

Abstract
Background and Aims
Silymarin, an extract from the seeds of the milk thistle plant Silybum marianum, has been used for centuries for the treatment of chronic liver diseases. Despite common use by patients with hepatitis C in the U.S., its clinical efficacy remains uncertain. The goal of this study was to determine if silymarin has in vitro effects on immune function that might have implications for its potential effect on HCV-induced liver disease.
Methods
Freshly isolated PBMC and T cells from HCV-infected and uninfected subjects were tested in vitro for responses to nonspecific and antigenic stimulation in the presence and absence of a standardized preparation of silymarin (MK001).
Results
Minimal MK001 toxicity on PBMC was found at concentrations between 5-40 μg/mL. MK001 dose-dependently inhibited the proliferation and secretion of TNF-α, IFN-γ, and IL-2 by PBMC stimulated with anti-CD3. In addition, MK001 inhibited proliferation by CD4+ T cells to HCV, Candida and Tetanus protein antigens, and by HLA-A2/HCV1406-1415-specific CD8+ T cells to allogeneic stimulation. MK001 inhibited T cell TNF-α and IFN-γ cytokine secretion to Tetanus and Candida protein antigens. Finally, MK001 inhibited NF-κB transcriptional activation after T cell receptor-mediated stimulation of Jurkat T cells, consistent with its ability to inhibit Jurkat T cell proliferation and secretion of IL-2.
Conclusion
Silymarin’s ability to inhibit the proliferation and pro-inflammatory cytokine secretion of T cells, combined with its previously described anti-viral effect suggests a possible mechanism of action that could lead to clinical benefit during HCV infection.
Keywords: Milk thistle, Silybum marianum, Silymarin, Silibinin, Immune, T cell, Hepatitis C, HCV, Anti-inflammatory, Proliferation, Cytokine
Chronic hepatitis C afflicts approximately 3 million individuals and is the leading cause of end-stage liver disease requiring transplantation in the United States. Liver fibrosis progression, the major consequence of chronic infection, generally occurs over decades in immunocompetent individuals.1
The current standard treatment for HCV infection, pegylated interferon and ribavirin, results in an overall sustained virologic response (SVR) rate of approximately 55%, which, in the vast majority of cases represents durable viral eradication.2, 3 However, significant numbers of patients do not achieve an SVR, or are intolerant/have contraindications to therapy. Complications of hepatitis C-induced liver fibrosis including liver failure and cancer are predicted to increase significantly over the next twenty years, and no other treatment options are currently available.4 Thus, many individuals have turned to herbal remedies. Two recent studies have reported that as many as 13% to 23% of U.S. patients with chronic liver disease are using herbal remedies, with silymarin being the most common preparation ingested for such purpose.5, 6
Silymarin, an extract from the seeds of the milk thistle plant, Silybum marianum, has been used for centuries as a “hepatoprotectant”. Silymarin is a mixture of seven flavonolignans (silybin A, silybin B, isosilybin A, isosilybin B, silychristin, silydianin, and isosilychristin) and one flavonoid (taxifolin). Although its mechanism of action is incompletely understood, silymarin has been described to possess anti-oxidant, immunomodulatory, anti-fibrotic, anti-proliferative and anti-viral activities.7, 8 Silymarin’s clinical efficacy in chronic liver disease has not yet been demonstrated,9, 10 as results have been inconsistent. Problems with the studies have included insufficient power, use of varying doses (including those that may be too low to have an effect), and the use of different nonstandardized preparations of silymarin, making it difficult to compare results among studies. Moreover, many reports have utilized silibinin or chemically modified silibinin, a ~50:50 mixture of silybin A and silybin B, rather than silymarin.
HCV is generally believed to be a noncytopathic virus and hepatic fibrosis in hepatitis C is thought to be the end result of long-standing inflammation, characterized by the accumulation of CD4+ and CD8+ T cells in the infected liver. A substantial amount of data suggests that HCV-specific T cell cytokine and cytolytic responses as well as “bystander” effects contribute to immune-mediated liver injury.11 As an important component of the inflammatory response to HCV infection, T cells are stimulated to proliferate, secrete cytokines, and kill infected cells. In this study, we investigated whether a standardized preparation of silymarin (MK001) can affect pro-inflammatory immune responses, such as T cell proliferation and cytokine secretion, as a means of providing hepatoprotection.
