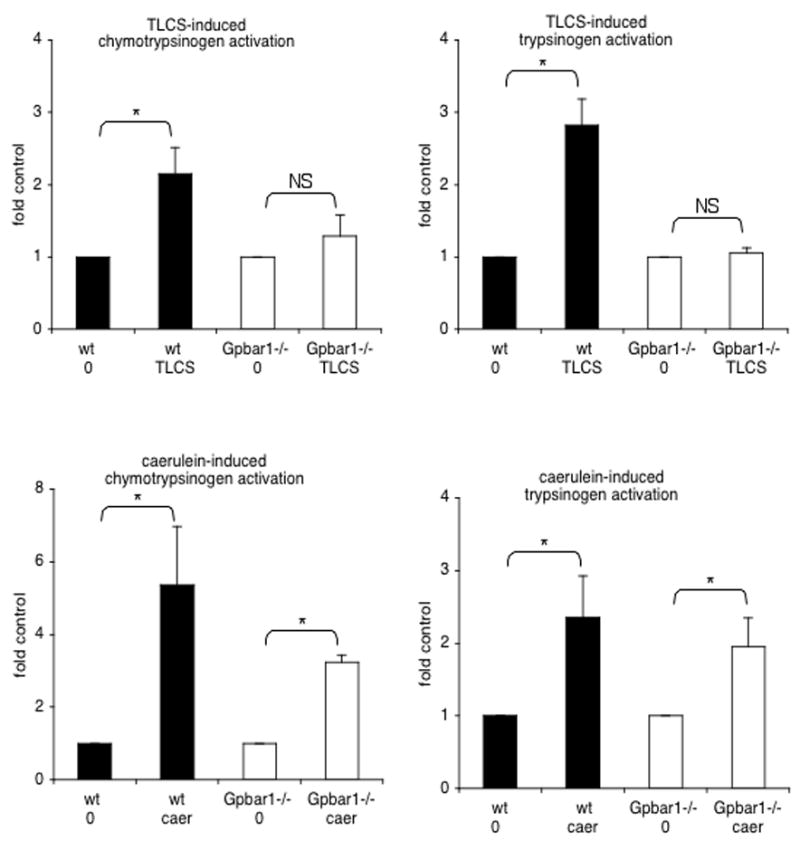
Large pancreatic acini from wild type (solid black columns) and Gpbar1−/− (open columns) mice were exposed to either 500 μM TLCS or 10 nM caerulein. Trypsinogen and chymotrypsinogen activation 15 and 30 min later, respectively, was quantitated as described in the text. Asterisks denote significant differences (p<0.05) between bracketed groups. NS = not significant.
Caerulein (10 nM) triggers digestive zymogen activation in wild type acini however, in contrast to the situation noted when TLCS is used, caerulein-induced activation of trypsinogen and chymotrypsinogen is also observed in acini prepared from Gpbar1−/− mice (Fig. 4). These observations indicate that Gpbar1 plays a major and critical role in mediating intracellular zymogen activation induced by 500 μM TLCS but that it plays little, if any, role in mediating the zymogen activation induced by caerulein.
TLCS-induced zymogen activation in acini from wild type mice is not altered by exposure to tetrodotoxin (Suppl. Fig. 4) indicating it is not neurally mediated. On the other hand, TLCS and caerulein-induced intra-acinar cell zymogen activation in acini from wild type mice is inhibited by exposure to the vacuolar ATPase inhibitor concanamycin A (Suppl. Fig. 4) suggesting zymogen activation in response to either TLCS or caerulein occurs by mechanisms that involve activation of vacuolar ATPases.
Genetic deletion of Gpbar1 reduces TLCS-induced leakage of LDH from pancreatic acini
LDH leakage is increased in pancreatic acini from wild type mice that are exposed, in vitro, to 500 μM TLCS (Fig. 5) but not when acini are exposed to either 100 μM or 200 μM TLCS (not shown). On the other hand, accelerated leakage of LDH is not observed when acini are exposed to 500 μM Na-taurocholate (Suppl. Fig. 1B) and LDH leakage induced by 500 μTLCS is significantly reduced in acini prepared from Gpbar1−/− mice when those acini are compared to acini prepared from wild type mice (Fig. 5). In contrast to the situation observed when TLCS is used, however, caerulein-induced LDH leakage is not altered by genetic deletion of Gpbar1 (Fig. 5). These observations indicate that Gpbar1 plays a role in mediating LDH leakage induced by 500 μM TLCS but not LDH leakage induced by caerulein.
