

Abstract
Botulinum neurotoxins (BoNTs) are the etiological agents responsible for botulism, a disease characterized by peripheral neuromuscular blockade and a characteristic flaccid paralysis of humans. The natural product toosendanin, a limonoid, is a traditional Chinese medicine that has reported antibotulinum properties in animal models. Toosendanin effectively inhibits the biological activity of BoNT/A in neuronal cells at concentrations of 200 nM, and partial inhibition can be observed with concentrations as low as 8 nM. Mechanistically, toosendanin's inhibition is due to prevention of transduction of the BoNT LC through the HC channel. Intriguing questions as to the molecular architecture of toosendanin as related to its anti-botulinum properties have focused our attention on a synthesis of toosendanin's unusual AB-ring, containing a unique bridged hemi-acetal. Within the current work, a synthetic strategy allowing access to the AB-fragment of toosendanin was achieved from a trans-decalin system. In addition, this fragment was examined for its modulation of BoNT/A intoxication in a rat spinal cord cellular assay.
Keywords: Botulinum neurotoxins, Toosendanin, Semi-synthesis, Rat spinal cord cellular assay
1. Introduction
Botulinum neurotoxins (BoNTs) are the most toxic poisons known to humans, with a lethal dose (LD50) of approximately 1 ng per kg of body weight.1 There are seven serologically distinct BoNTs (A–G); serotype A (BoNT/A) is the most potent, possessing a toxicity 106-fold higher than cobra toxin and 1011-fold greater than cyanide.2 BoNT intoxication is characterized by flaccid paralysis caused by the proteolytic cleavage of specific SNARE proteins critical for the release of the neurotransmitter acetylcholine from nerve cells.3
Limonoids (1 and 2, Fig. 1) are tetranortriterpenoids with a 4,4,8-trimethylfuranylsteroid skeleton derived from euphane or tirucallane triterpenoids.4–6 A major limonoid constituent found in Melia toosendan is the compound toosendanin (2, Fig. 1) which appears to have multiple modes of action in insects including damage to midgut tissues, inhibition of esterases, cytochrome P450-aldrin epoxidase and proteinase activities.7 Interest in the application of liminoid natural products, and in particular toosendanin, in pest management remains high, and while this area of toosendanin research remains fertile, we became intrigued by a series of reports over the past two decades, mainly originating from China detailing the special activity of toosendanin as it relates to BoNT. Most interesting were two reports both published in the early 1980's detailing toosendanin's in vivo activity.8 Indeed, these cryptic reports identified toosendanin's anti-botulinum properties in both monkey and mouse models spanning these serotypes. Unfortunately, these reports proved to be difficult to substantiate either due to a lack of pure material (i.e. toosendanin) or inconsistencies with toosendanin in animal assays.
Figure 1.
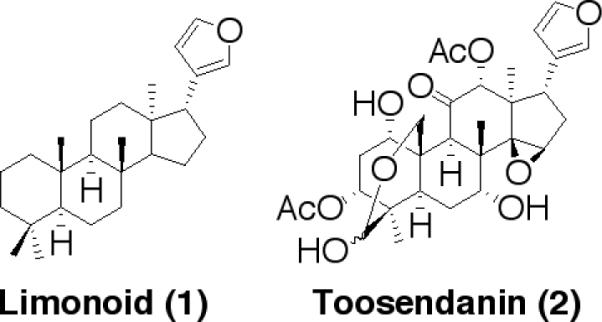
General structures of limonoid (1) and toosendanin (2).
To resolve this dilemma, we reported the effects of well characterized toosendanin, using a sensitive and specific spinal cord cell-based assay, which validated toosendanin's activity in both BoNT serotypes A and E.9 Thus, exposure of neurons to BoNT/A in presence of increasing concentrations of toosendanin resulted in the gradual preservation of intact, uncleaved synaptosomal associated protein of 25 kD (SNAP-25), the intracellular BoNT/A and BoNT/E substrate, becoming practically complete above 200 nM; excitingly, partial inhibition can be observed with concentrations as low as 8 nM for BoNT/A and 40 nM for BoNT/E, respectively. To further solidify toosendanin's BoNT inhibitory activity, we utilized single molecule channel forming experiments that clearly demonstrated toosendanin's inhibition mode is due to prevention of transduction of the BoNT light chain (LC) through the heavy chain (HC) channel.9,10 Thus, toosendanin selectively arrests the LC translocation step of intoxication with subnanomolar potency, and increases the unoccluded HC channel propensity to open with micromolar efficacy. Interestingly, these studies also provided strong evidence that toosendanin has an unprecedented dual mode of action within the protein-conducting channel acting both as a cargo-dependent inhibitor of translocation and as cargo-free channel activator. To further validate toosendanin's anti-botulinum properties, we have also investigated toosendanin's protective effects in a mouse lethality model. Positive findings have allowed us to begin to assign which chemical functionalities are important for the anti-botulinum properties found within toosendanin.9 As a starting point we initially turned to semi-synthesis to generate a set of rationally designed toosendanin analogues (3–6, Fig. 2) that were prepared so as to probe the most salient functionalities embedded within toosendanin without perturbing its gross chemical structure. These analogues were examined in the mouse lethality assay wherein only 4 proved to have equivalent activity to toosendanin.
Figure 2.

Structures of toosendanin's analogues (3–6).
To both potentially improve and ultimately decipher toosendanin's anti-botulinum properties, the next tactic we envisioned embraced Function-Oriented Synthesis (FOS) as advocated by Wender.11 The underpinnings of FOS are that the function of a biologically active lead structure can be emulated, tuned, or possibly improved by replacement with simpler scaffolds designed to encompass the key activity-determining structural features of the natural product. As stated (vide supra), through our semi-synthetic efforts, the epoxide and acetoxy moieties were found to be important, while the furan ring was not. To further define toosendanin, we dissected the molecule into two fragments consisting of AB- and CD-rings. As such, we reported our initial research exploring the synthesis of the CD-ring of toosendanin and its potential biological function.12 Our synthetic approach to the 4-acetoxy CD-ring of toosendanin was achieved starting from mesityl oxide and acetylacetone in 14 steps. This work represented the first semi-synthesis of the CD-ring of toosendanin correctly displaying all heteroatom substituents found within the natural product. In continuing with our general plan, we disclose our successful synthesis of toosendanin's AB-ring and testing of its biological activity (Fig. 3).
Figure 3.

AB-ring of toosendanin (7).
2. Results and Discussion
In the search for a suitable trans-decalin system as a starting material with an angular substituent and oxygen functionalities on both rings, we found ester (10) to be an ideal candidate for this study (Scheme 1). This compound is readily available in large scale, from commercially available starting materials (8 and 9) by a two-step modification as detailed by Jones and Dodds.13 Thus, upon benzylation of the hydroxy group found within 10 under acidic conditions,14 the resulting ketone was protected as the ketal (11) in 51% yield for the two steps. Reduction of 11 with LAH followed by acid-induced hydrolysis of the dimethyl ketal furnished a hydroxy ketone, which could then be utilized for protection of the primary alcohol with SEMCl affording 12 in 93% overall yield from 11. The preparation of the desired enone (13) was accomplished by KHMDS-promoted phenyselenylation at −78 °C followed by oxidation and elimination of the resulting selenide using hydrogen peroxide in 83% yield.15 Subsequent regioselective carboxymethylation with Mander's reagent16 in the presence of LHMDS at −78 °C afforded a β-ketomethylester; this was followed by methylation, thus methyl iodide in the presence of NaH granted the corresponding α-methyl-β-ketomethylester.17 Epoxidation of the enone moiety by TBHP in the presence of benzyltrimethylammonium hydroxide (triton B)18 furnished stereoselectively epoxide (14), in 65% yield for three steps from 13.19 The regioselective opening of the epoxide moiety20 found within 14 was established by PhSeNa (generated in situ from diphenyl diselenide and NaBH4) in 86% yield followed by silylation of 15 with SEMCl, which led to disilyl ether (16) in 85% yield from 14 (Scheme 1).
Scheme 1.