MATERIALS AND METHODS
Subjects
Healthy subjects and those with chronic HCV infection were recruited at Harborview Medical Center after written informed consent was obtained through a University of Washington (UW) Institutional Review Board-approved protocol.
Silymarin
A standardized preparation of silymarin (MK001)8 was used in all experiments. Using HPLC analyses with detection at 254 nm, all peaks more than 0.7% in quantity have been assigned, and the total flavonolignan content was 92%. MK001 was solubilized in DMSO, 95% ethanol or methanol. Control samples were treated with the same concentration of DMSO, 95% ethanol or methanol as used in the MK001-treated samples. Silybin A, silybin B, and silibinin were purified as previously described in detail.12
PBMC Isolation and Stimulation
All peripheral blood mononuclear cells (PBMC) were freshly isolated using standard Ficoll-Hypaque centrifugation within 24 hours of venipuncture and immediately applied to the assays described.
PBMC were stimulated for varying periods of time at 37°C 5%CO2 with plate-bound anti-CD3 (UCHT1, 10 μg/mL, BD Biosciences, San Jose, CA), anti-CD3 and anti-CD28 Dynabeads (Invitrogen, Carlsbad, CA), phytohemagglutinin (PHA, 1.6 μg/mL, Remel, Lenexa, KS) or PMA/ionomycin (50 ng/mL of PMA and 1μg/mL ionomycin, both, Sigma Aldrich, St. Louis, MO) in RPMI 1640 medium supplemented with 10% human serum (Gemini Bio-Products, Woodland, CA). Stimulation with Tetanus and Candida protein antigens was performed as described under proliferation assays (below).
Cellular Proliferation Assays
Cellular proliferation detected by 3H-thymidine incorporation into replicating DNA was measured by adding 1 μCi to each replicate well of 105 PBMC for 24 hours (for α-CD3 and PHA experiments) or 16-18 h (for protein antigen experiments) before quantitative analysis using a Topcount® Liquid Scintillation Counter (Perkin-Elmer, Waltham, MA). In general, data are presented as mean counts per minute (cpm) incorporated per condition tested. All experiments utilized four replicates per condition.
To measure HCV-specific proliferative responses, PBMC were incubated with superoxide dismutase (SOD)-recombinant HCV protein antigens (courtesy of Dr. Michael Houghton and Kevin Crawford, Chiron Corp.) at a final concentration of 10 μg/mL for 5 days prior to addition of 3H-thymidine as described above.13 Antigens included yeast-derived SOD (negative control), SOD-c22 (HCV aa 2-120), SOD-c100 (HCV aa 1569-1931), SOD-NS5 (HCV aa 2054-2995), and E. coli-derived SOD-c33c (HCV 1192-1457). PHA (1.6 μg/ml, Remel), Candida albicans (20 μg/mL, Greer Laboratories, Lenoir, NC), and Tetanus toxoid (12 Lf/mL, Wyeth-Ayerst Laboratories, Marietta, PA) were also tested. Assays were considered valid only when PHA responses were intact.
PBMC and T cell proliferation was also measured using carboxyfluoroscein succinimidyl ester (CFSE) dilution. PBMC were exposed to 1 μM CFSE (Invitrogen/Molecular Probes, Carlsbad, CA) for 10 min at 37°C, then washed and cultured with or without stimulation and with or without MK001 (20 μg/mL) at 37°C, 5% CO2. The next day, cells were labeled with the following fluorescent surface antibodies (all BD Biosciences): α-CD5-PerCP-Cy5.5, α-CD8-PE, α-CD45-APC and fixed with 1% paraformaldehyde and analysed by flow cytometry as described below. For T cell experiments, HCV 1406-1415-expanded T cells were cultured with allogeneic B-LCL at a ratio of 10:1 (T cells: BLCL), with or without MK001 (20 μg/mL) for 3 days before labeling with α-CD8-PerCP, α-CD3-APC (both BD Biosciences) and HLA-A2 HCV 1406-1415 pentamer-PE (ProImmune, Bradenton, FL).
