

Abstract
Proteins tagged with lysine (Lys, K) 48 polyubiquitins chains are destined for degradation by the 26S proteasomal system. Impairment of the ubiquitin proteasome system (UPS) function culminates in the accumulation of polyubiquitinated proteins in many neurodegenerative conditions including Parkinson’s disease (PD). Nevertheless, the cellular mechanisms underlying cell death induced by an impaired UPS are still not clear. Intriguingly, recent studies indicate that several proteins associated with familial PD are capable of promoting the assembly of Lys‐63 polyubiquitin chains. Therefore, the objective of this study was to examine the role of K48 and K63 ubiquitination in mitochondria‐mediated apoptosis in in vitro models of dopaminergic degeneration. Exposure of the widely used proteasome inhibitor MG‐132 to dopaminergic neuronal cell line (N27) induced a rapid accumulation of polyubiquitinated proteins in the mitochondria. This appears to result in the preferential association of ubiquitin conjugates in the outer membrane and polyubiquitination of outer membrane proteins. Interestingly, the ubiquitinK48R mutant effectively rescued cells from MG‐132‐induced mitochondrial apoptosis without altering the antioxidant status of cells; whereas the ubiqutinK63R mutant augmented the proapoptotic effect of MG‐132. Herein, we report a novel conclusion that polyubiquitinated proteins, otherwise subjected to proteasomal degradation, preferentially accumulate in the mitochondria during proteolytic stress; and that polyubiquitination of Lys‐48 and Lys‐63 are key determinants of mitochondria‐mediated cell death during proteasomal dysfunction. Together, these findings yield novel insights into a crosstalk between the UPS and mitochondria in dopaminergic neuronal cells.
Keywords: ubiquitin, proteasome, mitochondria, neurotoxicity, dopaminergic neurons, Parkinson’s disease, ubiquitin proteasomal system
Introduction
The ubiquitin proteasome system (UPS) is a vital cellular mechanism responsible for degradation of intracellular proteins participating in diverse biological processes [1]. The proteolytic function of UPS involves polyubiquitination of target proteins and proteolytic degradation of the ubiquitinated target proteins by the 26S proteasome. For polyubiquitination, ubiquitin first forms an isopeptidyl bond between its carboxyl group of glycine 76 and the ε‐amino group of an internal lysine residue of the target proteins. Then, a second ubiquitin is covalently linked to the internal lysine of the preceding ubiquitin via an isopeptidyl bond. Progressive additions of the ubiquitin result in the extension of polyubiquitin chains. All seven internal lysine residues of ubiquitin could potentially serve as polyubiquitination sites, with the K48 and K63 polyubiquitin being the two most abundant forms [2]. Normally, K48 polyubiquitin targets the substrates for degradation by the 26S proteasome, whereas K63 polyubiquitin is involved in signal pathways other than proteolysis [3].
Parkinson’s disease (PD) is a primary neurodegenerative movement disorder, pathologically characterized by relatively selective loss of nigral dopamine neurons. Mitochondrial dysfunction and oxidative stress have long been recognized as major events of nigral dopaminergic degeneration. A pathogenic role of mitochondrial dysfunction in PD is supported by PD modelling with mitochondrial complex I inhibitors and the observed mitochondrial complex I deficit in the substantia nigra and platelets of PD patients [4]. Mitochondrial dysfunction has been suggested to generate excessive free radical production, resulting in oxidative injury [5]. Furthermore, the recently discovered PD genes PINK1 and DJ‐1 have been tightly linked to mitochondrial dysfunction or oxidative stress [4]. In addition to mitochondrial dysfunction and oxidative stress, emerging evidence indicates a defective ubiquitin proteasome degradation pathway may also play an important role in the pathogenesis of PD. Compromised proteasome function has been reported in the substantia nigra of post mortem brain samples from subjects with sporadic PD [6] as well as in experimental PD models [2, 7, 8, 9]. Mutations in Parkin (a ubiquitin ligase) and Uch‐L1 (a deubiquitin enzyme) have been linked to early onset PD. Despite the observation of proteasomal inhibition by mitochondrial complex I inhibitors [10] and profound mitochondrial pathology in cells exposed to low doses of proteasome inhibitor [11], little is known about how UPS impairment and mitochondrial dysfunction are mutually related in the degenerative processes of dopaminergic neurons. Paradoxically, several PD‐related proteins including Parkin, α‐synuclein and Uch‐L1 (mutations of which are associated with familial PD) have been shown to facilitate the assembly of K63 polyubiquitin chains [12, 13, 14, 15], with the pathophysiological relevance of the observations to PD pathogenesis unknown. Recent finding by Bennett and the coworker revealed marked global alteration of ubiquitination systems in the mouse model of Huntington’s disease, including the significant accumulation of K48 and K63 polyubiquitin chains [16]. Herein, we report that inhibition of proteasome induces a rapid accumulation of polyubiquitinated proteins in the mitochondria during activation of an apoptotic cascade and that polyubiquitin sites Lys‐48 and Lys‐63 differentially regulate cell death and survival in dopaminergic cells following proteasomal dysfunction.
Materials and methods
Cell culture, plasmid construction and stable expression
The immortalized rat mesencephalic dopaminergic neuronal (N27) cells were cultured as described previously [17]. N27 cells are widely used as a model system for studying cellular mechanisms of neurodegeneration in PD [18, 19, 20, 21]. UbiquitinK48R and ubiquitinK63R plasmids (provided by Dr. Douglas Gray, Ottawa Health Research Institute, Ontario, Canada) encoding His6‐ubiquitin/green fluorescent protein (GFP) fusion proteins were subcloned into the pCEP4 vector at Xho I and Hind III sites. The linear fusion of His6‐ubiquitin and GFP are post‐translationally processed to release the functional ubiquitin monomer [22]. The AMAXA nucleofection kit was used to transfect the constructs in N27 cells. Single clones were selected and screened with hygromycin for stable expression.
DNA fragmentation and assays for proteasome and caspases
Quantification of DNA fragmentation using an ELISA kit and assays for proteasomal and caspase‐3 and caspase‐9 activities using fluorogenic substrates were conducted as described in our previous publications [17]. The fluorescence intensity was normalized to protein concentrations in each sample.