Synthesis of 16. Reagents and conditions: (a) NaOEt, EtOH; (b) H2, Pd/C, ether; (c) Benzyl 2,2,2-trichloroacetimidate, cat. TfOH, cyclohexane/CH2Cl2 (2:1, v/v); (d) cat. H2SO4, MeOH, 51% for 2 steps; (e) LAH, THF, reflux; (f) 5N HCl, THF; (g) SEMCl, DIPEA, CH2Cl2 93% for 3 steps; (h) KHMDS, THF, −78 °C, then TESCl, −78 °C; (i) PhSeCl, CH2Cl2, −78 °C; (j) 30% H2O2, THF, 83% for 3 steps; (k) LHMDS, THF, −78 °C, then NCCO2Me, −78 °C; (l) NaH, MeI, THF; (m) TBHP, triton B, THF, 65% for 3 steps; (n) (PhSe)2, NaBH4, EtOH, 86%; (o) SEMCl, DIPEA, CH2Cl2, 99%.
The relative stereochemistry of 15 was ascertained through analysis of its 2D-ROESY NMR. Thus, for 15, a ROESY correlation was observed between Me-1 (equatorial) and H-8a (axial); H-7 (axial) and H-8a; no long range interactions were observed between H-4 (equatorial) and H-8a protons (Fig. 4).
Figure 4.
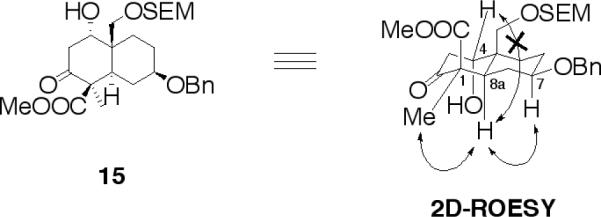
The relative stereochemistry of compound (15) as determined by 2D-ROESY experiment.
Synthesis of 7-epi AB-ring of toosendanin (TSDN)
To construct the diol directly, we attemped the stereoselective reduction of the ketone and methyl ester moiety of 16 with LAH or DIBAL at −78 °C, but unfortunately the undesired stereochemistry at C-2 position was obtained as a major isomer; the lone reaction that succeeded was NaBH4 in CH2Cl2/MeOH (15:1). Again, however, the desired compound 17 was obtained as a minor isomer yet, the total yield was quantitative, hence, we invoked a recycling method via oxidation of 2β-form (epi-17) with Dess-Martin reagent that granted 16 in 96% yield. Reduction of ester (17) to the primary alcohol using LAH proceeded in an excellent yield. The resulting diol was selectively oxidized with 2,2,6,6-tetramethyl-1-piperidinyloxy free radical (TEMPO) under biphasic conditions to furnish an aldehyde followed by acetylation of the free secondary alcohol granted 18 in 85% yield for three steps from 17.21 Hydrogenation of 18 with palladium hydroxide on carbon provided 19 as a hemi-acetal moiety in 94% yield; as expected 20 was not observed. While, removal of SEM group with TFA furnished 7-epi AB-ring of toosendanin (epi-7) in 80% yield (Scheme 2).
Scheme 2.

Synthesis of 7-epi AB-ring of toosendanin (epi-7). Reagents and conditions: (a) NaBH4, CH2Cl2/MeOH (15:1, v/v), 99% (ratio of epi-17/17 = 2/1); (b) Dess-Martin periodinane, CH2Cl2, 96%; (c) LAH, ether; (d) NaOCl, TEMPO, KBr, CH2Cl2/sat. NaHCO3 aq. (2:1, v/v); (e) Ac2O, pyridine, cat. DMAP, 85% for 3 steps; (f) H2, Pd(OH)2/C, CH2Cl2/MeOH (1:1, v/v), 94%; (g) TFA, CH2Cl2, 0 °C, 80%.
With the first hurdle in the synthesis in place, i.e. construction of the AB-ring, we were now faced with the challenge of inverting the stereochemistry at the C-7 position. Our initial attempt engaged the Mitsunobu reaction. Thus, 16's benzyl ether was removed by hydrogenation with palladium on carbon, and subsequent Mitsunobu reaction with 4-nitrobenzoic acid afforded 4-nitrobenzylester with the desired stereochemistry at C-7. Unfortunately, all attempts to hydrolyze this ester failed; we surmise that either the ester was resistant to hydrolysis or when hydrolysis was seen it coincided with elimination of SEM group resulting in the α,β-unsaturated ketone.
Synthesis of AB-ring of toosendanin (TSDN)
Since the necessary inversion of stereochemistry at C-7 was found untenable using a Mitsunobu sequence we were forced to rethink our strategy. We sought an advanced intermediate, and 19 appeared well-suited to meet this challenge. Thus, 19's hemi-acetal was protected by treatment with CbzCl, however, to improve yields due to over reaction, (21), was recycled providing the desired alcohol (22). Oxidation of 22 using Dess-Martin reagent afforded ketone (23) in 96% yield. Stereoselective reduction was attempted using a plethora of reagents, however, none were satisfactory. Hence, we simply relied on reduction using NaBH4 resulting in a mixture of 22 and the desired compound 24. These compounds were readily separated and again recycling was used on 22 to obtain the correct stereochemistry at C-7. The corresponding AB-ring (7), as embedded within toosendanin was finally secured by hydrogenation of 24, followed by SEM removal with TFA (Scheme 3).
Scheme 3.

Synthesis of AB-ring of toosendanin (7). Reagents and conditions: (a) CbzCl, TMEDA, CH2Cl2, −78 °C, 49% of 22; (b) H2, Pd/C, EtOH, 49% for 2 steps; (c) Dess-Martin periodinane, CH2Cl2, 96%; (d) NaBH4, CH2Cl2/MeOH (15:1, v/v), 95% (ratio of 22/24 = 2.6/1); (e) H2, Pd/C, EtOH; (f) TFA, CH2Cl2, 0 °C, 82% for 2 steps.
Testing toosendanin analogues 7 and epi-7 in an RSC assay
To examine the biological potency of toosendanin analogues 7 and epi-7, a primary rat spinal cord cellular assay (RSC assay) was engaged.22 The value of this assay is it avoids the use of animals and lethality as an end-point. Here western blot analysis can be employed where visualization of intact SNAP-25 is a measure of a compounds efficacy against botulinum neurotoxin A. In these regards and based on our previous findings in terms of data accrued from our semi-synthetic and CD-ring analogues, we anticipated little or no biological potency. Yet, we felt it incumbent to examine these structures in this cellular assay both to contrast, and validate our previous results/hypotheses as well as provide a solid grounding for future FOS studies focused upon utilizing the AB-ring nucleus. Thus, primary rat spinal cord cells were exposed to 5.6 pM BoNT/A1 combined with 200 μM toosendanin (2), 1 mM AB-ring (7), or 1 mM 7-epi AB-ring (epi-7). The controls contain toxin only, or no toxin. Cell lysates were analyzed by Western blot using a monoclonal anti-SNAP-25 antibody (Synaptic Systems). As expected, addition of toosendanin at 200 μM resulted in complete inhibition of BoNT/A1 activity, as evidenced by the appearance of uncleaved SNAP-25 only (Fig. 5). Addition of 7 or epi-7 at 1 mM concentrations consistently resulted in the same extent of SNAP-25 cleavage as the positive (toxin only) control. Thus, as expected, 7 and epi-7 did not result in any detectable inhibition of BoNT/A1 activity in primary neuronal cells.
Figure 5.
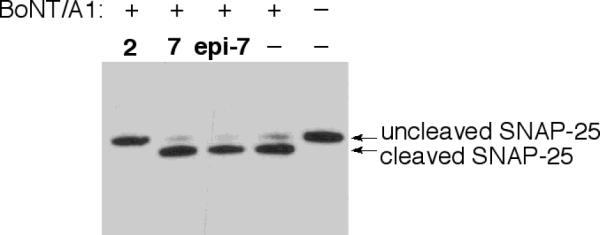
Western blot of RSC assay using toosendanin (2) and analogues 7/epi-7.
3. Conclusion
We have achieved the synthesis of the AB-ring (7) of toosendainin (5.8% overall yield in 23 steps from 10) and the 7-epi AB-ring (epi-7) of toosendanin (10% overall yield in 19 steps from 10). In accord with our previous findings neither structure afforded protection from BoNT/A intoxication within the RSC assay. However, noteworthy is that the merging of these studies with both our semi-synthetic and CD-ring analogues provides a unified picture emphasizing the importance of toosendanin's ABCD nucleus in the blocking of BoNT/A intoxication. Taken together a logical point for future studies, we will focus on the furan moiety found within toosendanin as its requirements appear more adaptable for analogues preparation and biological testing.