Flow Cytometric Analysis of Cell Viability
Cellular viability was measured after labeling with Live/Dead Fixable Dead Cell Stain Kit (Violet viability dye, Invitrogen/Molecular Probes, Carlsbad, CA) for 24 hours and analyzed using a Becton Dickinson FACS Calibur (Puget Sound Blood Center, Seattle, WA) and FlowJo software for Macintosh (version 6.3.3, Treestar, Inc., Ashland, OR).
Intracellular Cytokine Staining
PBMC were stimulated with negative controls (DMSO, 95% ethanol, or media) or plate-bound anti-CD3 (10 μg/mL), or PHA (1.6 μg/mL) in the presence and absence of MK001, and treated with 1 μg/mL Brefeldin A (Sigma-Aldrich, St Louis, MO). After a total of 16 hours, cells and labeled with the following surface markers: Live/Dead Fixable Dead Cell Stain Kit, α-CD5-PerCP-Cy5.5 (BD Biosciences), α-CD8-ECD (Beckman Coulter), α-CD4-APC-Cy7, and α-CD56-biotin (BD Biosciences) plus QDot605-streptavidin (Invitrogen, Molecular Probes). Cells were treated with BD Cytofix/Cytoperm (BD Biosciences), washed with BD Perm/Wash, and intracellular staining performed using TNF-α-APC (BD Biosciences) and IFN-γ-PE-Cy7 (BD Pharmingen). All samples were fixed at a final concentration of 1% paraformaldehyde and analyzed on a BD LSR II (BD Biosciences, Fred Hutchinson Flow Cytometry Center) with compensation and data analysis performed using FlowJo software.
Cytokine Detection by ELISA
Supernatants were collected 24 hours after stimulation at 37°C, 5% CO2, and immediately frozen at −80°C. These samples were subsequently tested for the presence of TNF-α, IFN-γ, IL-2, and IL-10 at the Fred Hutchinson Cancer Research Center Cytokine Analysis Facility, Seattle, WA, using standard ELISA kits or using multiplex immunobead ELISA (Biosource/Invitrogen) and the Luminex® xMAP system.
Jurkat T cell transfection and measurement of NF-κB-dependent transcription
Transient transfections of Jurkat T cells were performed using Cell Line Nucleofector Kit V according to manufacturer’s instructions (Amaxa, Gaithersburg, MD). Briefly, in 12 well plates, 5 × 106 cells/well were electroporated using program S-18 and 5 μg of a plasmid encoding a luciferase reporter gene under the control of an NF-κB responsive promoter. Forty-eight hours later, cells were treated or not treated with 20 μg/mL of MK001 in DMSO for 30 minutes. NF-κB transcription was activated via the TCR using plate-bound anti-CD3 (10 μg/mL), anti-CD3 (10 μg/mL) + soluble anti-CD28 (1 μg/mL) antibodies, or with rhTNF-α (15 ng/mL, Pierce Biotechnology, Rockford, IL). Four hours later, luciferase acitivity was measured on cell lysates using the Britelite assay system (Perkin-Elmer, Boston, MA).
RESULTS
Cellular Toxicity of MK001
PBMC from HCV-infected subjects were cultured for 24 hours with various concentrations of MK001, a standardized preparation of silymarin, to assess whether any toxic effects could be detected (Figure 1). Controls included co-culture with media alone and various DMSO, and ethanol concentrations, corresponding to various doses of MK001 tested. Viability of unstimulated (Figure 1A) and anti-CD3-stimulated (Figure 1B) PBMC as well as CD8+ and CD4+ T cell subsets was assessed using the Live/Dead Fixable Dead Cell Stain Kit and flow cytometric analysis as described in Materials and Methods. The percentage of non-viable cells taking up the dye is indicated in the upper right corner of each plot. These data demonstrate that the minor toxicity of co-culture with MK001 was due primarily to the effects of the vehicle, not MK001. Significant cellular toxicity consistently occurred at a dose of 100 μg/mL for 24 hours, a dose 5-fold higher than that used for most of the experiments included in this report (20 μg/mL). PBMC viability was also assessed using trypan blue exclusion after the addition of a single dose of various MK001 concentrations; viability was 99% and 97% after 24 hours and 5 days, respectively, at 20 μg/mL. In all subsequent experiments (excluding dose response experiments), MK001 was used at a concentration of 20 μg/mL or ~40 μM.