Subcellular fraction and Western blot
N27 cells were homogenized in buffer (250 mM sucrose, 1 mM EDTA, 50 mM Tris, 1 mM DTT, 1 mM PMSF and protease inhibitor cocktail [1 mM AEBSF, 800 nM aprotinin, 50 μM bestatin, 15 μM E64, 20 μM leupeptin, 10 μM pepstatin A, EDTA 5 mM; by Holt; Pierce, Rockford, IL, USA] pH 7.4). The resulting supernatant (1000 g × 10 min.) of homogenates was centrifuged at 10,000 ×g at 4°C to obtain a crude mitochondrial pellet. The supernatant was further centrifuged at 100,000 ×g to collect the supernatant and a pellet as cytosolic fraction and microsome fraction, respectively [51]. Nuclei were prepared using NE‐PER Nuclear and cytoplasmic extraction reagent (Pierce Biotech, Rockford, IL, USA). To improve purity, crude mitochondrial suspension was placed in a sucrose gradient (2 ml of 1.2 M and 1.6 M sucrose) and centrifuged at 40,000 ×g for 1 hr at 4°C [23]. Isolated mitochondria, microsome or nuclei were lysed with phosphate buffer solution (PBS) containing 0.2% Triton X‐100 and protease inhibitors.
To dissociate mitochondrial ubiquitin conjugates, mitochondria were washed with PBS and incubated in 0.1 M Na2CO3 at room temperature for 5 min. After spin‐down, the pellet and supernatant were collected for Western blot. The antibodies used included cytochrome c (BD Pharmingen, San Jose, CA, USA), β‐actin, (Sigma, St. Louis, MO, USA), COX IV (cytochrome c oxidase, subunit 4; Invitrogen, Carlsbad, CA, USA), ubiquitin (DAKO, Carpinteria, CA, USA) and voltage‐dependent anion channel (VDAC; Abcam, Cambridge, MA, USA).
Analysis of His6‐ubiquitin expression
Cells were homogenized in buffer (HEPES 20 mM, NaCl 300 mM, imidazole 5 mM and protease inhibitors, pH 8). The supernatant was incubated with Proaffinity Ni‐IMAC resin (Bio‐Rad, Hercules, CA, USA), and the bound proteins and the bounded proteins were eluted by boiling the resin in SDS loading buffer, and then separated on SDS‐PAGE for Coomassie blue staining.
In vitro ubiquitination
Ubiquitination kits (Boston Biochem, Cambridge, MA, USA) contain energy sources, ubiquitin and ubiquitination enzymes. Mitochondria were incubated with fraction A and B, ubiquitin aldehyde and ubiquitin for 2 hrs at 30°C, then washed with isolation buffer and lysed for Western blot analysis with ubiquitin and cytochrome c antibodies.
Confocal analysis of mitochondrial superoxide
MitoSOX Red (Invitrogen) can be selectively targeted to mitochondria. The oxidization of MitoSOX Red by superoxide stains mitochondrial DNA and exhibits Red fluorescence. N27 cells are incubated with MitoSOX Red (5 μM), then washed with Hank’s buffered salt solution (HBSS) before confocal analysis (Nikon, Model TE‐2000U).
Glutathione assay
Briefly, cells were lysed with buffer (50 mM Tris, 1 mM EDTA, 10 mM EGTA and 1% NP‐40, pH 7.4). The supernatant (16,000 g for 10 min.) of cell lysates was incubated with 2 mM monochlorobimane for 15 min. at 37°C. The fluorescence intensity was monitored with Ex/Em at 380 nm/460 nm.
Results
Proteasome inhibitor MG‐132 activates mitochondrial apoptosis
To determine whether proteasome inhibition has any effect on mitochondrial function, we measured early mitochondrial proapoptotic markers: cytochrome c release and caspase‐9 activation, in rat mesencephalic dopaminergic neuronal cells (N27 cells) following treatment with the proteasome inhibitor MG‐132. As shown in Figure 1A, 2.5‐μM MG‐132 rapidly inhibited more than 80% of proteasomal activity within 5 min. This was followed by cytochrome c release from mitochondria to cytosol (Fig. 1B) and a dramatic activation of caspase‐9 (Fig. 1C). No significant changes in cytochrome c release and caspase‐9 activation were observed until 15 min. of MG‐132 treatment (data not shown). These results indicate that proteasome inhibition triggers a mitochondrial apoptotic cascade in dopaminergic cells.
Figure 1.
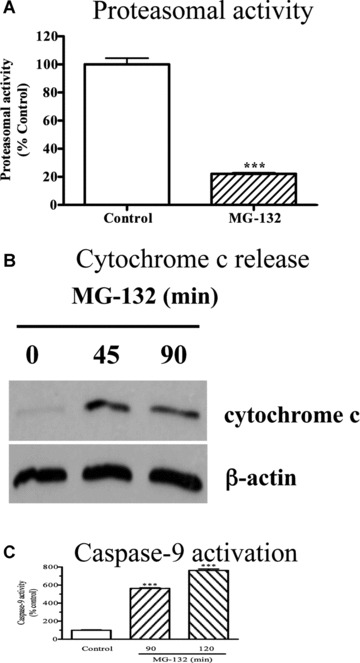
(A) Activation of mitochondrial apoptosis by MG‐132. N27 cells were exposed to 2.5 μM MG‐132 for 5 min. and then chymotrypsin‐like proteasomal activity was assayed as described in the methods. The data represent mean ± S.E.M., n= 6, ***P < 0.001. (B) Cytochrome c release. N27 cells were treated with 2.5 μM MG‐132 for 45 or 90 min., and the level of cytosolic cytochrome c was examined by Western blot using cytochrome c antibody. The membranes were reprobed with β‐actin antibody. (C) Caspase‐9 activation. Caspase‐9 activity was assayed in the cells exposed to MG‐132 for 90 or 120 min. as described in the methods section. The activity was expressed as the percentage of vehicle‐treated cells. n= 6, ***P < 0.001 as compared to control group.
Proteasome inhibition by MG‐132 causes mitochondrial accumulation of ubiquitinated proteins
To understand how proteasome inhibition triggers mitochondrial apoptosis in dopaminergic neuronal cells, we first examined the relative accumulation of polyubiquitinated proteins in subcellular fractions following proteasome inhibition. Surprisingly, Western blot analysis for the subcellular fractions revealed a dramatic elevation of ubiquitin conjugates in the mitochondrial fraction, whereas only a slight increase in the cytosolic and nuclear fractions, and a relatively moderate increase in the microsomal fraction were noted in N27 cells following MG‐132 exposure (Fig. 2A). Detailed time‐course analysis (Fig. 2B) further confirmed the novel finding of a dramatic elevation of the ubiquitinated protein in the mitochondria following exposure to proteasome inhibitor. To improve the purity, the crude mitochondria were further separated on a sucrose gradient. Western blot analysis of the fractions collected showed that the mitochondrial marker COX IV was predominantly detected in fractions 3 to 5, and especially in fraction 4 (Fig. 2C), at the interface approximately midway through the gradient, which is consistent with a previous study that used a similar mitochondrial purification procedure [23]. Importantly, ubiquitin conjugates of high molecular weights were distributed in a pattern similar to that of COX IV, with the strongest immunoreactivity revealed in fraction 4 (Fig. 2C).