Figure 5. Genetic deletion of Gpbar1 prevents acinar cell injury induced by TLCS but not caerulein.
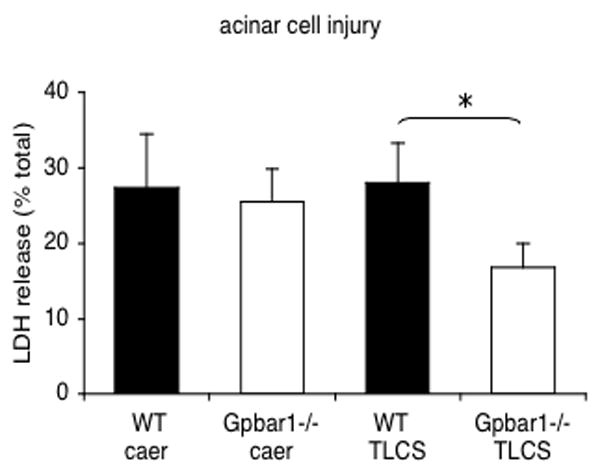
Large pancreatic acini from wild type (wt) and Gpbar1−/− mice were exposed to either 500 μM TLCS or 10 nM caerulein. LDH leakage from the acini was quantitated as described in the text. Results reflect net LDH leakage, expressed as a percent of total LDH content, after subtracting leakage noted in the absence of both TLCS and caerulein. That subtracted value was 11.6 +/− 0.9% and 12.8 +/− 0.6 % of total LDH content for wild type and Gpbar1−/− acini, respectively. Asterisks denote significant differences when results obtained with Gpbar1−/− mice are compared to those obtained with wild type acini.
Genetic deletion of Gpbar1 does not alter calcium-dependent TLCS-stimulated amylase discharge from acinar cells
TLCS triggers concentration-dependent increases in amylase discharge from both wild type and Gpbar1−/− acini (Fig. 6). TLCS-induced amylase discharge, induced by 500 μM TLCS, is largely prevented by pre-loading the acini with BAPTA. A higher concentration of TLCS (1 mM) also accelerates amylase discharge from both wild type and Gpbar1−/− acini but that increased rate of amylase discharge is not altered by pre-loading the acini with BAPTA. Caerulein triggers concentration-dependent increases in amylase discharge from wild type and Gpbar1−/− acini and that discharge is largely prevented by pre-loading the acini with BAPTA. These observations indicate that calcium-dependent amylase secretion, induced by either TLCS or caerulein stimulation, is not altered by genetic deletion of Gpbar1 (Fig. 6).
Figure 6. TLCS- and caerulein-induced calcium-dependent acinar cell discharge of amylase is not altered by genetic deletion of Gpbar1.
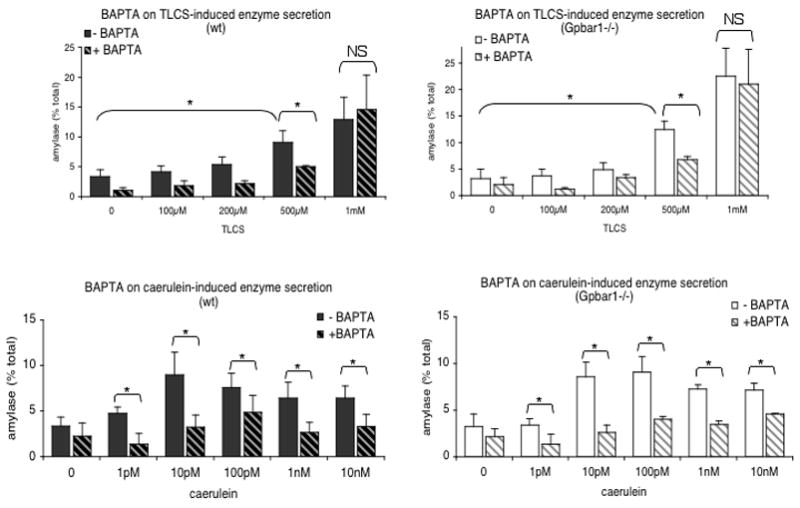
Large pancreatic acini from wild type (left plots) and Gpbar1−/− (right plots) mice were exposed to varying concentrations of either TLCS (upper plots) or caerulein (lower plots). Others (hatched columns) were pre-loaded with BAPTA and similarly treated. Amylase discharge, over 15 min, was measured as described in the text. Asterisks denote significant differences when acini pre-loaded with BAPTA are compared with those not pre-loaded with BAPTA or when acini exposed to TLCS were compared to those not exposed to TLCS.