Figure 1. Minimal effects of silymarin on cell viability.


PBMC from representative HCV-infected subjects were cultured with media, and with various concentrations of MK001 dissolved in DMSO or ethanol vehicle or with corresponding vehicle controls (DMSO or ethanol alone) for 24 hours. PBMC were unstimulated (A) or stimulated with anti-CD3 (B) at culture initiation. Cells were labeled with Live/Dead Fixable Dead Cell Stain Kit (Violet viability dye, x-axis) and analyzed by flow cytometry. Non-viable cells take up the dye, and the percentage of non-viable cells is indicated in the upper right corner of each plot. Positive control samples treated with PMA/ionomycin for 24 hours exhibited significant cell death (bottom left, panels A and B). Similar results were found upon analysis of the CD4+ and CD8+ T cell compartments for both unstimulated and stimulated cells. Two additional experiments from uninfected subjects demonstrated similar results (data not shown).
MK001 dose-dependently inhibits PBMC and T cell proliferation
During an immune response to viral infection, an important component of the response is the proliferation of antigen-specific T cells. We asked whether MK001 had the ability to inhibit antigen-specific and non-specific proliferation of freshly isolated PBMC and T cells from HCV-infected and uninfected subjects. A single dose of MK001 at culture initiation was able to dose-dependently inhibit proliferation of freshly isolated PBMC stimulated nonspecifically with anti-CD3 (Figure 2A) or PHA (Figure 2C). As shown in Figure 2B, inhibition of proliferation by MK001 (20 μg/mL) ranged from 54-78% (mean 70%) in 5 unique individuals (2 HCV-uninfected and 3 HCV-infected) using a standard 3H-thymidine incorporation assay and plate-bound anti-CD3 as the stimulant. A similar degree of inhibition was detected using PHA (23-79%, mean 48%) to stimulate cells from the same subjects (Figure 2D). In addition, purified preparations of milk thistle extract, silibinin, silybin A and silybin B induced similar inhibitory effects on PBMC proliferation (Supplemental Figure 1A).
Figure 2. MK001 inhibition of PBMC proliferation is dose dependent.
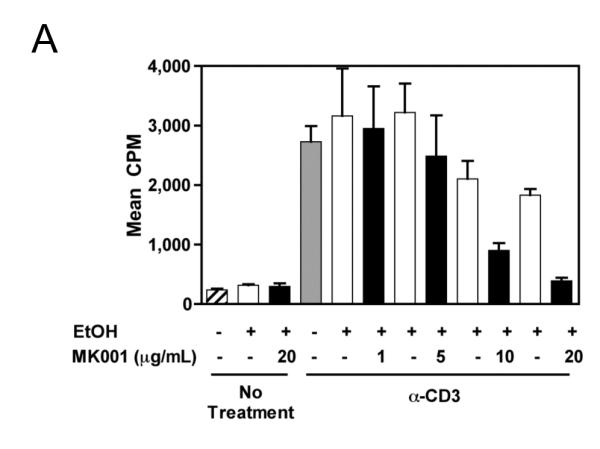

For all assays, a single dose of MK001 was added at culture initiation and compared to parallel cultures treated with a single dose of MK001 vehicle (DMSO or EtOH). Freshly isolated PBMC were tested in quadruplicate for proliferative responses to plate-bound anti-CD3 (10 μg/mL) (A, B) or PHA (1.6 μg/mL) (C, D) stimulation measured at 24 hours using 3H-thymidine incorporation. Results are shown as mean cpm incorporated in cultures treated with MK001 at various doses (black bars) or with the corresponding vehicle control (Ethanol or DMSO, open bars). Gray bars represent proliferation of PBMC stimulated with anti-CD3 or PHA alone. Error bars indicate 1 standard deviation among 4 replicates for each condition. PBMC for representative dose response experiments shown were obtained from HCV-infected (A) and HCV-uninfected (C) subjects. Similar dose response results were obtained in 5 additional HCV-infected and 1 additional HCV-uninfected subjects. Results from individual HCV-infected and uninfected subjects tested with MK001 (20 μg/mL) are also shown (B, D). Results from MK001-treated samples were statistically different from vehicle-treated samples (Wilcoxon matched pairs test, p=0.03 for anti-CD3 or PHA separately).