Figure 2.
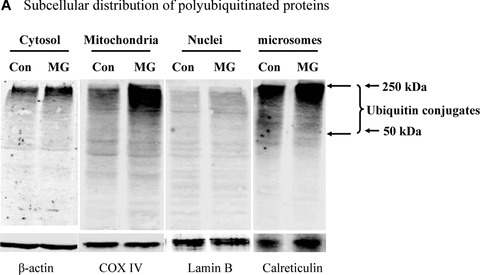
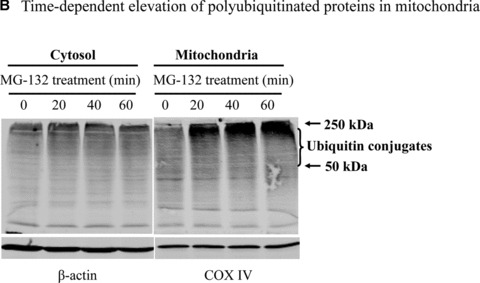
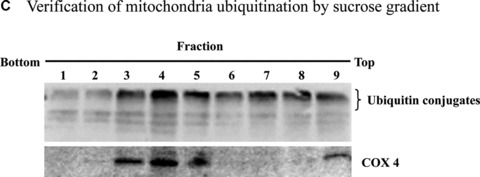
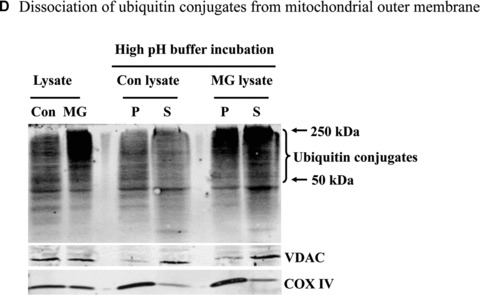
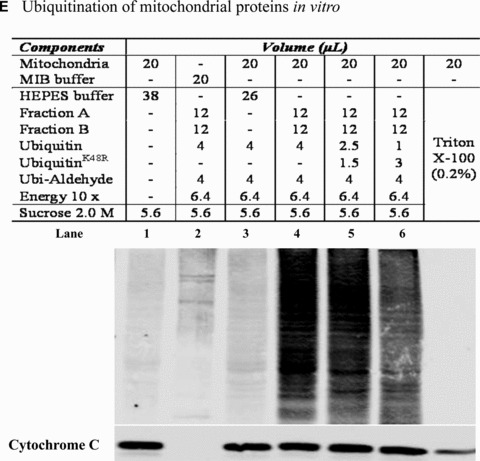
Mitochondrial accumulation of ubiquitinated proteins. (A) Subcellular distribution of polyubiquitin proteins. N27 cells were treated with 2.5 μM MG‐132 for 40 min. The cytosolic, mitochondrial, nuclear and microsomal fractions were prepared as described in the methods. Equal amounts of proteins were used for Western blot analysis using antibodies against ubiquitin, and subcellular markers β‐actin (cytosol), COX IV (mitochondria), lamin B (Nuclei) and calreticulin (microsome). (B) Time‐dependent elevation of polyubiquitinated proteins in mitochondria. Cytosolic and mitochondrial fractions were prepared from N27 cells exposed to 2.5 μM MG‐132 for 20, 40 or 60 min., and processed for ubiquitin immunoblotting. (C) Verification of mitochondria ubiquitination by sucrose gradient. Crude mitochondria isolated from N27 cells exposed to MG‐132 (2.5 μM for 40 min.) were subjected to sucrose gradient separation as described in the methods section. All the fractions collected were resolved on SDS‐PAGE and blotted with ubiquitin, β‐actin and/or COX IV antibodies. (D) Dissociation of ubiquitin conjugates from mitochondrial outer membrane. Mitochondria isolated from vehicle (Con) or MG‐132‐treated cells (MG) were incubated with a high pH buffer as described in the methods section, and then mitochondria were spun down. Both the mitochondrial pellets (P) and the equal proportion of the supernatant (S) were separated on SDS‐PAGE and immunoblotted with antibodies for ubiquitin, VDAC and COX IV. (E) Ubiquitination of mitochondrial proteins in vitro. The reaction was carried out by incubating the mitochondrial suspension (4 mg/ml) with ubiquitination enzymes (9.6 μg for fraction A and B included in the kits), ubiquitin (8 μg), ubiquitin aldehyde and an energy source. Mitochondria were then recovered from the reaction mixture for Western blotting using ubiquitin or cytochrome c antibodies.
Since the mitochondrial outer membrane pore VDAC is inaccessible to free ubiquitin (MW 8 kD), mitochondrial polyubiquitinated proteins could result either from direct hyperubiquitination of mitochondrial outer membrane proteins and/or preferential mitochondrial association of cytosolic polyubiquitinated proteins. In order to determine the amount of ubiquitinated proteins loosely associated with the mitochondria, we used a high pH buffer (0.1 M Na2CO3, pH 10) that disturbs loosely associated membrane proteins [24]. Incubation of mitochondria with the high pH buffer released the mitochondrial outer membrane marker VDAC together with ubiquitin conjugates, but not the inner membrane marker COX IV, into the supernatant (Fig. 2D). However, a substantial portion of the ubiquitin conjugates was resistant to the high pH buffer and remained associated with the mitochondrial pellet (Fig. 2D). This observation raises the possibility that some mitochondrial membrane proteins with tight membrane interaction could undergo hyperubiquitination.
To determine further whether mitochondrial membrane proteins readily undergo ubiquitination, we performed an in vitro cell‐free ubiquitination assay in isolated mitochondria. As shown in Figure 2E, ubiquitin immunoreactivity dramatically increased in themitochondria incubated with ubiquitinating enzymes (lane 4), but not in the absence of ubiquinating enzymes E1, E2 and E3 in fractions A and B (lane 3). Notably, substitution of wild‐type (wt) ubiquitin with ubiquitinK48R partially reduced mitochondrial ubiquitin immunoreactivity, suggesting that an appreciable amount of ubiquitination occurred as a result of the extension of K48 polyubiquitin chains (lane 5 and 6).