DISCUSSION
Recent reports have indicated that, in addition to playing an essential role in dietary lipid absorption and cholesterol metabolism, bile acids can also act as signaling molecules. In this capacity, after being taken up by target cells, bile acids can regulate the expression of bile acid transporters by activating nuclear hormone receptors such as the Farnesoid X receptor and the pregnane X receptor26–28 and they can directly activate MAPK pathways29, 30. Maruyama et al31 and Kawamata et al32 have independently shown that bile acids can activate the recently discovered G protein-coupled receptor Gpbar1 and subsequent studies, by several groups, have shown that bile acid-induced activation of Gpbar1 can (a) regulate energy expenditure by promoting intracellular thyroid hormone activation in brown adipose tissue and muscle33, (b) promote secretion of glucagon-like peptide-1 by enteroendocrine cells34, (c) promote hepatocyte apoptosis by activating JNK35, and (d) inhibit macrophage function36.
Gpbar1, also known as M-BAR/BG37 (membrane-bile acid receptor) and TGR5 (Takeda G-protein coupled receptor clone 5), is a Class 1 or rhodopsin-like 7-transmembrane G protein-coupled cell surface receptor that is widely expressed and highly conserved in mammals. A homolog of this receptor has also been identified in aquatic vertebrates36. In mice, the Gpbar1 gene is located on chromosome 1qc3 and knockout mice created by targeted genetic deletion of Gpbar1 have been shown to be resistant to cholesterol gallstone disease when fed a lithogenic diet17. To our knowledge, no previously reported studies have explored the possibility that Gpbar1 might play an important role in mediating biliary acute pancreatitis.
In this communication, we report the seminal observations that (a) Gpbar1 is expressed at the apical pole of mouse pancreatic acinar cells and (b) Gpbar1−/− mice are protected against bile acid-induced pancreatitis. While our studies indicate that Gpbar1 is expressed in pancreatic acinar cells, they do not exclude the possibility, or even the likelihood, that other, non-acinar, pancreatic cells (e.g. duct cells, immune cells, etc) may also express Gpbar1 and that Gpbar1 expression by these non-acinar cells may also play a role in mediating or regulating the severity of biliary pancreatitis.
Our pancreatitis studies were performed using a recently developed model of biliary pancreatitis that involves the retrograde infusion of bile acids such as TLCS or taurocholate into the pancreatic duct of anesthetized mice5. This model is extremely useful for studies such as ours because it allows the investigator to utilize any of the currently available large number of genetically manipulated mouse strains and, in addition, the model replicates events thought to underlie the development of clinical biliary, or gallstone, pancreatitis. Furthermore, studies utilizing this model are facilitated by its remarkable reproducibility and the fact that it yields pancreatitis that reaches its maximal severity within 24 hours of induction5.
Acute pancreatitis, induced by retrogradely infusing 3 mM TLCS into the mouse pancreatic duct, is characterized by hyperamylasemia, pancreatic edema, pancreatic inflammation, and pancreatic acinar cell injury/death. These changes also characterize pancreatitis induced by supramaximal caerulein stimulation and, in both models, the magnitude of these changes is conventionally used to define the severity of pancreatitis. Genetic deletion of Gpbar1 leads to a marked reduction in each of these severity parameters when pancreatitis is induced by infusion of TLCS but none of these severity parameters is altered by Gpbar1 deletion when pancreatitis is induced by a non-biliary mechanism (i.e. supramaximal stimulation with the cholecystokinin analog caerulein). Our observations suggest that Gpbar1-mediated events play a critical role in regulating the severity of bile acid-induced, but not non-biliary pancreatitis in mice.