CFSE dilution by flow cytometry is another method of measuring cellular proliferation; CFSE is bound by cytoplasmic proteins and progressively diluted as daughter cells divide. Proliferation of both CD8+ and CD8− (equivalent to CD4+) T cells was demonstrated using this method with anti-CD3 stimulation (Figure 3A). The addition of MK001 led to inhibition of proliferation measured 24 hours later. Finally, the addition of MK001 (20 μg/mL) to PBMC stimulated by HCV protein antigens as well as Tetanus and Candida antigens (Figure 3B) resulted in inhibition of proliferative responses measured using 3H-thymidine incorporation. In this latter assay, proliferative responses are found exclusively in the CD4+ T cell subset (data not shown).
Figure 3. MK001 Inhibits antigen-specific and nonspecific T cell proliferation.



For all assays, a single dose of MK001 (20 μg/mL) was added at culture initiation and compared to parallel cultures treated with a single dose of vehicle alone (DMSO). (A) Fresh PBMC from a representative HCV-infected subject was exposed to CFSE for 10 minutes, then washed and cultured with media, anti-CD3 (platebound,10 μg/mL) alone, anti-CD3 + DMSO, or anti-CD3 + MK001 in DMSO overnight. Flow cytometric analyses of live CD45-expressing lymphocytes that were also CD5-positive allowed the detection of cell proliferation in both CD8− (left panel) and CD8+ (right panel) T cell populations. Y-axis indicates the percentage of maximum cell number (note offset histograms). CD8−CD5+ T cells were confirmed to be CD4+CD5+ T cells, equivalent to CD8−CD3+ and CD4+CD3+, respectively (data not shown). (B) Freshly isolated PBMC from two HCV-infected subjects were exposed to HCV and recall antigens, and PHA for 5 days, as described.13 Results are shown for 3H-thymidine uptake measured in mean cpm (counts per minute) incorporated into replicate cultures treated (black bars) or untreated (open bars) with MK001. HCV-specific responses considered to be significantly inhibited (HCV-positive to HCV-negative) are indicated by the asterisks. Similar results were obtained from 4 additional HCV-infected subjects. The threshhold for a positive HCV-specific response was a stimulation index of >4.0. Error bars indicate 1 standard deviation among 4 replicates for each condition. (C) T cells from an HCV-infected HLA-A2+ subject were expanded from PBMC using the 9-mer HCV 1406-1415 and rhIL-2 at 37°C, 5% CO2 for 12 days. Cells were exposed to CFSE for 10 minutes, then washed and cultured with allogeneic B-LCL with MK001 (20 μg/mL) in DMSO or with DMSO alone. Three days later the cells were surface labeled with fluorescently conjugated anti-CD3, anti-CD8 and pentamer HLA-A2-HCV NS3 1406-1415 and analyzed using a BD FACS Calibur and FlowJo software. Live lymphocytes that express CD3 and CD8 were gated upon. Shown in the upper right quadrant are cells that bind the pentamer and contain CFSE. Cells that have divided are CFSE-negative and are found in the left quadrants. Offset histogram plots indicate the difference in cellular proliferation between MK001 (gray) and DMSO (vehicle, white)-treated pentamer-positive (top) or pentamer-negative (bottom) T cells.
To demonstrate that MK001 can also inhibit the proliferation of HCV antigen-specific cells, HCV1406-1415-expanded PBMC from an HLA-A2+ HCV-infected subject were evaluated. HCV-antigen-specific T cells proliferated in response to allogeneic stimulation (Figure 3C, left panel), as evidenced by the loss of CFSE fluorescence. Addition of MK001 at culture initiation resulted in an inhibition of CFSE dilution in both HCV-1406-1415-specific and non-specific T cells (Figure 3C, histograms).