4. Experimental
4.1. Chemistry
In general, CH2Cl2 and MeOH were distilled from CaH2. Tetrahydrofuran (THF) was distilled from Sodium. Reagents were purchased from commercial sources and used without further purification. Synthetic reactions were monitored by analytical thin layer chromatography (TLC) carried out on 0.25 mm silica gel plates (60F-254). All flash column chromatography was performed using silica gel 60 (230–400 mesh). Preparative TLC was also performed using Merck Kieselgel 60F-254 silica gel plates (0.5 or 1 mm). 1H NMR spectra were recorded on Bruker DRX-600 (600 MHz) or DRX-500 (500 MHz) spectrometers, and 13C NMR spectra was recorded on Bruker DRX-600 (150 MHz) or DRX-500 (125 MHz) spectrometers. Chemical shifts were reported in parts per million (ppm) on the δ scale from an internal standard (NMR descriptions: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet). Electrospray ionization (ESI) time-of-flight reflectron experiments were performed at The Scripps Research Institute on an Agilent ESI-TOF mass spectrometer. Samples were electrosprayed into the TOF reflectron analyzer at a ESI voltage of 4,000 V and a flow rate of 200 μL/min. Toosendanin was purchased from AvaChem Scientific. Subsequently, the purity of the toosendanin was confirmed by both TLC and HPLC. Toosendanin by TLC with EtOAc–hexane (3:1, v/v) runs as two spots with Rf values of 0.38 and 0.56 representing a mixture of endo and exo isomers. Finally, we note that reported NMR data displays compound 7, epi-7, and 19 as a mixture of both endo and exo isomers.
4.1.1. (4aRS,7RS,8aSR)-Ethyl 7-(Benzyloxy)-2,2-dimethoxydecahydronaphthalene-4a-carboxylate (11)
Catalytic amount of trifluoromethanesulfonic acid was added to a solution of (2RS,4aRS,8aSR)-ethyl 2-hydroxy-7-oxodecahydronaphthalene-4a-carboxylate (10)13 (1.37 g, 5.7 mmol) and benzyl 2,2,2-trichloroacetimidate (1.2 mL, 6.3 mmol) in cyclohexane (20 mL) and CH2Cl2 (10 mL) at room temperature, and the mixture was stirred at room temperature for 9 h. The precipitate was filtered off, and the filtrate was washed with sat. NaHCO3 aq. and brine, dried over Na2SO4. The solvent was evaporated under reduced pressure to leave an oil. The crude was dissolved in dry MeOH (40 mL), then addition of catalytic amount of concentrated H2SO4 at room temperature. The mixture was stirred at room temperature for 14 h (over night). The reaction mixture was poured into sat. K2CO3 aq. under ice-cooling. The mixture was extracted with CHCl3, washed with brine, dried over Na2SO4. The solvent was evaporated under reduced pressure to leave an oil, which was purified by flash chromatography on SiO2 with EtOAc–hexane (1:4, v/v) to give 11 (1.10 g, 51%, for 2 steps from 10) as a yellow oil: 1H NMR (CDCl3, 600 MHz) δ 7.33–7.30 (m, 4H), 7.27–7.23 (m, 1H), 4.54 (q, J = 12.4 Hz, 2H), 4.17–4.12 (m, 2H), 3.39 (sept, J = 5.1 Hz, 1H), 3.18 (s, 3H), 3.12 (s, 3H), 2.17–2.14 (m, 1H), 2.06 (t, J = 13.2 Hz, 1H), 1.98–1.79 (m, 5H), 1.75–1.71 (m, 1H), 1.48–1.43 (m, 1H), 1.30–1.12 (m, 7H, including 1.25 (t, J = 7.2 Hz, 3H)); 13C NMR (CDCl3, 150 MHz) δ 174.6, 138.9, 128.3, 127.5, 127.4, 100.1, 77.1, 69.8, 60.0, 47.6, 47.5, 47.3, 38.9, 36.2, 35.5, 34.8, 33.6, 29.6, 29.4, 14.3; ESI-TOF MS (m/z): [MNa]+ calcd for C22H32O5Na 399.2142, Found 399.2144.
4.1.2. (4aRS,7RS,8aSR)-7-(Benzyloxy)-4a-(((2-(trimethylsilyl)ethoxy)methoxy)methyl)octahy dronaphthalen-2(1H)-one (12)
LiAlH4 (2.0 M in THF, 2.1 mL, 4.1 mmol) was added to a solution of 11 (779 mg, 2.1 mmol) in dry THF (8 mL) at 0 °C, and the mixture was stirred at 100 °C (reflux condition) for 4 h. After addition of EtOAc, 1 N HCl, and Rochelle salt, the precipitate was filtered off, then the whole was washed with 1 N HCl, sat. NaHCO3 aq., and brine, dried over Na2SO4. The solvent was evaporated under reduced pressure to leave an oil. The crude was dissolved in THF (16 mL), then addition of 5 N HCl (4 mL) at room temperature. The mixture was stirred at room temperature for 1 h. After addition of solid NaHCO3, water was added to the mixture until the sediment dissolved (pH 9). The whole was extracted with CH2Cl2, washed with brine, dried over Na2SO4. The solvent was evaporated under reduced pressure to leave an oil. The crude was dissolved in dry CH2Cl2 (8 mL), then DIPEA (0.75 mL, 4.6 mmol) and SEMCl (0.73 mL, 4.1 mmol) were added at 0 °C. The mixture was stirred at room temperature for 15 h (over night). After addition of EtOAc, the whole was washed with 1 N HCl, sat. NaHCO3 aq., and brine, then dried over Na2SO4. The solvent was evaporated under reduced pressure to leave an oil, which was purified by flash chromatography on SiO2 with EtOAc–hexane (1:5, v/v) to give 12 (809 mg, 93%, for 3 steps from 11) as a pale yellow oil. 1H NMR (CDCl3, 600 MHz) δ 7.34–7.32 (m, 4H), 7.29–7.26 (m, 1H), 4.69 (s, 2H), 4.56 (s, 2H), 3.80 (s, 2H), 3.62–3.59 (m, 2H), 3.41 (sept, J = 5.3 Hz, 1H), 2.52–2.46 (m, 1H), 2.34–2.29 (m, 2H), 2.22–2.14 (m, 2H), 2.06–1.99 (m, 2H), 1.80–1.78 (m, 1H), 1.73–1.62 (m, 1H), 1.61–1.56 (m, 1H), 1.40 (q, J = 12.2 Hz, 1H), 1.27–1.21 (m, 1H), 0.98–0.91 (m, 3H), 0.02 (s, 9H); 13C NMR (CDCl3, 150 MHz) δ 210.8, 138.7, 128.4, 127.5, 95.1, 76.9, 70.1, 65.2, 63.6, 44.2, 42.4, 37.9, 36.1, 34.6, 34.5, 32.7, 27.6, 18.1, −1.5; ESI-TOF MS (m/z): [MNa]+ calcd for C24H38O4SiNa 441.2431, Found 441.2448.