TLCS-induced pancreatitis in mice is also associated with acinar cell apoptosis but genetic deletion of Gpbar1 does not significantly alter the percent of injured or dead acinar cells that are TUNEL-positive even though Gpbar1 deletion does reduce the extent of acinar cell injury/death and the severity of bile acid-induced pancreatitis. Together, these findings suggest that Gpbar1 deletion does not down-regulate the severity of TLCS-induced pancreatitis by altering the balance between apoptotic and necrotic cell death.
We performed a series of in vitro studies, using pancreatic acini prepared from wild type and Gpbar−/− mice, to examine the mechanisms by which activation of the bile acid receptor might lead to pancreatitis. We used TLCS as the bile acid for our studies for several reasons including the fact that it is a naturally occurring bile acid in mice and humans. The critical micellar concentration for naturally occurring bile acids is 1–5 mM and the concentration of bile acids within the gallbladder as well as within the intestinal lumen has been reported to be in the millimolar range as well37. In order to minimize detergent- or ionophore-like effects of TLCS, we performed our in vitro studies using TLCS concentrations that did not exceed 500 μM. These are concentrations that are believed to be pathologically relevant and they have been employed by others for recently reported in vitro studies designed to evaluate the effects of TLCS on pancreatic acinar cells11–14.
In accord with findings previously reported by others11–14, we found that a sub-micellar concentration of TLCS (500 μM) triggers pathological calcium transients in acinar cells. We also found that it causes both intra-acinar cell activation of digestive zymogens and acinar cell injury. Similar responses were also observed when acinar cells were exposed to a supramaximally stimulating concentration of caerulein. Most importantly, however, we found that genetic deletion of Gpbar1 markedly down-regulates TLCS-induced generation of pathological calcium transients, TLCS-induced zymogen activation, and TLCS-induced cell injury. In contrast, Gpbar1 deletion does not alter any of these responses when they are elicited by supramaximal stimulation with caerulein.
Our in vivo and in vitro observations, made using TLCS, lead us to hypothesize that biliary pancreatitis results from Gpbar1-mediated promotion of acinar cell events (e.g. zymogen activation, accelerated LDH leakage from acinar cells) generally thought to play an important mechanistic role in the evolution of acute pancreatitis. To further test this hypothesis, we have performed additional studies using an alternative bile acid (Na-taurocholate) rather than TLCS. Retrograde intraductal infusion of either bile acid triggers pancreatitis but the concentration of Na-taurocholate needed to elicit this response (37 mM)5 greatly exceeds the concentration of TLCS that is required (3 mM). When 3 mM Na-taurocholate in infused, pancreatitis is not observed. Similarly, when equimolar concentrations (500 μM) of TLCS and Na-taurocholate are used for in vitro studies, only TLCS and not Na-taurocholate induces zymogen activation and accelerated LDH leakage. Presumably, the differing dose/response relationships for the 2 bile acids reflect their differing affinities for Gpbar1. Our observation that 3 mM TLCS causes pancreatitis and 500 μM TLCS causes zymogen activation/accelerated LDH leakage while the same concentrations of Na-taurocholate elicits neither of these responses and the fact that Gpbar1 deletion protects against both TLCS-induced pancreatitis and those TLCS-induced cellular changes support our hypothesis.
Our studies indicating that Gpbar1 mediates bile acid-induced generation of pathological calcium transients, intra-cellular zymogen activation, and cell injury might suggest a sequential model whereby bile acid-induced zymogen activation and cell injury are the down-stream results of bile acid-induced activation of Gpbbar1 and Gpbar1-mediated generation of pathological calcium transients. However, the mechanisms responsible for bile acid-induced cell injury may be more complex than that suggested by this simple model since, as reported by Kim et al11 pre-loading acini with BAPTA does not prevent TLCS-induced cell injury (i.e. LDH leakage). If correct, their findings would suggest that TLCS-induced cell injury is not the result of TLCS-induced pathological calcium transients but, rather, that TLCS-induced cell injury is triggered by calcium-independent mechanisms. Further studies will be needed to define those calcium-independent mechanisms.