MK001 inhibits pro-inflammatory cytokine secretion by stimulated human T cells in vitro
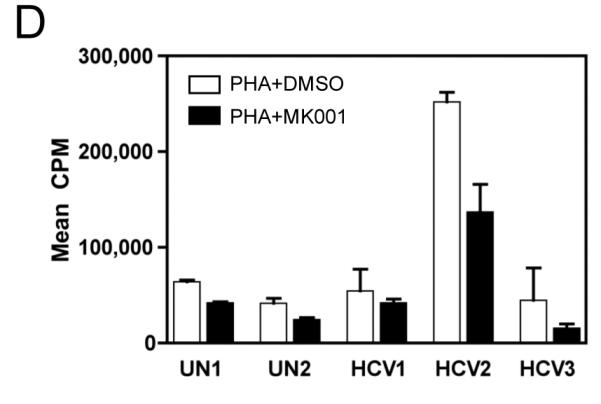
Pro-inflammatory cytokine secretion can lead to the amplification of the immune response. Thus, we asked whether MK001 could affect the secretion of TNF-α, IFN-γ and IL-2 from PBMC, freshly isolated from HCV-infected and uninfected donors. As shown in Figure 4 (A, C and E), TNF-α, IFN-γ and IL-2 secretion by anti-CD3 stimulated PBMC was dose-dependently inhibited by MK001. Furthermore, testing of PBMC from an additional 3 HCV-infected and 2 uninfected individuals using MK001 (20 μg/mL) demonstrated consistent results (Figure 4: B, D, and F). Inhibition of cytokine secretion ranged from 47-86% (mean 70%) for TNF-α, 58-83% (mean 68%) for IFN-γ and 53-87% (mean 71%) for IL-2. Treatment with MK001 alone had no effect on unstimulated PBMC (data not shown). PBMC treated with components purified from milk thistle extract, silibinin, silybin A and silybin B also inhibited anti-CD3-stimulated IFN-γ responses in a dose-dependent manner (Supplemental Figure 1B).
Figure 4. MK001 dose-dependently inhibits TNF-α, IFN-γ, and IL-2 secretion by stimulated human PBMC in vitro.


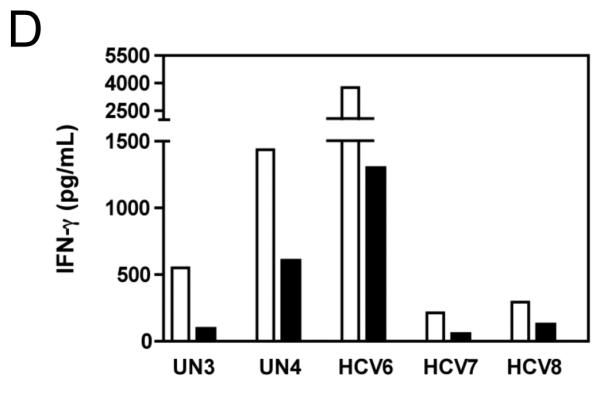

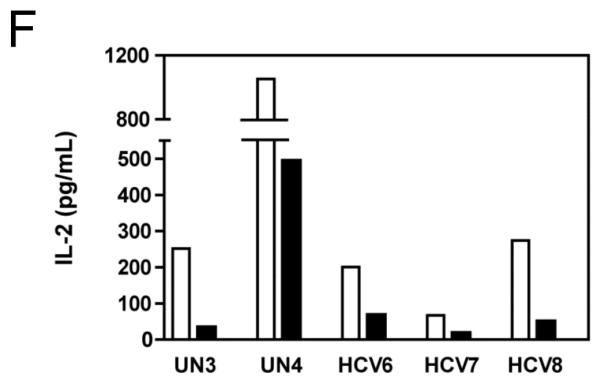
PBMC from an HCV-infected subject were stimulated with plate-bound anti-CD3 (10 μg/mL) or negative controls (95% EtOH or media) in the presence of MK001 (in 95% EtOH) or vehicle alone. Supernatants collected 24 hours after stimulation were tested by multiplex ELISA for the presence of TNF-α (A), IFN-γ (C), or IL-2 (E) as described. White bars indicate samples treated without MK001 but with vehicle, black bars indicated samples treated with MK001 in vehicle. Gray bars represent proliferation of PBMC stimulated with anti-CD3 alone. PBMC from additional HCV-infected and uninfected control subjects were tested similarly and the consistent effect of MK001 (20 μg/mL) on TNF-α (B), IFN-γ (D), and IL-2 (F) secretion is shown. Similar dose response results to those shown were found using PBMC from both an HCV-infected and an uninfected subject. Results from MK001-treated samples were statistically different from vehicle-treated samples (Wilcoxon matched pairs test, p=0.03 for each cytokine separately).