Stable expression of wild‐type or mutant ubiquitin and their effect on cellular redox status
To determine whether polyubiquitin sites have any role in mitochondria‐mediated apoptosis during proteasome inhibition, we established dopaminergic cells stably expressing ubiquitin with polyubiquitination sites Lys‐48 or Lys‐63 mutated using the plasmids encoding His6‐ubiquitin/GFP fusion proteins. The expressed His6‐ubiquitin/GFP fusion proteins have been previously shown to be readily processed to yield functional His6‐ubiquitin and GFP [22, 25]. To achieve a stable and constitutive expression, the coding sequence for His6‐ubiquitin/GFP (wt, K48R or K63R ubiquitin, a kind gift from Dr. Douglas Gray, Ottawa Health Research Institute, Ontario, Canada) was subcloned into a pCEP4 vector with CMV promoter for mammalian expression. After prolonged hygromycin B screening, the majority of cells derived from single clones were positive, as indicated by the notable GFP expression levels in wild‐type (wt‐Ub), lysine‐48 mutant (K48R‐Ub) and lysine‐63 mutant (K63R‐Ub) transfected cells (Fig. 3A). Analysis of the His6‐tagged proteins enriched from the transfected cells by SDS‐PAGE and Coomassie blue staining revealed the presence of His6‐tagged ubiquitin only in transfected cells, with comparable expression levels observed among wt‐Ub, K48R‐Ub and K63R‐Ub expressing cells (Fig. 3B). This corroborates the precise processing of the fusion proteins into His6‐tagged ubiquitin, as reported previously [22, 25].
Figure 3.
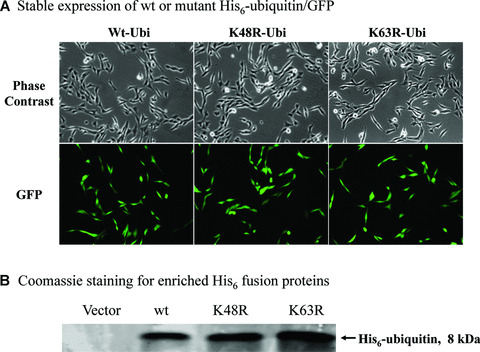
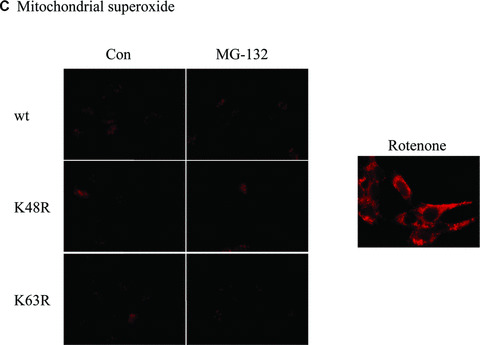
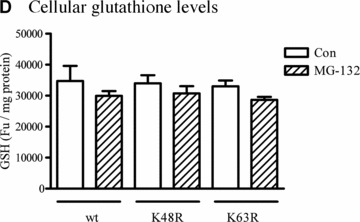
Effect of wt or mutant ubiquitin on cellular Redox status. (A) Stable expression of wt or mutant His6‐ubiquitin/GFP. Phase contrast and GFP images for cells stably expressing the linear fusion of wt, K48R or K63R ubiquitin/GFP. (B) Coomassie staining for enriched His6 fusion proteins. His6‐tagged proteins were enriched from three lines of stable cells using Ni‐IMAC resin, and resolved on SDS‐PAGE and then processed for Coomassie blue staining. The arrow indicates the protein of about 8 kD expressed in the cells, and the size roughly matches the molecular weight of His6‐ubiquitin. (C) Mitochondrial superoxide. Cells stably expressing His6‐tagged ubiquitin/GFP were treated with either 2.5 μM MG‐132 or 1 μM rotenone (positive control) for 1 hr and then incubated with the MitoSOX for 15 min. The live images were then analysed with confocal microscopy. (D) Cellular glutathione levels. Cells were treated with 2.5 μM MG‐132 for 1 hr. Cellular glutathione levels were determined as described in the methods. Data represent results of two experiments performed in triplicate.
Next, we examined whether expression of ubiquitin mutants alters cellular redox status by analysing mitochondrial superoxide production and cellular GSH levels in wt‐Ub, K48R‐Ub and K63R‐Ub expressing cells. Confocal analysis of mitochondrial superoxide using MitoSOX Red showed that neither the expression of ubiquitin mutants nor MG‐132 treatment altered mitochondrial reactive oxygen species (ROS) generation (Fig. 3C), whereas 1‐hr exposure to 1‐μM rotenone induced mitochondrial superoxide production (positive control). Similarly, analysis of cellular glutathione revealed no significant difference in the three different mutant cells (Fig. 3D).
Effect of wild‐type, K48R‐Ub and K63R‐Ub on proteasomal inhibitor‐induced mitochondrial apoptosis
The MG‐132‐induced mitochondrial apoptosis cascade was examined in the cells expressing wt, K48R‐Ub or K63R‐Ub in order to determine the role of different polyubiquitination sites in cell death. As shown in Figure 4A, MG‐132 treatment triggered a profound release of cytochrome c from mitochondria to cytosol in the wt‐Ub and K63R‐Ub mutants, but only minimal cytosolic cytochrome c release in K48R‐Ub expressing cells. In addition to cytochrome c release, activation of caspase‐9 (Fig. 4B), caspase‐3 (Fig. 4C) and DNA fragmentation (Fig. 4D) was also significantly attenuated in K48R‐Ub mutant cells compared to cells expressing wt‐Ub alone. This suggests the assembly of K48 polyubiquitin chains plays a causal role in activation of mitochondrial apoptosis. Of note, the anti‐apoptotic effect of the K48R‐Ub mutant is Lys48 ubiquitination site‐specific, since K63R‐Ub mutation did not provide similar protection. On the contrary, K63R‐Ub mutant render the dopaminergic cells more susceptible to MG‐132‐induced apoptosis (Fig. 4B–D), suggesting that K63 may be neuroprotective in dopaminergic cells.
Figure 4.