The mechanisms responsible for intra-acinar cell activation of digestive enzyme zymogens during pancreatitis are poorly understood but recently completed studies by others (41), have suggested that vacuolar ATPases may play an important role in mediating caerulein-induced intracellular zymogen activation. Our studies, using acini prepared from wild type mice, suggest that vacuolar ATPases may also play an important role in mediating bile acid-induced intra-acinar cell activation of zymogens.
The studies reported in this communication are the first to evaluate the role of Gpbar1 in pancreatic acinar cells. Studies on other cell types have concluded that activation of Gpbar1 leads to activation of adenylate cyclase and that the cellular responses to Gpbar1 activation are mediated by a rise in cellular cAMP levels31–33, 35, 36. Our studies do not exclude the possibility that TLCS-induced activation of Gpbar1 triggers pathological calcium transients in acinar cells by events that involve adenylate cyclase activation and generation of cAMP but that would seem unlikely given the robust literature that has shown that receptor-mediated generation of acinar cell calcium transients occurs in response to activation of the phospholipase/IP3 pathway and that it involves activation of PI3K. Future studies, employing both pharmacological and genetic tools, should address this issue.
We have noted that amylase discharge is accelerated when acinar cells are exposed to 500 μM TLCS and that this response is prevented by chelation of calcium with BAPTA. Amylase discharge is also accelerated by exposure to 1 mM TLCS but that response is not prevented by calcium chelation and, presumably, it reflects the detergent-like effects of a micellar or near-micellar concentration (1 mM) of TLCS. Our finding that neither TLCS- nor caerulein-induced amylase secretion are altered by genetic deletion of Gpbar1 indicates that Gpbar1 does not play a critical role in mediating amylase secretion in response to either agent. It is widely known that caerulein-stimulated amylase secretion from acinar cells is mediated via receptors for cholecystokinin but the mechanisms responsible for bile acid-stimulated enzyme secretion from acinar cells is not known and our results do not exclude the possibility that it may be mediated by as yet unidentified cell surface or intracellular bile acid receptors.
It is perhaps noteworthy that TLCS triggers intracellular zymogen activation in wild type acini and pancreatitis in wild type mice yet it stimulates, rather than inhibits, amylase secretion. Studies by our group as well as others, primarily using caerulein as the stimulus, have suggested that zymogen activation, cell injury, and pancreatitis occur when secretory stimulation is combined with blockade of secretion and support for this concept has come from the observation that interventions which promote enzyme discharge from acinar cells can reduce the severity of caerulein-induced pancreatitis38–40. Our currently reported findings suggest that this concept may not be operative when cell injury or, indeed, pancreatitis is triggered by exposure to TLCS since the inciting agent, itself, promotes secretion in that setting.
In summary, we have shown that the cell surface bile acid receptor Gpbar1 is expressed by pancreatic acinar cells, that genetic deletion of the cell surface bile acid receptor Gpbar1 protects mice from bile acid-induced pancreatitis and that Gpbar1 deletion interferes with in vitro bile acid-induced generation of acinar cell pathological calcium transients, intracellular activation of digestive enzyme zymogens, and acinar cell injury. These findings lead us to the potentially paradigm-shifting conclusion that biliary acute pancreatitis may be a receptor-mediated disease and/or that Gpbar1 may function as a severity-modifying gene in biliary pancreatitis. As such, variations in Gpbar1 expression and/or function could be responsible for the frequently made observation that only a subset of potentially susceptible patients actually develop either gallstone pancreatitis or ERCP-induced pancreatitis and, within that subset, the severity of a pancreatitis attack may vary considerably. Furthermore, our findings lead us to speculate that interventions which interfere with Gpbar1 activation could favorably effect the course and outcome of biliary pancreatitis. Although our observations are compatible with the conclusion that Gpbar1 expression by pancreatic acinar cells mediates bile acid-induced pancreatitis, our findings do not exclude the possibilities that (a) Gpbar1 expression by non-acinar pancreatic cells or (b) pancreatic expression of other bile acid receptors (e.g., the nuclear farnesoid X receptor) might also contribute to the mediation of biliary pancreatitis and/or the regulation of its severity.