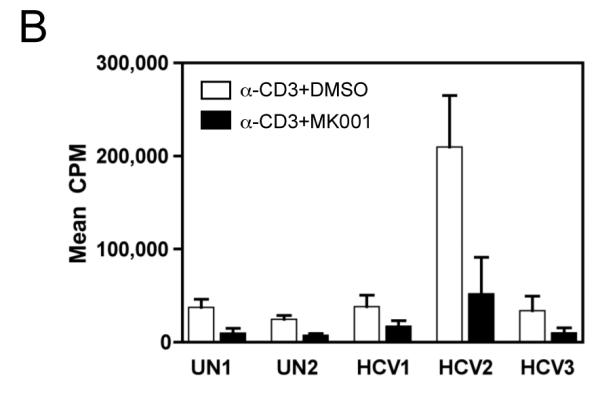
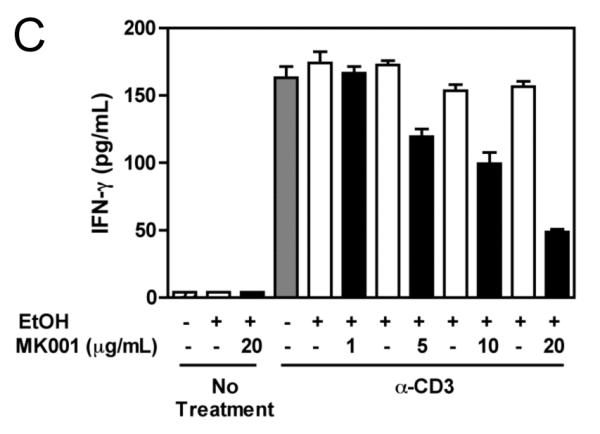
To specifically investigate whether T cell secretion of these cytokines was inhibited, we utilized a highly enriched population of T cells (99.4% purity, Figure 5B) and compared the levels of TNF-α, IFN-γ and IL-2 in culture supernatants after 24 hours (Figure 5A). These data demonstrated a strong inhibition of pro-inflammatory cytokine secretion specifically by T cells as a result of MK001 treatment. Furthermore, intracellular detection of cytokines TNF-α and IFN-γ was reduced in CD8+ and CD8− (CD4+) T cells after culturing in the presence of MK001 compared to the DMSO vehicle control using flow cytometric analysis (Figure 5C).
Figure 5. T cell secretion of TNF-α, IFN-γ, and IL-2 is inhibited by MK001.