4.1.3. (4aSR,7RS,8aSR)-7-(Benzyloxy)-4a-(((2-(trimethylsilyl)ethoxy)methoxy)methyl)-4a,5,6 ,7,8,8a-hexahydronaphthalen-2(1H)-one (13)
KHMDS (0.5 M in toluene, 26.4 mL, 13.2 mmol) was added (dropwise over the course of 10 min) to a solution of 12 (3.67 g, 8.8 mmol) in dry THF (75 mL) at −78 °C. After 40 min of stirring at −78 °C, TESCl (2.2 mL, 13.2 mmol) was added, and the mixture was stirred at −78 °C for 40 additional min. After addition of sat. NaHCO3 aq., the whole was extracted with ether, washed with brine, dried over Na2SO4. The solvent was evaporated under reduced pressure to leave an oil. The crude was dissolved in dry CH2Cl2 (75 mL), cooled to −78 °C, and addition of a solution of PhSeCl (98%, 2.1 g, 10.5 mmol) in dry CH2Cl2 (15 mL). The mixture was stirred at −78 °C for 30 min. After 30 min, the mixture was washed with sat. NaHCO3 aq. and brine, dried over Na2SO4. The solvent was evaporated under reduced pressure to leave an oil. The crude was dissolved in THF (75 mL), cooled at 0 °C, then H2O2 (30 wt. % in water, 2.7 mL, 26.3 mmol) was added. The mixture was stirred at room temperature for 1 h. After addition of EtOAc, the mixture was washed with 5% Na2CO3 aq. and water, the aqueous layer was extracted with EtOAc. The combined organic layer was washed with brine, dried over Na2SO4. The solvent was evaporated under reduced pressure to leave an oil, which was purified by flash chromatography on SiO2 with EtOAc–hexane (1:5, v/v) to give 13 (3.02 g, 83%, for 3 steps from 12) as a pale yellow oil. 1H NMR (CDCl3, 600 MHz) δ 7.34–7.33 (m, 4H), 7.29–7.24 (m, 1H), 6.83 (d, J = 10.0 Hz, 1H), 5.97 (d, J = 10.0 Hz, 1H), 4.60–4.54 (m, 4H), 3.81 (d, J = 9.4 Hz, 1H), 3.77 (d, J = 9.4 Hz, 1H), 3.56–3.53 (m, 2H), 3.45 (sept, J = 5.1 Hz, 1H), 2.54 (dd, J = 17.7, 14.6 Hz, 1H), 2.24 (dd, J = 17.7, 4.3 Hz, 1H), 2.11–1.97 (m, 3H), 1.92–1.89 (m, 1H), 1.67–1.61 (m, 1H), 1.51 (q, J = 12.3 Hz, 1H), 1.30–1.24 (m, 1H), 0.97–0.90 (m, 2H), 0.01 (s, 9H); 13C NMR (CDCl3, 150 MHz) δ 199.2, 157.7, 138.5, 128.8, 128.3, 127.5, 127.4, 94.8, 76.5, 70.1, 67.9, 65.1, 40.7, 40.1, 39.2, 33.4, 31.2, 27.7, 17.9, −1.5; ESI-TOF MS (m/z): [MNa]+ calcd for C24H36O4SiNa 439.2275, Found 439.2285.
4.1.4. (1aRS,3RS,3aRS,5RS,7aRS,7bRS)-Methyl 5-(Benzyloxy)-3-methyl-2-oxo-7a-(((2-(trimethylsilyl)ethoxy)methoxy)methyl)decahyd ronaphtho[2,1-b]oxirene-3-carboxylate (14)
LiHMDS (1.0 M in THF, 10.5 mL, 10.5 mmol) was added in a single portion to a solution of 13 (2.18 g, 5.2 mmol) in dry THF (80 mL) at −78 °C. After 40 min of stirring at −78 °C, methyl cyanoformate (0.63 mL, 7.7 mmol) was added, and the mixture was stirred at −78 °C for 4 h. After addition of water and EtOAc, the whole was extracted with EtOAc, washed with brine, dried over Na2SO4. The solvent was evaporated under reduced pressure to leave an oil, which was purified by flash chromatography on SiO2 with EtOAc-hexane (1:4, v/v) to give β-ketomethylester (2.33 g, 94%) as a pale yellow oil. The β-ketomethylester (2.33 g, 4.9 mmol) was dissolved in dry THF (80 mL), cooled at 0 °C, and addition of NaH (60%, 0.24 g, 5.9 mmol). After 5 min of stirring at 0 °C, methyl iodide (1.5 mL, 24.6 mmol) was added, and the mixture was stirred at room temperature for 14 h (over night). After addition of sat. NH4Cl aq., the whole was extracted with EtOAc, washed with brine, dried over Na2SO4. The solvent was evaporated under reduced pressure to leave an oil. The crude was dissolved in THF (80 mL), cooled at 0 °C, and addition of triton B (40 wt. % in water, 3.9 mL, 9.8 mmol) and TBHP (70 wt. % in water, 1.4 mL, 9.8 mmol). The mixture was stirred at room temperature for 3 h. After addition of EtOAc, the whole was washed with 10% Na2S2O3 aq., then the aqueous layer was extracted with EtOAc. The combined organic layer was washed with brine, dried over Na2SO4. The solvent was evaporated under reduced pressure to leave an oil, which was purified by flash chromatography on SiO2 with EtOAc-hexane (1:5, v/v) to give 14 (1.71 g, 65%, for 3 steps from 13) as a very pale yellow oil. 1H NMR (CDCl3, 600 MHz) d 7.34–7.24 (m, 5H), 4.63 (s, 2H), 4.56–4.51 (m, 2H), 3.85 (d, J = 10.4 Hz, 1H), 3.66–3.58 (m, 6H, including 3.64 (s, 3H)), 3.55–3.51 (m, 1H), 3.42 (d, J = 4.4 Hz, 1H), 3.34 (sept, J = 5.2 Hz, 1H), 2.32–2.29 (m, 1H), 2.06–2.03 (m, 2H), 1.84–1.82 (m, 1H), 1.56–1.49 (m, 1H), 1.41–1.36 (m, 4H, including 1.40 (s, 3H)), 1.24–1.18 (m, 1H), 0.95–0.89 (m, 2H), 0.01 (s, 9H); 13C NMR (CDCl3, 150 MHz) d 205.3, 170.9, 138.4, 128.4, 127.6, 127.5, 95.0, 77.0, 70.3, 66.9, 65.2, 64.2, 57.4, 55.9, 52.2, 42.6, 38.8, 29.4, 28.9, 27.1, 23.8, 18.1, −1.4; ESI-TOF MS (m/z): [MNa]+ calcd for C27H40O7SiNa 527.2435, Found 527.2441.
4.1.5. (1R,4S,4aR,7R,8aR)-methyl 7-(benzyloxy)-4-hydroxy-1-methyl-2-oxo-4a-(((2-(trimethylsilyl)ethoxy)methoxy)meth yl)decahydronaphthalene-1-carboxylate (15)
Acetic acid (7.1 μL, 0.12 mmol) was added to an ethanolic solution of PhSeNa which prepared by the reduction of (PhSe)2 (98%, 119 mg, 0.37 mmol) with NaBH4 (28 mg, 0.75 mmol) in EtOH (2 mL), and the mixture was stirred at room temperature for few minutes until the bright yellow solution turned colorless. The resulting solution was added to a solution of 14 (125 mg, 0.25 mmol) in EtOH (3 mL), and stirred at room temperature for 30 min. After addition of EtOAc, the whole was washed with sat. NH4Cl aq. and brine, dried over Na2SO4. The solvent was evaporated under reduced pressure to leave an oil, which was purified by flash chromatography on SiO2 with EtOAc–hexane (1:3, v/v) to give 15 (108 mg, 86%) as a yellow oil. 1H NMR (CDCl3, 600 MHz) d 7.37–7.33 (m, 4H), 7.29–7.26 (m, 1H), 4.63 (s, 2H), 4.59 (s, 2H), 4.26 (br s, 1H), 3.85 (d, J = 10.4 Hz, 1H), 3.68 (s, 3H), 3.62 (d, J = 10.4 Hz, 1H), 3.58–3.55 (m, 2H), 3.34–3.31 (m, 1H), 3.27 (dd, J = 15.1, 2.9 Hz, 1H), 2.55 (dd, J = 15.1, 3.3 Hz, 1H), 2.22–2.20 (m, 1H), 2.05–2.02 (m, 2H), 1.89–1.80 (m, 2H), 1.71 (br s, 1H), 1.59–1.54 (m, 2H), 1.36 (s, 3H), 0.98–0.89 (m, 2H), 0.02 (s, 9H); 13C NMR (CDCl3, 150 MHz) d 206.6, 173.5, 138.6, 128.4, 127.6, 127.5, 94.9, 77.8, 71.7, 70.2, 65.2, 64.7, 57.3, 52.3, 45.2, 44.4, 42.0, 29.4, 27.2, 26.7, 20.8, 18.1, −1.5; ESI-TOF MS (m/z): [MNa]+ calcd for C27H42O7SiNa 529.2592, Found 529.2596.