Supplementary Material
Acknowledgments
Supported by NIH Grant RO-1 31396 from NIDDK (MLS), P30-NS047423 and by a fellowship from Sigrid Jusélius Foundation, Finland (JML)
Footnotes
Financial Disclosure: Nothing to declare
Author contribution: GP designed, performed the experiments, analyzed the data and wrote the manuscript
JL designed, performed the experiments and analyzed the data
GV analyzed the data and edited the manuscript
MS designed the experiments, analyzed the data and wrote the manuscript
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Opie EL. The etiology of acute hemorrhagic pancreatitis. Bull Johns Hopkins Hosp. 1901;12:182–8. [Google Scholar]
- 2.Opie EL. The relationship of cholelithiasis to disease of the pancreas and fat necrosis. Bull Johns Hopkins Hosp. 1901;12:19–21. [Google Scholar]
- 3.Lerch MM, Saluja AK, Runzi M, et al. Pancreatic duct obstruction triggers acute necrotizing pancreatitis in the opossum. Gastroenterology. 1993;104:853–861. doi: 10.1016/0016-5085(93)91022-a. [DOI] [PubMed] [Google Scholar]
- 4.Aho HJ, Koskensalo SM, Nevalainen TJ. Experimental pancreatitis in the rat. Sodium taurocholate-induced acute hemorrhagic pancreatitis. Scand J Gastroenterol. 1980;15:411–416. doi: 10.3109/00365528009181493. [DOI] [PubMed] [Google Scholar]
- 5.Laukkarinen JM, van Acker GJ, Weiss ER, et al. A mouse model of acute biliary pancreatitis induced by retrograde pancreatic duct infusion of Na-taurocholate. Gut. 2007;56:1590–1598. doi: 10.1136/gut.2007.124230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sherman S, Lehman GA. Endoscopic retrograde cholangiopancreatography- and endoscopic sphincterotomy-induced pancreatitis. In: Beger HG, Warshaw AL, Buchler MW, et al., editors. The Pancreas. Blackwell; Oxford: 1998. pp. 291–310. [Google Scholar]
- 7.Zamniak P, Little JM, Radominska A, Oelberg DG, et al. Taurine-conjugated bile acids act as Ca2+ ionophores. Biochemistry. 1991;30:8598–8604. doi: 10.1021/bi00099a015. [DOI] [PubMed] [Google Scholar]
- 8.Bouscarel B, Kroll SD, Fromm H. Signal transduction and hepatocellular bile acid transport: cross talk between bile acids and second messengers. Gastroenterology. 1999;117:433–452. doi: 10.1053/gast.1999.0029900433. [DOI] [PubMed] [Google Scholar]
- 9.Voronina S, Longbottom R, Sutton R, et al. Bile acids induce calcium signals in mouse pancreatic acinar cells: implications for bile-induced pancreatic pathology. J Physiol. 2002;540:49–55. doi: 10.1113/jphysiol.2002.017525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mauricio AC, Sllawik M, Heitzman D, et al. Deoxycholic acid (DOC) affects the transport properties of distal colon. Pflugers Arch. 2000;439:532–540. doi: 10.1007/s004249900226. [DOI] [PubMed] [Google Scholar]
- 11.Kim JY, Kim KH, Lee JA, et al. Transporter-mediated bile acid uptake causes Ca2+-dependent cell death in rat pancreatic acinar cells. Gastroenterology. 2002;122:1941–1953. doi: 10.1053/gast.2002.33617. [DOI] [PubMed] [Google Scholar]
- 12.Fischer L, Gukovskaya AS, Penninger JM, Mareninova OA, Friess H, Gukovsky I, Pandol SJ. Phosphatidylinositol 3 kinase facilitates bile acid-induced Ca2+ responses in pancreatic acinar cells. Am J Physiol Gastrointest Liver Physiol. 2006;292:G875–G886. doi: 10.1152/ajpgi.00558.2005. [DOI] [PubMed] [Google Scholar]