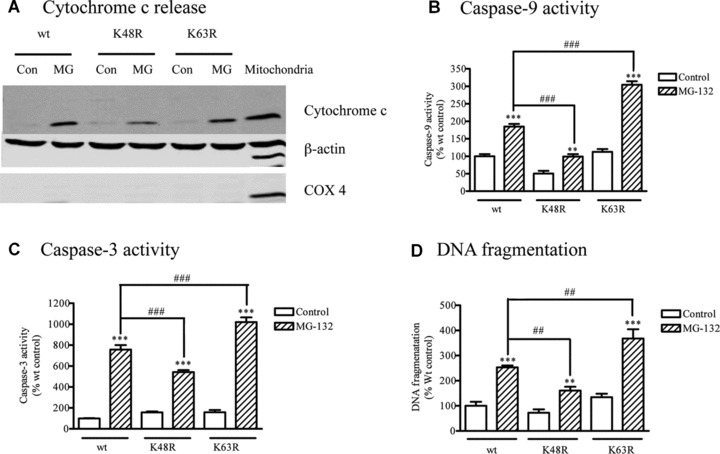
Effect of mutant ubiquitin on apoptosis. (A) Cytochrome c release. N27 cells stably expressing wt or mutant ubiquitins (K48R or K63R) were treated with 2.5 μM MG‐132 for 45 min., and the cytosolic fractions were subjected to Western blot analysis using cytochrome c. The membranes were reprobed with β‐actin and COX 4 antibodies. (B) Caspase‐9 activity, (C) Caspase‐3 activity and (D) DNA fragmentation. N27 cells stably expressing wt or mutant ubiquitins (K48R or K63R) were treated with MG‐132 for 120 min. Enzymatic activities of caspase‐9 and ‐3 and DNA fragmentation were determined as described previously. Data for caspase‐9 represent results of two separate experiments with a total of 11 individual samples, respectively; the data for caspase‐3 were derived from three experiments with a total of 16 individual samples, respectively. Two separate experiments with a total of six separate samples were analysed for DNA fragmentation. Values were expressed as the percentage of vehicle‐treated wt ubiquitin control group. **P < 0.01, ***P < 0.001 compared with individual control groups; ##P < 0.01, ###P < 0.001, comparison between indicated groups.
Discussion
Impairment in the ubiquitin‐proteasome system and mitochondrial dysfunction have both been implicated in the pathogenesis of PD. This study revealed a prominent accumulation of polyubiquitinated proteins in the mitochondria during the activation of mitochondrial apoptosis in dopaminergic neuronal cells following proteasome inhibition. Intriguingly, the same Lys to Arg mutation at different polyubiquitination sites, Lys‐48 or −63, appeared to exert opposite effects on the activation of the mitochondrial apoptosis cascade, indicating distinctly different roles for these two types of polyubiquitin in dopaminergic cell survival.
Proteasome inhibitors have been previously shown to mimic some key features of PD, including dopamine cell death [2], one of the fundamental pathological features of PD, in vivo and in vitro. In this dopaminergic cell model of PD, we found that proteasome inhibition triggers the activation of the mitochondrial apoptotic cascade (Fig. 1A). The mechanisms underlying activation of mitochondrial apoptotic signalling during proteasome impairment are not currently understood. A number of proteins modulate mitochondrial apoptosis (e.g. IAPs, Mcl‐1, flip, Bax, Smac, p53). All have been shown to be the proteolytic substrates of UPS [26]. A study by Rideout and colleagues showed that dopamine neurons selectively undergo apoptosis upon proteasomal inhibition in mesencephalic primary culture [27]. Recently, we observed that MPP+, a widely used Parkinsonian toxin, induces oxidative stress and inhibits proteasome activities in dopaminergic cell models. Importantly, mutant ubiquitin K48R effectively attenuates MPP+ ‐triggered apoptosis in N27 cells, similarly to that of observed in MG132 treatment (unpublished observations). Furthermore, we observed extracellular dopamine can reduce proteasome activity in our cell model but the effect of ubiquitin mutants on dopamine‐induced neurotoxicity is yet to be characterized.
The functional consequence of proteasome inhibition is the inadequate clearance and resulting intracellular accumulation of K48‐tagged polyubiquitinated proteins. Western blot analysis of subcellular fractions and the sucrose gradient purified mitochondrial fraction revealed a marked elevation of polyubiquitinated proteins in the mitochondria from cells exposed to proteasome inhibitor (Fig. 2A–C), indicative of the potential role of the mitochondrion as an early key sensor of polyubiquitinated protein stress following proteasome dysfunction in dopaminergic cells. The similar elevation of ubiquitin conjugates was also manifested in the microsomal, cytosolic and nuclear fractions, although to a much lower extent (Fig. 2A). In addition to oxidative stress, ER stress may play a role in the cell death processes in PD [28, 29, 30]. However, we did not observe any significant activation of caspase‐12, a primary effector marker of ER stress, following MG‐132 in our cell model (unpublished observations). Recently, we showed that MG‐132 induces cytochrome c release from the mitochondria and activates caspase‐9 and caspase‐3 in a time‐dependent manner [19], demonstrating mitochondria‐dependent apoptotic cell death in dopaminergic cells. Together, our results suggest that a mitochondria‐dependent apoptotic cell death pathway, not an ER stress pathway, may be the primary contributor in dopaminergic cell death during proteasomal dysfunction. Since the mitochondrial outer membrane pore VDAC is permeable only to molecules smaller than 5 kD, it is thus inaccessible to free ubiquitin (MW 8 kD). Therefore, it is conceivable that mitochondrial accumulation of polyubiquitinated proteins could result either from direct hyperubiquitination of the mitochondrial outer membrane proteins or preferential association of cytosolic polyubiquitinated proteins with the mitochondrial outer membrane. In fact, both factors appeared to be involved in the observed elevation of mitochondrial ubiquitin conjugates. Release of mitochondrial ubiquitin conjugates, together with the outer membrane marker, VDAC (but not inner membrane marker COX IV), by a high pH buffer that disturbs loosely associated membrane proteins [24], implied a preferential association of a significant amount of polyubiquitinated proteins with the mitochondrial outer membrane (Fig. 2D). However, a substantial portion of the ubiquitin conjugates that are resistant to the high pH buffer and, as a result, remain associated is reminiscent of some integral membrane proteins or of proteins with tight membrane interactions (Fig. 2D). Taken together with the presence of several mitochondrial‐associated E3 ligases [31, 32, 33] and deubiquitinating enzymes [34, 35], this observation raises the possibility that some mitochondrial membrane proteins could undergo hyperubiquitination. The idea was substantiated by using an in vitro cell‐free ubiquitination assay, demonstrating that mitochondrial proteins are ready for ubiquitination/polyubiquitination modification (Fig. 2E). The mitochondrial proteins that are targets of hyperubiquitination are not currently well characterized. Some mitochondrial proteins previously shown to undergo ubiquitination modification are DJ‐1 [15], prohibitin [36], aconitate hydratase, ATP synthase alpha chain, isocitrate dehydrogenase precursor, aspartate aminotransferase precursor, malate dehydrogenase precursor [37], mitofusin [38], and mitochondrial protein hFis1 and Drp1 [33]. The mitochondrial outer membrane protein, Fzo 1, which is subjected to proteasome dependent degradation, remains associated even after ubiquitination [39]. A previous study suggested neuroprotective roles for both Parkin, an E3 ligase associated with the mitochondrial outer membrane [31], and the mitochondrial resident proteins, DJ1 and PINK1, in mitochondria‐mediated apoptosis [31, 40, 41], indicating that proteasome inhibitors may impair the functions of these proteins in the mitochondria. Additionally, Parkin‐knockout mice revealed gross mitochondrial pathology [42]. A recent study demonstrated that polyubiquitination of misfolded DJ‐1 by Parkin facilitates its targeting to aggresomes [15].