4.1.6. (1RS,4SR,4aRS,7RS,8aRS)-Methyl 7-(benzyloxy)-1-methyl-2-oxo-4-((2-(trimethylsilyl)ethoxy)methoxy)-4a-(((2-(trimethyl silyl)ethoxy)methoxy)methyl)decahydronaphthalene-1-carboxylate (16)
The 15 (108 mg, 0.21 mmol) was dissolved in dry CH2Cl2 (3 mL), then DIPEA (0.19 mL, 1.2 mmol) and SEMCl (0.19 mL, 1.1 mmol) were added at 0 °C. The mixture was stirred at room temperature for 39 h. After addition of EtOAc, the whole was washed with 1 N HCl, sat. NaHCO3 aq., and brine, then dried over Na2SO4. The solvent was evaporated under reduced pressure to leave an oil, which was purified by flash chromatography on SiO2 with EtOAc-hexane (1:5, v/v) to give 16 (134 mg, 99%) as a colorless oil. 1H NMR (CDCl3, 600 MHz) d 7.37–7.33 (m, 4H), 7.29–7.27 (m, 1H), 4.70 (d, J = 7.2 Hz, 1H), 4.62 (s, 2H), 4.60 (d, J = 7.2 Hz, 1H), 4.59 (s, 2H), 4.09 (br t, J = 2.9 Hz, 1H), 3.86 (d, J = 10.4 Hz, 1H), 3.69–3.58 (m, 5H, including 3.67 (s, 3H)), 3.57–3.53 (m, 3H), 3.35–3.29 (m, 1H), 3.11 (dd, J = 15.3, 2.8 Hz, 1H), 2.72 (dd, J = 15.3, 3.3 Hz, 1H), 2.20–2.18 (m, 1H), 2.01–1.96 (m, 2H), 1.85–1.75 (m, 2H), 1.60–1.54 (m, 2H), 1.35 (s, 3H), 0.98–0.89 (m, 4H), 0.03 (s, 9H), 0.03 (s, 9H); 13C NMR (CDCl3, 150 MHz) δ 205.9, 173.5, 138.7, 128.3, 127.5, 127.5, 94.9, 94.2, 77.7, 77.4, 70.1, 65.4, 65.2, 64.3, 57.1, 52.2, 45.1, 42.2, 42.0, 29.5, 27.2, 26.6, 20.9, 18.1, 18.0, −1.5; ESI-TOF MS (m/z): [MNa]+ calcd for C33H56O8Si2Na 659.3406, Found 659.3406.
4.1.7. (1RS,2RS,4SR,4aRS,7RS,8aRS)-Methyl 7-(Benzyloxy)-2-hydroxy-1-methyl-4-((2-(trimethylsilyl)ethoxy)methoxy)-4a-(((2-(trim ethylsilyl)ethoxy)methoxy)methyl)decahydronaphthalene-1-carboxylate (17)
NaBH4 (226 mg, 5.8 mmol) was added to a solution of 16 (929 mg, 1.5 mmol) in CH2Cl2 (15 mL) and MeOH (1 mL) at 0 °C, the mixture was stirred at room temperature for 24 h. After addition of CH2Cl2, the whole was washed with sat. NH4Cl aq. and brine, then dried over Na2SO4. The solvent was evaporated under reduced pressure to leave an oil, which was purified by flash chromatography on SiO2 with EtOAc–hexane (1:5, v/v) to give 17 (286 mg, 31%) as a colorless oil and epi-17 (644 mg, 68%) as a colorless oil in the order of elution. Compound 17: 1H NMR (CDCl3, 600 MHz) δ 7.38–7.34 (m, 4H), 7.29–7.26 (m, 1H), 4.75–4.74 (m, 1H), 4.62–4.53 (m, 5H), 4.10 (br s, 1H), 4.07 (br s, 1H), 3.94 (br s, 1H), 3.76–3.71 (m, 1H), 3.67–3.54 (m, 5H, including 3.65 (s, 3H)), 3.53–3.48 (m, 2H), 3.36–3.32 (m, 1H), 3.08 (d, J = 10.2 Hz, 1H), 2.25–2.18 (m, 2H), 2.09–2.06 (m, 1H), 2.03–1.84 (m, 3H), 1.79–1.77 (m, 1H), 1.53–1.44 (m, 2H), 1.29 (s, 3H), 0.96–0.90 (m, 4H), 0.03–0.01 (m, 18H); 13C NMR (CDCl3, 150 MHz) δ 177.2, 138.9, 128.4, 127.6, 127.5, 95.2, 94.8, 78.5, 77.7, 72.0, 70.1, 66.0, 65.0, 64.0, 51.5, 48.2, 42.5, 38.5, 29.3, 29.1, 27.4, 26.9, 23.6, 18.1, 18.0, −1.4; ESI-TOF MS (m/z): [MNa]+ calcd for C33H58O8Si2Na 661.3562, Found 661.3562. Compound epi-17: 1H NMR (CDCl3, 600 MHz) δ 7.37–7.33 (m, 4H), 7.29–7.26 (m, 1H), 4.73 (d, J = 7.0 Hz, 1H), 4.62–4.54 (m, 5H), 3.89 (br s, 1H), 3.68 (s, 3H), 3.65–3.60 (m, 3H), 3.57–3.50 (m, 3H), 3.29 (br sept, J = 5.0 Hz, 1H), 3.22 (d, J = 9.9 Hz, 1H), 2.23–2.15 (m, 2H), 2.07–1.95 (m, 2H), 1.78–1.64 (m, 4H), 1.54–1.37 (m, 6H, including 1.40 (s, 3H)), 0.94–0.92 (m, 4H), 0.03–0.01 (m, 18H); 13C NMR (CDCl3, 150 MHz) δ 178.2, 138.8, 128.4, 127.6, 127.5, 94.8, 94.2, 78.2, 75.5, 73.1, 70.1, 65.4, 64.9, 63.9, 51.6, 49.2, 44.6, 42.2, 32.7, 29.7, 27.1, 26.7, 23.2, 18.1, 18.0, −1.4; ESI-TOF MS (m/z): [MNa]+ calcd for C33H58O8Si2Na 661.3562, Found 661.3576. The epi-17 (826 mg, 1.3 mmol) was dissolved in dry CH2Cl2 (10 mL), then Dess-Martin periodinane (97%, 679 mg, 1.6 mmol) was added at 0 °C. The mixture was stirred at room temperature for 1 h. After addition of sat. NaHCO3 aq, the whole was extracted with CH2Cl2, washed with brine, dried over Na2SO4. The solvent was evaporated under reduced pressure to leave an oil, which was purified by flash chromatography on SiO2 with EtOAc–hexane (1:5, v/v) to give 16 (790 mg, 96%) as a colorless oil.
4.1.8. (1RS,2RS,4SR,4aRS,7RS,8aRS)-7-(Benzyloxy)-1-formyl-1-methyl-4-((2-(trimethylsilyl)ethoxy)methoxy)-4a-(((2-(trimethylsilyl)ethoxy)methoxy)methyl)decahydronaphthalen-2-yl Acetate (18)
LiAlH4 (1.0 M in Et2O, 3.6 mL, 3.6 mmol) was added to a solution of 17 (1.16 g, 1.8 mmol) in dry Et2O (12 mL) at 0 °C, and the mixture was stirred at room temperature for 1.5 h. After careful addition of 1 mL of water, 3mL of 15% NaOH aq., and 1 mL of water, the white precipitate was filtrated off. The aqueous layer was extracted with ether, washed with brine, dried over Na2SO4. The solvent was evaporated under reduced pressure to leave an oil, which was purified by flash chromatography on SiO2 with EtOAc–hexane (1:2, v/v) to give diol (1.02 g, 91%) as a colorless oil. 1H NMR (CDCl3, 600 MHz) δ 7.35–7.33 (m, 4H), 7.29–7.26 (m, 1H), 4.75 (d, J = 7.0 Hz, 1H), 4.62–4.54 (m, 5H), 3.95 (br s, 1H), 3.83 (br s, 1H), 3.76–3.71 (m, 2H), 3.64–3.56 (m, 3H), 3.50 (d, J = 11.0 Hz, 1H), 3.46–3.43 (m, 2H), 3.41–3.37 (m, 1H), 2.06–1.97 (m, 4H), 1.89–1.82 (m, 2H), 1.51–1.42 (m, 2H), 1.34 (br q, J = 12.3 Hz, 1H), 1.25 (s, 1H), 1.12 (s, 3H), 0.96–0.92 (m, 4H), 0.03–0.02 (m, 18H); 13C NMR (CDCl3, 150 MHz) δ 138.8, 128.4, 127.6, 127.5, 95.0, 95.0, 78.4, 77.4, 71.1, 70.1, 66.0, 65.9, 65.8, 65.2, 43.1, 42.5, 39.7, 28.4, 28.0, 27.7, 27.4, 22.2, 18.2, 18.1, −1.4; ESI-TOF MS (m/z): [MH]+ calcd for C32H59O7Si2 611.3794, Found 611.3795. The diol (1.02 g, 1.7 mmol) was dissolved in CH2Cl2 (40 mL), then TEMPO (21.0 mg, 0.13 mmol), KBr (31.7 mg, 0.27 mmol), and sat. NaHCO3 aq. (20 mL) ware added at room temperature. The resulting biphasic mixture was cooled at 0 °C, and then treated with NaOCl (4% in water, 3.0 mL, 1.8 mmol), the mixture was stirred at room temperature for 20 min. After addition of brine and sat. NaHCO3 aq. the whole was extracted with CH2Cl2, washed with brine, dried over Na2SO4. The solvent was evaporated under reduced pressure to leave an oil. The crude was dissolved in pyridine (6 mL), and then Ac2O (3 mL) and catalytic amount of DMAP were added at room temperature. The mixture was stirred at room temperature for 17 h (over night). The solvent was removed under reduced pressure to leave an oil, which was purified by flash chromatography on SiO2 with EtOAc–hexane (1:4, v/v) to give 18 (1.01 g, 85% for 3 steps from 17) as a colorless oil. 1H NMR (CDCl3, 600 MHz) δ 9.65 (s, 1H), 7.37–7.32 (m, 4H), 7.31–7.28 (m, 1H), 5.36 (br s, 1H), 4.65–4.63 (m, 2H), 4.59–4.52 (m, 4H), 3.82 (br s, 1H), 3.68 (d, J = 9.8 Hz, 1H), 3.62–3.48 (m, 4H), 3.44 (br sept, J = 5.1 Hz, 1H), 3.18 (d, J = 9.8 Hz, 1H), 2.23–2.16 (m, 3H), 2.08–2.02 (m, 4H, including 2.07 (s, 3H)), 1.95–1.91 (m, 1H), 1.76–1.66 (m, 3H), 1.54–1.47 (m, 1H), 1.04 (s, 3H), 0.94–0.88 (m, 4H), 0.02 (s, 9H), 0.01 (s, 9H); 13C NMR (CDCl3, 150 MHz) δ 201.7, 170.4, 138.6, 128.4, 127.6, 127.6, 94.7, 93.6, 77.9, 73.1, 71.2, 70.2, 65.3, 65.2, 64.8, 51.0, 42.0, 39.3, 27.2, 27.1, 27.1, 26.2, 21.3, 19.3, 18.1, 18.0, −1.4, −1.4; ESI-TOF MS (m/z): [MNa]+ calcd for C34H58O8Si2Na 673.3562, Found 673.3562.