- 13.Voronina S, Longbottom R, Sutton R, et al. Bile acids induce calcium signals in mose pancreatic acinar cells: implications for bile-induced pancreatic pathology. J Physiol. 2008;540:49–55. doi: 10.1113/jphysiol.2002.017525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gerasimenko JV, Flowerdew SE, Voronina SG, et al. Bile acids induce Ca2+ release from both the endoplasmic reticulum and acidic intracellular calcium stores through activation of inositol trisphosphate receptors and ryanodine receptors. J Biol Chem. 2006;281:40154–40163. doi: 10.1074/jbc.M606402200. [DOI] [PubMed] [Google Scholar]
- 15.Maruyama T, Miyamoto Y, Nakamura T, et al. Identification of membrane-type receptor for bile acids (M-BAR) Biochem Biophys Res Commun. 2002;298:714–719. doi: 10.1016/s0006-291x(02)02550-0. [DOI] [PubMed] [Google Scholar]
- 16.Kawamata Y, Fujii R, Hosoya M, et al. A G protein-coupled receptor responsive to bile acids. J Biol Chem. 2003;278:9435–9440. doi: 10.1074/jbc.M209706200. [DOI] [PubMed] [Google Scholar]
- 17.Vassileva G, Golovko A, Markowitz L, et al. Targeted deletion of Gpbar1 protects mice from cholesterol gallstone formation. Biochem J. 2006;398:423–430. doi: 10.1042/BJ20060537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maruyama T, Tanaka K, Suzuki J, et al. Targeted disruption of G protein-coupled bile acid receptor 1 (Gpbar1/M-Bar) in mice. J Endocrinol. 2006;191:197–205. doi: 10.1677/joe.1.06546. [DOI] [PubMed] [Google Scholar]
- 19.Lampel M, Kern HF. Acute interstitial pancreatitis in the rat induced by excessive doses of a pancreatic secretagogue. Virchows Arch A Pathol Anat Histol. 1977;37:97–117. doi: 10.1007/BF00432156. [DOI] [PubMed] [Google Scholar]
- 21.Frossard JL, Hadengue A, Spahr L. Natural history of long-term lung injury in mouse experimental pancreatitis. Crit Care Med. 2002;30:1541–1546. doi: 10.1097/00003246-200207000-00024. [DOI] [PubMed] [Google Scholar]
- 22.Van Acker GJ, Perides G, Weiss ER, et al. Tumor progression locus-2 is a critical regulator of pancreatic and lung inflammation during acute pancreatitis. J Biol Chem. 2008;282:22140–22149. doi: 10.1074/jbc.M702225200. [DOI] [PubMed] [Google Scholar]
- 23.Laukkarinen JM, Weiss ER, van Acker GJ, et al. Protease-activated receptor-2 exerts contrasting model-specific effects on acute experimental pancreatitis. J Biol Chem. 2008;283:20703–207012. doi: 10.1074/jbc.M801779200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sharma A, Tao X, Gopal A, et al. Calcium dependene of proteinase-activated receptor 2 and cholecystokinin-mediated amylase secretion from pancreatic acini. Am J Physiol Gastrointest Liver Physiol. 2005;289:G686–695. doi: 10.1152/ajpgi.00342.2004. [DOI] [PubMed] [Google Scholar]
- 25.Sharma A, Tao X, Gopal A, et al. Protection against acute pancreatitits by activation of protease-activated receptor-2. Am J Physiol Gastrointest Liver Physiol. 2005;288:G388–395. doi: 10.1152/ajpgi.00341.2004. [DOI] [PubMed] [Google Scholar]
- 26.Makishima M, Okamoto AY, Repa JJ, et al. Identification of a nuclear receptor for bile acids. Science. 1999;284:1362–65. doi: 10.1126/science.284.5418.1362. [DOI] [PubMed] [Google Scholar]
- 27.Parks DJ, Blanchard SG, Bledsoe RK, et al. Bile acids are ligands for an orphan nuclear receptor. Science. 1999;284:1365–68. doi: 10.1126/science.284.5418.1365. [DOI] [PubMed] [Google Scholar]