Accumulation of mitochondrial ubiquitin conjugates likely represents a key early cellular response during neuronal stress since ubiquitin conjugates have also been reported to accumulate in the mitochondria of cortical and hippocampal neurons following cerebral ischaemia [43]. In this dopaminergic cell line, stable expression of dominant negative ubiquitinK48R, which prevents the polyubiquitin chains from extension via Lys‐48, tremendously attenuated the MG‐132‐triggerred mitochondrial apoptosis. This suggests the assembly of K48 polyubiquitin chains plays a causal role in activation of mitochondrial apoptosis. Of note, the anti‐apoptotic effect of the K48R‐Ub mutant is Lys48 ubiquitination site‐specific, since K63R‐Ub mutation did not provide similar protection, but rather rendered the dopaminergic cells more susceptible to MG‐132‐induced apoptosis (Fig. 4B–D). Very likely, assembly of K63 polyubiquitin chains is neuroprotective in dopaminergic cells upon stress. A previous study by Tsirigotis et al. showed that stable expression of both K48R‐Ub and K63R‐Ub mutants enhanced toxicity of cadmium and canavanine in mouse HT4 neuroblastoma cells [22]. In contrast, our results reveal an opposing differential regulation of apoptotic cell death by the two mutants in which the K48R‐Ub mutant showed an anti‐apoptotic effect, while the K63R‐Ub mutant was proapoptotic. Although the reasons for the apparent difference remain elusive, the distinct effect could reflect the fact that the role of ubiquitin sites might depend on cell types and stimuli. In line with this, ubiquitinK48R caused elevated oxidative damage in HT4 neuroblastoma for reasons yet to be understood [25]. However, we found that mitochondrial superoxide production (Fig. 3C) and glutathione levels (Fig. 3D) appear to be comparable in dopaminergic neuronal cells expressing wt, ubiquitinK48R, or ubiquitinK63R. Of note, lentivirus‐mediated transfection of K48R‐Ub mutant was also effective in suppressing MG‐132 ‐induced caspase−9 and −3 activation, indicating that the type of vector used is not a primary factor in the observed neuroprotection (data not shown). The protective effect of K48R‐Ub was supported by a recent study in which genetic crosses between K48R‐Ub mutant mice and spinocerebellar ataxic mice showed a delayed neuronal loss in the cerebellum [44].
Elevation of mitochondrial ubiquitin conjugates and suppression of mitochondrial apoptosis by the K48R‐Ub mutant together suggest that preferential mitochondrial accumulation of ubiquitinated proteins could be proapoptotic. In this context, suppression of ubiquitination by the dominant negative yeast ubiquitin conjugating, enzyme cdc34 (Ubc3), protected against proteasome inhibitor‐induced apoptotic cell death [45]. However, it remains to be determined whether mitochondrial accumulation of polyubiquitinated proteins could be a direct causal event sufficient to activate mitochondrial apoptosis, since a similar elevation of ubiquitin conjugates was also observed in other fractions, though to a much lower extent.
It has been observed that proteasomal inhibition might be beneficial. A recent study by Fueller et al. showed that C‐RAF activation promotes phosphorylation and ubiquitination of BAD protein leading to increased turnover and degradation leading to increased cell survival [46].However, in the same study authors reported that proteasomal inhibition by MG‐132 causes intracellular buildup of BAD which leads to toxicity. Another study reported that oxidative stress can induce apoptosis by regulating phosphorylation and ubiquitination of Bcl‐2 family proteins, resulting in increased proapoptotic protein levels and decreased antiapoptotic protein expression [47]. Thus, ubiquitination of proapoptotic protein such as BAD can be beneficial if it is targeted for degradation or harmful if it is sequestered or accumulated during proteasomal inhibition.
It is also likely that activation of mitochondrial apoptosis requires mitochondrial translocation of some ubiquitinated cytosolic factors such as p53 [48]. An elevated CREB binding protein (CBP) level, due to its insufficient UPS degradation, was hypothesized to underlie the neuroprotection of ubiquitinK48R in transgenic mice [44]. However, no appreciable change in the CBP level was observed in our cell model transfected with K48R‐Ub mutant, although proteasome inhibition by MG‐132 effectively increased the cellular CBP level (data not shown), consistent with ubiquitin‐dependent proteasome degradation of CBP [49].
Another interesting observation of our study is the sensitization of the mitochondrial apoptotic cascade by K63R‐Ub mutant, suggesting that the Lys‐63 ubiquitination site has an important anti‐apoptotic function in dopaminergic cells. The cellular mechanisms underlying the protective role of the Lys‐63 ubiquitination site is yet to be characterized. The Lys‐63 polyubiquitin chains are involved in cellular events such as DNA damage repair and NF‐κB activation [50]. Presumably, interference with the cellular processes by mutation of Lys‐63 poses additional neuronal stress, predisposing cells to apoptosis. Furthermore, assembly of K63 polyubiquitin chains might facilitate the delivery of misfolded proteins, such as DJ‐1, to aggresomes, thus sequestrating the neurotoxic misfolded proteins [15]. Interestingly, several PD genes, including Parkin, α‐synuclein and Uch‐L1, have been shown to promote the assembly of K63 polyubiquitin chains [12, 13, 14, 15]. A recent study in a mouse model of Huntington’s disease demonstrated brain accumulation of both K48 and K63 polyubiquitin chains [16]. The antiapoptotic capacity of Lys‐63 polyubiquitination sites observed here appeared to present the first line of evidence for the potential pro‐survival role of K63 polyubiquitin chains during dopaminergic degeneration in PD.
In conclusion, we demonstrate a novel finding that mitochondrial proteins are preferentially hyperubiquitinated in a dopaminergic neuronal cell model during proteasome dysfunction. Additionally, our results reveal a unique site‐specific regulation of the mitochondrial apoptotic cascade by polyubiquitination sites, in which Lys‐48 plays a proapoptotic role while Lys‐63 has an anti‐apoptotic function during impairment of proteasome function. Future efforts focusing on characterization of mitochondrial‐associated polyubiquitinated proteins are anticipated to further unravel the relationship between UPS impairment and mitochondrial apoptosis. This line of research will have important implications for elucidating the cellular mechanisms of neurodegenerative processes in Parkinson’s and Huntington diseases and will thereby facilitate the development of therapeutic strategies for these disorders.