4.1.9. 4-((2-Trimethylsilyl)ethoxy)methoxy-7-epi AB-ring of TSDN (19)
A solution of 18 (135 mg, 0.21 mmol) in dry CH2Cl2 (2 mL) and dry MeOH (2 mL) was hydrogenated in the presence of Pd(OH)2 on carbon (20 wt. %, 60 mg) at room temperature, and 1 atom for 1 h. Pd(OH)2 on carbon was filtered off, and the filtrate was evaporated under reduced pressure to leave an oil, which was purified by flash chromatography on SiO2 with EtOAc–hexane (2:1, v/v) to give 19 (84.0 mg, 94%) as a colorless oil. Compound 19 is an inseparable mixture of diastereomers (dr = 5:2). 1H NMR (CDCl3, 500 MHz) δ 5.22 (d, J = 3.7 Hz, 1H), 4.95 (s, 1H), 4.74 (s, 1H), 4.72 (d, J = 3.7 Hz, 1H), 4.70 (d, J = 7.0 Hz, 1H), 4.69 (d, J = 7.0 Hz, 1H), 4.53 (d, J = 7.0 Hz, 1H), 4.53 (d, J = 7.0 Hz, 1H), 4.28 (d, J = 12.0 Hz, 1H), 4.12 (d, J = 12.0 Hz, 1H), 3.85 (br s, 1H), 3.66–3.44 (m, 8H), 3.41 (br s, 1H), 3.30 (d, J = 12.0 Hz, 1H), 3.07 (d, J = 12.0 Hz, 1H), 2.56–2.51 (m, 2H), 2.36–1.24 (m, 20H, including 2.04 (s, 3H) and 2.03 (s, 3H)), 1.06–1.03 (m, 2H), 0.95–0.83 (m, 10H, including 0.91 (s, 3H) and 0.86 (s, 3H)), 0.00 (s, 18H); 13C NMR (CDCl3, 125 MHz) δ 170.7, 96.3, 95.8, 93.3, 93.2, 78.1, 78.0, 76.0, 72.7, 71.1, 70.9, 66.7, 65.4, 61.8, 40.1, 39.6, 36.5, 36.5, 33.9, 32.4, 31.8, 31.7, 31.5, 31.1, 30.8, 30.1, 27.8, 27.0, 21.3, 21.3, 18.9, 18.2, 18.0, −1.5; ESI-TOF MS (m/z): [MNa]+ calcd for C21H38O7SiNa 453.2279, Found 453.2283.
4.1.10. 7-Epi AB-ring of TSDN (epi-7)
TFA (2 mL) was added to a solution of 19 (45.4 mg, 0.11 mmol) in CH2Cl2 (2 mL) at 0 °C, the mixture was stirred at 0 °C for 1.5 h. The solvent was removed under reduced pressure to leave an oil, which was purified by flash chromatography on SiO2 with EtOAc–MeOH (95:5, v/v) to give epi-7 (25.2 mg, 80%) as a colorless oil. Compound epi-7 is an inseparable mixture of diastereomers (dr = 7:1). 1H NMR (CD3OD, 600 MHz) δ 5.17 (d, J = 3.6 Hz, 1H), 4.87 (s, 1H), 4.67 (d, J = 3.6 Hz, 1H), 4.62 (s, 1H), 4.27 (d, J = 12.0 Hz, 1H), 4.11 (d, J = 12.0 Hz, 1H), 3.63–3.62 (m, 1H), 3.49–3.43 (m, 3H), 3.24 (d, J = 12.0 Hz, 1H), 2.98 (d, J = 12.0 Hz, 1H), 2.69–2.63 (m, 2H), 2.33 (dd, J = 13.5, 3.8 Hz, 1H), 2.23–1.72 (m, 15H, including 2.03 (s, 3H) and 2.02 (s, 3H)), 1.52–1.46 (m, 1H), 1.37–1.24 (m, 3H), 1.05–1.01 (m, 2H), 0.87 (s, 3H), 0.81 (s, 3H); 13C NMR (CD3OD, 150 MHz) δ 172.8, 172.7, 97.4, 96.8, 78.2, 74.8, 74.2, 74.2, 72.2, 72.0, 67.8, 62.7, 41.7, 41.2, 38.0, 37.9, 36.9, 36.7, 34.7, 33.4, 32.5, 32.0, 31.7, 31.2, 29.2, 28.4, 21.4, 21.3, 19.6, 19.0; ESI-TOF MS (m/z): [MNa]+ calcd for C15H24O6Na 323.1465, Found 323.1452.
4.1.11. Hemi-acetal-protected 4-((2-trimethylsilyl)ethoxy)methoxy-7-epi AB-ring of TSDN (22)
CbzCl (95%, 0.10 mL, 0.70 mmol) was added to a solution of 19 (177 mg, 0.41 mmol) in dry CH2Cl2 (5 mL) and TMEDA (55.5 μL, 0.37 mmol) at −78 °C, and the mixture was stirred at −78 °C for 2 h. After addition of CH2Cl2, The whole was washed with water, 1 N HCl aq., and brine, dried over Na2SO4. The solvent was evaporated under reduced pressure to leave an oil, which was purified by flash chromatography on SiO2 with EtOAc–hexane (1:1, v/v) to give 21 (165 mg, including di-Cbz and/or undesired mono-Cbz products) as a colorless oil, 22 (115 mg, 49%) as a colorless oil, and incompletely purified recovery of 19 (14.3 mg) as a colorless oil in the order of elution. Compound 22: 1H NMR (CDCl3, 600 MHz) δ 7.38–7.33 (m, 5H), 5.71 (s, 1H), 5.19–5.15 (m, 3H), 4.71 (d, J = 7.0 Hz, 1H), 4.54 (d, J = 7.0 Hz, 1H), 4.22 (d, J = 12.4 Hz, 1H), 3.68–3.54 (m, 3H), 3.49 (d, J = 3.6 Hz, 1H), 3.39 (d, J = 12.4 Hz, 1H), 2.54–2.43 (m, 2H), 2.10–1.83 (m, 7H, including 2.04 (s, 3H)), 1.60–1.54 (m, 1H), 1.39–1.32 (m, 1H), 1.10–1.08 (m, 1H), 0.96–0.84 (m, 5H, including 0.86 (s, 3H)), 0.01 (s, 9H); 13C NMR (CDCl3, 150 MHz) δ 170.3, 154.2, 134.7, 128.6, 128.6, 128.4, 97.9, 93.2, 77.7, 72.8, 70.8, 70.1, 67.4, 65.5, 38.9, 36.4, 34.1, 31.2, 30.9, 30.3, 26.8, 21.3, 18.2, 18.1, −1.4; ESI-TOF MS (m/z): [MNa]+ calcd for C29H44O9SiNa 587.2647, Found 587.2648. A solution of 21 (165 mg) in dry EtOH (5 mL) was hydrogenated in the presence of Pd on carbon (10 wt. %, 80 mg) at room temperature, and 1 atom for 1.5 h. Pd on carbon was filtered off, and the filtrate was combined with incompletely purified recovery of 19 (14.3 mg), then evaporated under reduced pressure to leave an oil, which was purified by flash chromatography on SiO2 with EtOAc–hexane (2:1, v/v) to give 19 (86.1 mg, 49% for 2 steps from 19 via 21) as a colorless oil.