- 28.Kliewer SA, Moore JT, Wade L, et al. An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell. 1998;92:73–82. doi: 10.1016/s0092-8674(00)80900-9. [DOI] [PubMed] [Google Scholar]
- 29.Qin P, Tang X, Elloso MM, et al. Bile acids induce adhesion molecule expression in endothelial cells trough activation of reactive oxygen species, NF-kappaB, and p38. Am J Physiol Heart Circ Physiol. 2006;291:H741–747. doi: 10.1152/ajpheart.01182.2005. [DOI] [PubMed] [Google Scholar]
- 30.Brady LM, Beno DWA, Davis BH. Bile acid stimulation of early growth response gene and mitogen-activated protein kinase is protein kinase C-dependent. Biochem J. 1996;316:765–769. doi: 10.1042/bj3160765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maruyama T, Miyamoto Y, Nakamura T, Tamai Y, Okada H, Sugiyama E, Itadani H, Tanaka K. Identification of membrane-type receptor for bile acids (M-BAR) Biochem Biophys Res Commun. 2002;298:714–719. doi: 10.1016/s0006-291x(02)02550-0. [DOI] [PubMed] [Google Scholar]
- 32.Kawamata Y, Fuji R, Hosoya M, et al. A G-protein coupled receptor responsive to bile acids. J Biol Chem. 2003;278:9435–9440. doi: 10.1074/jbc.M209706200. [DOI] [PubMed] [Google Scholar]
- 33.Houten SM, Watanabe M, Auwerx J. Endocrine functions of bile acids. EMBO J. 2006;25:1419–1425. doi: 10.1038/sj.emboj.7601049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Katsuma S, Hirasawa A, Tsujimoto G. Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem Biophys Res Commun. 2005;329:386–390. doi: 10.1016/j.bbrc.2005.01.139. [DOI] [PubMed] [Google Scholar]
- 35.Yang JI, Yoon JH, Myung SJ, et al. Bile acid-induced TGR5-dependent cJun-N terminal kinase activation leads to enhanced caspase 8 activation in hepatocytes. Biochem Biophys Res Commun. 2007;361:156–161. doi: 10.1016/j.bbrc.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 36.Thomas C, Auwerx J, Schoonjans K. Bile acids and the membrane bile acid receptor TGR5–connecting nutrition and metabolism. Thyroid 2007. 2008;18:167–174. doi: 10.1089/thy.2007.0255. [DOI] [PubMed] [Google Scholar]
- 37.Hofman AF. Biliary secretion and excretion. In: West JB, Baltimore MD, editors. Physiological Basis of Medical Practice. 11. Williams and Wilkins; 1990. pp. 675–691. [Google Scholar]
- 38.Saluja AK, Saluja M, Printz H, Zavertnik A, Sengupta A, Steer ML. Experimental pancreatitis is mediated by low-affinity cholecystokinin receptors that inhibit digestive enzyme secretion. Proc Natl Acad Sci USA. 1989;86:8968–8971. doi: 10.1073/pnas.86.22.8968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saluja AK, Dawra RK, Lerch MM, Steer ML. CCK-JMV-180, an analog of cholecystokinin, releases intracellular calcium from an inositol trisphosphate-independent pool in rat pancreatic acini. J Biol Chem. 1992;267:11202–11207. [PubMed] [Google Scholar]
- 40.Grady T, Mah’Moud M, Otani T, et al. Zymogen proteolysis within the pancreatic acinar cell is associated with cellular injury. Am J Physiol. 1998;275:G1010–1017. doi: 10.1152/ajpgi.1998.275.5.G1010. [DOI] [PubMed] [Google Scholar]
- 41.Bhoomagoud M, Jung T, Atladotir J, Kolodecik TR, Shugrue C, Chaudhuri A, Thrower EC, Gorelick FS. Reducing extracellular pH sensitizes the acinar cell to secretagogue-induce pancreatitis responses in rats. Gastroenterology. 2009;137:1083–1092. doi: 10.1053/j.gastro.2009.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.