Acknowledgements
This study was supported by grants from the National Institute of Health NS38644, ES10586 and NS65167. The W. Eugene and Linda Lloyd Endowed Chair and Professorship to AGK is also acknowledged. The authors acknowledge Dr. Douglas Gray, Ottawa Health Research Institute, Ontario, Canada, for providing the ubiquitin mutant constructs for the study. The authors also acknowledge Ms. Keri Henderson and Ms. Mary Ann deVries for their assistance in the preparation of this manuscript.
†Current institutional affiliation: Washington University School of Medicine, St Louis, MO, USA
References
- 1. Glickman MH, Ciechanover A. The ubiquitin‐proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002; 82: 373–428. [DOI] [PubMed] [Google Scholar]
- 2. Sun F, Kanthasamy A, Anantharam V, et al . Environmental neurotoxic chemicals‐induced ubiquitin proteasome system dysfunction in the pathogenesis and progression of Parkinson’s disease. Pharmacol Ther. 2007; 114: 327–44. [DOI] [PubMed] [Google Scholar]
- 3. Pickart CM, Eddins MJ. Ubiquitin: structures, functions, mechanisms. Biochim Biophys Acta. 2004; 1695: 55–72. [DOI] [PubMed] [Google Scholar]
- 4. Abou‐Sleiman PM, Muqit MM, Wood NW. Expanding insights of mitochondrial dysfunction in Parkinson’s disease. Nat Rev Neurosci. 2006; 7: 207–19. [DOI] [PubMed] [Google Scholar]
- 5. Jenner P. Oxidative stress in Parkinson’s disease. Ann Neurol. 2003; 53: S26–36; discussion S36–8. [DOI] [PubMed] [Google Scholar]
- 6. Olanow CW, McNaught KS. Ubiquitin‐proteasome system and Parkinson’s disease. Mov Disord. 2006; 21: 1806–23. [DOI] [PubMed] [Google Scholar]
- 7. Betarbet R, Canet‐Aviles RM, Sherer TB, et al . Intersecting pathways to neurodegeneration in Parkinson’s disease: effects of the pesticide rotenone on DJ‐1, alpha‐synuclein, and the ubiquitin‐proteasome system. Neurobiol Dis. 2006; 22: 404–20. [DOI] [PubMed] [Google Scholar]
- 8. Halliwell B. Proteasomal dysfunction: a common feature of neurodegenerative diseases? Implications for the environmental origins of neurodegeneration. Antioxid Redox Signal. 2006; 8: 2007–19. [DOI] [PubMed] [Google Scholar]
- 9. Zeng BY, Iravani MM, Lin ST, et al . MPTP treatment of common marmosets impairs proteasomal enzyme activity and decreases expression of structural and regulatory elements of the 26S proteasome. Eur J Neurosci. 2006; 23: 1766–74. [DOI] [PubMed] [Google Scholar]
- 10. Hoglinger GU, Carrard G, Michel PP, et al . Dysfunction of mitochondrial complex I and the proteasome: interactions between two biochemical deficits in a cellular model of Parkinson’s disease. J Neurochem. 2003; 86: 1297–307. [DOI] [PubMed] [Google Scholar]
- 11. Sullivan PG, Dragicevic NB, Deng JH, et al . Proteasome inhibition alters neural mitochondrial homeostasis and mitochondria turnover. J Biol Chem. 2004; 279: 20699–707. [DOI] [PubMed] [Google Scholar]
- 12. Doss‐Pepe EW, Chen L, Madura K. Alpha‐synuclein and parkin contribute to the assembly of ubiquitin lysine 63‐linked multiubiquitin chains. J Biol Chem. 2005; 280: 16619–24. [DOI] [PubMed] [Google Scholar]
- 13. Lim KL, Dawson VL, Dawson TM. Parkin‐mediated lysine 63‐linked polyubiquitination: a link to protein inclusions formation in Parkinson’s and other conformational diseases Neurobiol Aging. 2006; 27: 524–9. [DOI] [PubMed] [Google Scholar]
- 14. Liu C, Fei E, Jia N, et al . Assembly of lysine 63‐linked ubiquitin conjugates by phosphorylated alpha‐synuclein implies Lewy body biogenesis. J Biol Chem. 2007; 282: 14558–66. [DOI] [PubMed] [Google Scholar]
- 15. Olzmann JA, Li L, Chudaev MV, et al . Parkin‐mediated K63‐linked polyubiquitination targets misfolded DJ‐1 to aggresomes via binding to HDAC6. J Cell Biol. 2007; 178: 1025–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bennett EJ, Shaler TA, Woodman B, et al . Global changes to the ubiquitin system in Huntington’s disease. Nature. 2007; 448: 704–8. [DOI] [PubMed] [Google Scholar]
- 17. Sun F, Anantharam V, Zhang D, et al. Proteasome inhibitor MG‐132 induces dopaminergic degeneration in cell culture and animal models. Neurotoxicology. 2006; 27: 807–15. [DOI] [PubMed] [Google Scholar]
- 18. Chinta SJ, Andersen JK. Reversible inhibition of mitochondrial complex I activity following chronic dopaminergic glutathione depletion in vitro: implications for Parkinson’s disease. Free Radic Biol Med. 2006; 41: 1442–8. [DOI] [PubMed] [Google Scholar]
- 19. Sun F, Kanthasamy A, Song C, et al . Proteasome inhibitor‐induced apoptosis is mediated by positive feedback amplification of PKCdelta proteolytic activation and mitochondrial translocation. J Cell Mol Med. 2008; 12: 2467–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zafar KS, Inayat‐Hussain SH, Ross D. A comparative study of proteasomal inhibition and apoptosis induced in N27 mesencephalic cells by dopamine and MG132. J Neurochem. 2007; 102: 913–21. [DOI] [PubMed] [Google Scholar]
- 21. Zhang X, Li L, Prabhakaran K, et al . Uncoupling protein‐2 up‐regulation and enhanced cyanide toxicity are mediated by PPARalpha activation and oxidative stress. Toxicol Appl Pharmacol. 2007; 223: 10–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tsirigotis M, Zhang M, Chiu RK, et al . Sensitivity of mammalian cells expressing mutant ubiquitin to protein‐damaging agents. J Biol Chem. 2001; 276: 46073–8. [DOI] [PubMed] [Google Scholar]
- 23. Kim TH, Zhao Y, Ding WX, et al . Bidcardiolipin interaction at mitochondrial contact site contributes to mitochondrial cristae reorganization and cytochrome C release. Mol Biol Cell. 2004; 15: 3061–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cavalli V, Kujala P, Klumperman J, et al . Sunday Driver links axonal transport to damage signaling. J Cell Biol. 2005; 168: 775–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hyun DH, Gray DA, Halliwell B, et al . Interference with ubiquitination causes oxidative damage and increased protein nitration: implications for neurodegenerative diseases. J Neurochem. 2004; 90: 422–30. [DOI] [PubMed] [Google Scholar]