4.1.12. Hemi-acetal-protected 4-((2-trimethylsilyl)ethoxy)methoxy-7-keto AB-ring of TSDN (23)
The 22 (134 mg, 0.24 mmol) was dissolved in dry CH2Cl2 (5 mL), then Dess-Martin periodinane (97%, 311 mg, 0.71 mmol) was added at 0 °C. The mixture was stirred at room temperature for 17 h (over night). After addition of 10% Na2S2O3 aq., the whole was extracted with CH2Cl2, washed with sat. NaHCO3 aq. and brine, dried over Na2SO4. The solvent was evaporated under reduced pressure to leave an oil, which was purified by flash chromatography on SiO2 with EtOAc–hexane (1:2, v/v) to give 23 (127 mg, 96%) as a colorless oil. 1H NMR (CDCl3, 600 MHz) δ 7.39–7.33 (m, 5H), 5.70 (s, 1H), 5.20–5.16 (m, 3H), 4.70 (d, J = 7.1 Hz, 1H), 4.54 (d, J = 7.1 Hz, 1H), 4.27 (d, J = 12.6 Hz, 1H), 3.64–3.59 (m, 3H), 3.55–3.50 (m, 1H), 2.90 (dd, J = 14.2, 4.7 Hz, 1H), 2.55–2.51 (m, 2H), 2.45–2.33 (m, 3H), 2.25–2.20 (m, 1H), 2.16–2.04 (m, 1H), 2.02 (s, 3H), 1.37–1.33 (m, 1H), 0.95–0.83 (m, 5H, including 0.84 (s, 3H)), 0.00 (s, 9H); 13C NMR (CDCl3, 150 MHz) δ 210.3, 170.1, 154.1, 134.5, 128.7, 128.6, 128.4, 97.6, 92.9, 77.7, 72.3, 70.2, 67.4, 65.7, 39.0, 37.4, 36.5, 36.4, 35.5, 31.0, 28.3, 21.2, 18.1, 18.0, −1.5; ESI-TOF MS (m/z): [MNa]+ calcd for C29H42O9SiNa 585.2490, Found 585.2501.
4.1.13. Hemi-acetal-protected 4-((2-trimethylsilyl)ethoxy)methoxy AB-ring of TSDN (24)
NaBH4 (17.3 mg, 0.45 mmol) was added to a solution of 23 (127 mg, 0.23 mmol) in CH2Cl2 (5.6 mL) and MeOH (0.4 mL) at 0 °C, the mixture was stirred at room temperature for 16 h (over night). After addition of CH2Cl2, the whole was washed with sat. NH4Cl aq. and brine, then dried over Na2SO4. The solvent was evaporated under reduced pressure to leave an oil, which was purified by flash chromatography on SiO2 with CH2Cl2–MeOH (93:7, v/v) to give mixture of 22 and 24 (122 mg, 95%, 22/24 = 2.6/1 by analytical HPLC) as a colorless oil. The mixture was completely separated by p-TLC and HPLC technique. Compound 24: 1H NMR (CDCl3, 600 MHz) δ 7.38–7.33 (m, 5H), 5.68 (s, 1H), 5.19–5.15 (m, 3H), 4.74 (d, J = 7.1 Hz, 1H), 4.58 (d, J = 7.1 Hz, 1H), 4.21–4.19 (m, 2H), 3.67–3.63 (m, 1H), 3.60–3.55 (m, 1H), 3.44 (d, J = 3.7 Hz, 1H), 3.40 (d, J = 12.2 Hz, 1H), 2.80–2.77 (m, 1H), 2.54–2.50 (m, 1H), 2.34–2.28 (m, 1H), 2.11–2.01 (m, 5H, including 2.04 (s, 3H)), 1.80–1.64 (m, 3H), 0.95–0.79 (m, 6H, including 0.83 (s, 3H)), 0.01 (s, 9H); 13C NMR (CDCl3, 150 MHz) δ 170.5, 154.3, 134.7, 128.6, 128.6, 128.3, 98.1, 93.2, 78.1, 73.1, 70.0, 67.1, 66.0, 65.3, 38.5, 36.8, 31.2, 29.0, 27.9, 27.2, 21.9, 21.4, 18.1, −1.4; ESI-TOF MS (m/z): [MNa]+ calcd for C29H44O9SiNa 587.2647, Found 587.2646.
4.1.14. AB-ring of TSDN (7)
A solution of 24 (12.0 mg) in dry EtOH (2 mL) was hydrogenated in the presence of Pd on carbon (10 wt. %, 5 mg) at room temperature, and 1 atom for 1 h. Pd on carbon was filtered off, then evaporated under reduced pressure to leave an oil. The crude was dissolved in CH2Cl2 (2 mL), cooled at 0 °C, then TFA (2 mL) was added. The mixture was stirred at 0 °C for 1.5 h. The solvent was removed under reduced pressure to leave an oil, which was purified by flash chromatography on SiO2 with EtOAc–MeOH (95:5, v/v) to give 7 (5.2 mg, 82%) as a colorless oil. Compound 7 is an inseparable mixture of diastereomers (dr = 2:1). 1H NMR (CDCl3, 600 MHz) δ 5.44 (d, J = 3.3 Hz, 1H), 4.96 (d, J = 2.5 Hz, 1H), 4.94 (d, J = 3.3 Hz, 1H), 4.79 (d, J = 2.5 Hz, 1H), 4.29 (d, J = 12.1 Hz, 1H), 4.21 (br s, 1H), 4.17 (br s, 1H), 4.12 (d, J = 12.1 Hz, 1H), 3.67 (br s, 2H), 3.47 (br s, 2H), 3.32 (d, J = 12.1 Hz, 1H), 3.09 (d, J = 12.1 Hz, 1H), 2.85–2.79 (m, 2H), 2.69 (d, J = 3.5 Hz, 1H), 2.59 (dd, J = 13.3, 4.2 Hz, 1H), 2.47 (dd, J = 13.3, 4.2 Hz, 1H), 2.41–2.33 (m, 3H), 2.12 (s, 3H), 2.11 (s, 3H), 1.93–1.62 (m, 10H), 0.92–0.84 (m, 8H, including 0.91 (s, 3H) and 0.87 (s, 3H)); 13C NMR (CDCl3, 150 MHz) δ 169.9, 169.7, 96.5, 95.9, 74.6, 74.5, 74.1, 66.3, 66.1, 66.1, 61.4, 60.4, 40.3, 39.6, 37.5, 35.7, 35.6, 29.7, 29.1, 28.2, 28.1, 28.1, 26.9, 25.8, 22.9, 22.2, 21.5, 21.4, 18.6, 18.0; ESI-TOF MS (m/z): [MNa]+ calcd for C15H24O6Na 323.1465, Found 323.1463.
4.2. Biological evaluation
4.2.1. Details of assay procedures
Pure botulinum neurotoxin type A1 was purified from C. botulinum strains Hall A hyper as described.23 The specific activity was determined via the mouse bioassay24 to be 7.8 pg / mouse LD50. The toxins were stored in 40% glycerol, 15 mM sodium phosphate, 90 mM NaCl at −20 °C.