- 26. Zhang HG, Wang J, Yang X, et al . Regulation of apoptosis proteins in cancer cells by ubiquitin. Oncogene. 2004; 23: 2009–15. [DOI] [PubMed] [Google Scholar]
- 27. Rideout HJ, Lang‐Rollin IC, Savalle M, et al . Dopaminergic neurons in rat ventral midbrain cultures undergo selective apoptosis and form inclusions, but do not up‐regulate iHSP70, following proteasomal inhibition. J Neurochem. 2005; 93: 1304–13. [DOI] [PubMed] [Google Scholar]
- 28. Wang HQ, Takahashi R. Expanding insights on the involvement of endoplasmic reticulum stress in Parkinson’s disease. Antioxid Redox Signal. 2007; 9: 553–61. [DOI] [PubMed] [Google Scholar]
- 29. Ryu EJ, Harding HP, Angelastro JM, et al . Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson’s disease. J Neurosci. 2002; 22: 10690–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Holtz WA, O’Malley KL. Parkinsonian mimetics induce aspects of unfolded protein response in death of dopaminergic neurons. J Biol Chem. 2003; 278: 19367–77. [DOI] [PubMed] [Google Scholar]
- 31. Darios F, Corti O, Lucking CB, et al . Parkin prevents mitochondrial swelling and cytochrome c release in mitochondria‐dependent cell death. Hum Mol Genet. 2003; 12: 517–26. [DOI] [PubMed] [Google Scholar]
- 32. Li W, Bengtson MH, Ulbrich A, et al . Genome‐wide and functional annotation of human E3 ubiquitin ligases identifies MULAN, a mitochondrial E3 that regulates the organelle’s dynamics and signaling. PLoS ONE. 2008; 3: e1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yonashiro R, Ishido S, Kyo S, et al . A novel mitochondrial ubiquitin ligase plays a critical role in mitochondrial dynamics. EMBO J. 2006; 25: 3618–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kinner A, Kolling R. The yeast deubiquitinating enzyme Ubp16 is anchored to the outer mitochondrial membrane. FEBS Lett. 2003; 549: 135–40. [DOI] [PubMed] [Google Scholar]
- 35. Nakamura N, Hirose S. Regulation of mitochondrial morphology by USP30, a deubiquitinating enzyme present in the mitochondrial outer membrane. Mol Biol Cell. 2008; 19: 1903–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Thompson WE, Ramalho‐Santos J, Sutovsky P. Ubiquitination of prohibitin in mammalian sperm mitochondria: possible roles in the regulation of mitochondrial inheritance and sperm quality control. Biol Reprod. 2003; 69: 254–60. [DOI] [PubMed] [Google Scholar]
- 37. Weekes J, Morrison K, Mullen A, et al . Hyperubiquitination of proteins in dilated cardiomyopathy. Proteomics. 2003; 3: 208–16. [DOI] [PubMed] [Google Scholar]
- 38. Cohen MM, Leboucher GP, Livnat‐Levanon N, et al . Ubiquitin‐proteasome‐dependent degradation of a mitofusin, a critical regulator of mitochondrial fusion. Mol Biol Cell. 2008; 19: 2457–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Neutzner A, Youle RJ, Karbowski M. Outer mitochondrial membrane protein degradation by the proteasome. Novartis Found Symp. 2007; 287: 4–14; discussion 14–20. [PubMed] [Google Scholar]
- 40. Gorner K, Holtorf E, Waak J, et al . Structural determinants of the C‐terminal helix‐kink‐helix motif essential for protein stability and survival promoting activity of DJ‐1. J Biol Chem. 2007; 282: 13680–91. [DOI] [PubMed] [Google Scholar]
- 41. Petit A, Kawarai T, Paitel E, et al . Wild‐type PINK1 prevents basal and induced neuronal apoptosis, a protective effect abrogated by Parkinson disease‐related mutations. J Biol Chem. 2005; 280: 34025–32. [DOI] [PubMed] [Google Scholar]
- 42. Palacino JJ, Sagi D, Goldberg MS, et al . Mitochondrial dysfunction and oxidative damage in parkin‐deficient mice. J Biol Chem. 2004; 279: 18614–22. [DOI] [PubMed] [Google Scholar]
- 43. Hayashi T, Takada K, Matsuda M. Post‐transient ischemia increase in ubiquitin conjugates in the early reperfusion. Neuroreport. 1992; 3: 519–20. [DOI] [PubMed] [Google Scholar]
- 44. Tsirigotis M, Tang MY, Beyers M, et al . Delayed spinocerebellar ataxia in transgenic mice expressing mutant ubiquitin. Neuropathol Appl Neurobiol. 2006; 32: 26–39. [DOI] [PubMed] [Google Scholar]
- 45. Rideout HJ, Stefanis L. Proteasomal inhibition‐induced inclusion formation and death in cortical neurons require transcription and ubiquitination. Mol Cell Neurosci. 2002; 21: 223–38. [DOI] [PubMed] [Google Scholar]
- 46. Fueller J, Becker M, Sienerth AR, et al . C‐RAF activation promotes BAD poly‐ubiquitylation and turn‐over by the proteasome. Biochem Biophys Res Commun. 2008; 370: 552–6. [DOI] [PubMed] [Google Scholar]
- 47. Li D, Ueta E, Kimura T, et al . Reactive oxygen species (ROS) control the expression of Bcl‐2 family proteins by regulating their phosphorylation and ubiquitination. Cancer Sci. 2004; 95: 644–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Marchenko ND, Wolff S, Erster S, et al . Monoubiquitylation promotes mitochondrial p53 translocation. EMBO J. 2007; 26: 923–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sanchez‐Molina S, Oliva JL, Garcia‐Vargas S, et al . The histone acetyltransferases CBP/p300 are degraded in NIH 3T3 cells by activation of Ras signalling pathway. Biochem J. 2006; 398: 215–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pickart CM, Fushman D. Polyubiquitin chains: polymeric protein signals. Curr Opin Chem Biol. 2004; 8: 610–6. [DOI] [PubMed] [Google Scholar]
- 51. Qin ZH, Wang Y, Kikly KK, et al . Pro‐caspase‐8 is predominantly localized in mitochondria and released into cytoplasm upon apoptotic stimulation. J Biol Chem. 2001; 276: 8079–86. [DOI] [PubMed] [Google Scholar]