4.2.2. RSC assay
Primary rat spinal cord cells were prepared as described previously,22 and seeded into collagen coated 96-well plates (BD Biosciences) at a density of 75,000 cells/well. The RSC assay was essentially performed as described,9,22 after the cells had matured for at least three weeks. In short, the cells were exposed to a mixture of the indicated concentrations of toosendanin or analogues and 5.6 pM BoNT/A1 (42 pg or about 5.5 mouse LD50 Units) in 50 μL of culture medium containing 1% DMSO. The positive control contained toxin and 1% DMSO, and the negative control contained 1% DMSO only in culture medium. All samples were tested in replicates of three. After 24 h of incubation at 37 °C, 5% CO2, the cells were harvested by lysis in 1 × LDS sample buffer (Invitrogen), and analyzed by western blot as described.9,22
5. Acknowledgment
This work was supported by National Institutes of Health Grant (AI 072358) and The Skaggs Institute for Chemical Biology.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
6. References and notes
- 1.(a) Johnson EA, Bradshaw M. Toxicon. 2001;39:1703. doi: 10.1016/s0041-0101(01)00157-x. [DOI] [PubMed] [Google Scholar]; (b) Schantz EJ, Johnson EA. Microbiol. Rev. 1992;56:80. doi: 10.1128/mr.56.1.80-99.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Singh BR. Nat. Struct. Biol. 2000;7:617. doi: 10.1038/77900. [DOI] [PubMed] [Google Scholar]
- 3.(a) Willis B, Eubanks LM, Dickerson TJ, Janda KD. Angew. Chem. Int. Ed. 2008;47:8360. doi: 10.1002/anie.200705531. [DOI] [PubMed] [Google Scholar]; (b) Dickerson TJ, Janda KD. ACS Chem. Biol. 2006;1:359. doi: 10.1021/cb600179d. [DOI] [PubMed] [Google Scholar]; (c) Simpson L. Annu. Rev. Pharmacol. Toxicol. 2004;44:167. doi: 10.1146/annurev.pharmtox.44.101802.121554. [DOI] [PubMed] [Google Scholar]
- 4.Nakatani M. Heterocycles. 1999;50:595. [Google Scholar]
- 5.Carpinella C, Ferrayoli C, Valladares G, Defago M, Palacios S. Biosci. Biotechnol. Biochem. 2002;66:1731. doi: 10.1271/bbb.66.1731. [DOI] [PubMed] [Google Scholar]
- 6.Xie YS, Fields PG, Isman MB, Chen WK, Zhang X. J. Stored Prod. Res. 1995;31:259. [Google Scholar]
- 7.(a) Akhtar Y, Isman MB. J. Chem. Ecol. 2003;29:1853. doi: 10.1023/a:1024802328458. [DOI] [PubMed] [Google Scholar]; (b) Carpinella MC, Defago MT, Valladares G, Palacios SM. J. Agric. Food Chem. 2003;51:369. doi: 10.1021/jf025811w. [DOI] [PubMed] [Google Scholar]; (c) Zhou J-B, Minami Y, Yagi F, Tadera K, Nakatani M. Heterocycles. 1997;45:1781. [Google Scholar]
- 8.(a) Zou J, Miao W, Ding F, Meng J, Ye H, Jia G, He X, Sun G, Li P. J. Tradit. Chin. Med. 1985;5:29. [PubMed] [Google Scholar]; (b) Li PZ, Zhou J, Miao WY, Ding FH, Meng J-Y, Ye HJ, Jia GR, He XY. Chin. Tradit. Herb Drugs. 1982;17:28. [Google Scholar]
- 9.Fischer A, Nakai Y, Eubanks LM, Clancy CM, Tepp WH, Pellett S, Dickerson TJ, Johnson EA, Janda KD, Montal M. Proc. Natl. Acad. Sci. USA. 2009;106:1330. doi: 10.1073/pnas.0812839106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Čapcová K, Yoneda Y, Dickerson TJ, Janda KD. Bioorg. Med. Chem. Lett. 2007;17:6463. doi: 10.1016/j.bmcl.2007.09.103. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Eubanks LM, Hixon MS, Jin W, Hong S, Clancy CM, Tepp WH, Baldwin MR, Malizio CJ, Goodnough MC, Barbieri JT, Johnson EA, Boger DL, Dickerson TJ, Janda KD. Proc. Natl. Acad. Sci. USA. 2007;104:2602. doi: 10.1073/pnas.0611213104. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Silvaggi NR, Boldt GE, Hixon MS, Kennedy JP, Tzipori S, Janda KD, Allen KN. ACS Chem Biol. 2007;14:533. doi: 10.1016/j.chembiol.2007.03.014. [DOI] [PubMed] [Google Scholar]; (d) Boldt GE, Eubanks LM, Janda KD. Chem. Commun. 2006:3063. doi: 10.1039/b603099h. [DOI] [PubMed] [Google Scholar]; (e) Boldt GE, Kennedy JP, Janda KD. Org. Lett. 2006;8:1729. doi: 10.1021/ol0603211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wender PA, Verma VA, Paxton TJ, Pillow TH. Acc. Chem. Res. 2008;41:40. doi: 10.1021/ar700155p. [DOI] [PubMed] [Google Scholar]
- 12.Nakai Y, Tepp WH, Dickerson TJ, Johnson EA, Janda KD. Bioorg. Med. Chem. 2009;17:1152. doi: 10.1016/j.bmc.2008.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones JB, Dodds DR. Can. J. Chem. 1987;65:2397. [Google Scholar]
- 14.Nicolaou KC, Follmann M, Roecker AJ, Hunt KW. Angew. Chem. Int. Ed. 2002;41:2103. [PubMed] [Google Scholar]
- 15.(a) Ghosh S, Rivas F, Fischer D, González MA, Theodorakis EA. Org. Lett. 2004;6:941. doi: 10.1021/ol036492c. [DOI] [PubMed] [Google Scholar]; (b) Ziegler FE, Hwang K-J, Kadow JF, Klein SI, Pati UK, Wang T-F. J. Org. Chem. 1986;51:4573. [Google Scholar]
- 16.Crabtree SR, Alex Chu WL, Mander LN. Synlett. 1990:169. [Google Scholar]
- 17.Fairweather KA, Mander LN. Org. Lett. 2006;8:3395. doi: 10.1021/ol061228f. [DOI] [PubMed] [Google Scholar]
- 18.Rodeschini V, Van de Weghe P, Salomon E, Tarnus C, Eustache J. J. Org. Chem. 2005;70:2409. doi: 10.1021/jo047858h. [DOI] [PubMed] [Google Scholar]
- 19.Nicolaus KC, Sasmal PK, Roecker AJ, Sun X-W, Mandal S, Converso A. Angew. Chem. Int. Ed. 2005;44:3443. doi: 10.1002/anie.200500216. [DOI] [PubMed] [Google Scholar]
- 20.(a) Rodeschini V, Boiteau J-G, Van de Weghe P, Tarnus C, Eustache E. J. Org. Chem. 2004;69:357. doi: 10.1021/jo035065+. [DOI] [PubMed] [Google Scholar]; (b) Miyashita M, Suzuki T, Hoshino M, Yoshikoshi A. Tetrahedron. 1997;53:12469. [Google Scholar]; (c) Miyashita M, Suzuki T, Yoshikoshi A. Tetrahedron Lett. 1987;28:4293. [Google Scholar]
- 21.(a) Smith AB, III, Davulcu AH, Cho YS, Ohmoto K, Kürti L, Ishiyama H. J. Org. Chem. 2007;72:4596. doi: 10.1021/jo062422i. [DOI] [PubMed] [Google Scholar]; (b) Nicolaus KC, Jennings MP, Dagneau P. Chem. Commun. 2002:2480. [Google Scholar]; (c) De Mico A, Margarita R, Parlanti L, Vescovi A, Piancatelli G. J. Org. Chem. 1997;62:6974. [Google Scholar]
- 22.Pellett S, Tepp WH, Clancy CM, Borodic GE, Johnson EA. FEBS Lett. 2007;581:4803. doi: 10.1016/j.febslet.2007.08.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malizio CJ, Goodnough MC, Johnson EA. Methods Mol. Biol. 2000;145:27. doi: 10.1385/1-59259-052-7:27. [DOI] [PubMed] [Google Scholar]
- 24.(a) Hatheway CL. Botulism. in: Laboratory Diagnosis of Infectious Diseases. Principles and Practise. Springer-Verlag; New York: 1988. pp. 111–113. [Google Scholar]; (b) Schantz EJ, Kautter DA. J. Assoc. Off. Anal. Chem. 1978;61:96. [Google Scholar]