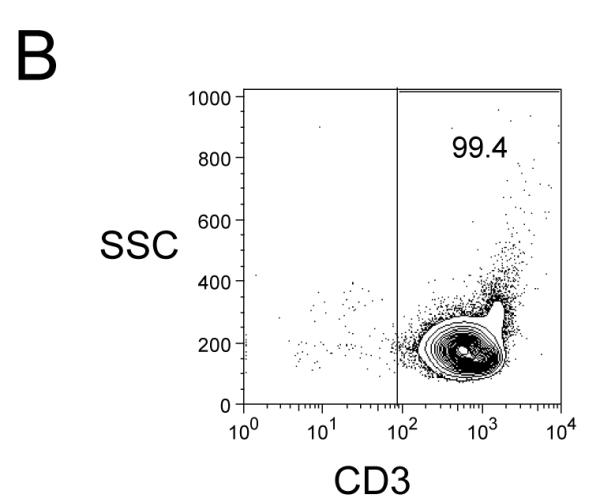

(A) PBMC from an HCV-infected subject were enriched for T cells and cultured with 0.5% DMSO or media (not shown) or plate-bound anti-CD3 (10 μg/mL) in the presence of MK001 (20 μg/mL) or vehicle (DMSO) for 24 hours. TNF-α, IFN-γ, and IL-2 levels in culture supernatants were evaluated using multiplex ELISA. Detection limits were 2 pg/mL, 2 pg/mL, and 4 pg/mL, respectively. (B) Purity of enriched CD3+ T cells used in (A) was assessed using flow cytometric analysis, gating on CD45+ live lymphocytes (NB: purity was 97.5% without gating). (C) PBMC from a second HCV-infected subject were stimulated with negative controls (DMSO or media) or plate-bound anti-CD3 (10 μg/mL), or PHA (1.6 μg/mL) in the presence of MK001 (20 μg/mL) or vehicle (DMSO) for 16 hours. Intracellular TNF-α and IFN-γ cytokine expression were evaluated in the presence of 1 μg/mL brefeldin A. Violet viability dye was used to distinguish live cells (from dead), and cells were further selected on the basis of CD5 expression and the absence of CD56 expression. TNF-α expression (x-axis, left panels) and IFN-γ (x-axis, right panels) from live CD5+CD8+CD56- lymphocytes are shown in the upper right quadrant. Percentages of cells in each quadrant are displayed. Cytokines detected in the CD5+CD8-CD56- population were confirmed to derive from CD5+CD4+CD56- cells in the same experiment, and CD5+CD8+ T cells previously confirmed to represent the same population as CD3+CD8+ T cells (data not shown).
The effect of MK001 on cytokine secretion by antigen specific T cells was examined using fresh PBMC from two unique HCV-infected subjects (Figure 6). Tetanus and Candida-specific TNF-α and IFN-γ responses, quantified by ELISA, were similarly inhibited by treatment with MK001 (20 μg/mL).
Figure 6. Inhibition of TNF-α and IFN-γ secretion by Tetanus toxoid and Candida albicans-specific T cells.




Fresh PBMC from two representative HCV-infected subjects (A, C and B, D) were cultured with Tetanus and Candida protein antigens and PHA, with or without MK001 (20 μg/mL) for 4 days. Culture supernatants were frozen at −80°C and subsequently tested by multiplex ELISA for the presence of TNF-α and IFN-γ (sensitivity of detection: 2 pg/mL for both). No cytokine secretion was detected in the absence of protein antigens. Similar results were obtained in 5 HCV-infected and 2 HCV-uninfected subjects.
MK001 inhibits proliferation, IL-2 secretion and NF-κB-mediated transcription in the Jurkat T cell line
Since silymarin has been shown to inhibit NF-κB activation,7 we asked whether NF-κB blockade could account for the inhibition of cellular proliferation and cytokine secretion. To investigate this possibility, we utilized the transformed Jurkat T cell line, which proliferates spontaneously. A single dose of MK001 was able to inhibit Jurkat T cell proliferation measured by 3H-thymidine incorporation 24 hours later (Figure 7A). Stimulation with anti-CD3 alone was insufficient to induce cytokine secretion in Jurkat cells (data not shown), but stimulation with anti-CD3 and anti-CD28 antibodies together resulted in IL-2 secretion. (NB: Stimulation of Jurkat T cells with anti-CD3 and anti-CD28 together does not induce TNF-α or IFN-γ secretion, data not shown). Therefore, we tested whether MK001 could inhibit IL-2 secretion from Jurkat T cells. Our data indicate that MK001 was indeed able to inhibit IL-2 secretion from Jurkat T cells as measured by standard ELISA (Figure 7B). Finally, to determine whether inhibition of NF-κB might also be occurring, Jurkat T cells were transfected with a plasmid containing a luciferase reporter gene under the control of an NF-κB responsive promoter. These data, shown in Figure 7C, indicate that treatment with MK001 resulted in a dose-dependent inhibition of T cell receptor-induced NF-κB activation. We also investigated whether MK001 could affect IL-10 secretion, indirectly limiting pro-inflammatory responses, but found that IL-10 secretion by PBMC was also inhibited in the presence of MK001 (Supplemental Figure 2).
Figure 7. MK001 inhibits proliferation, IL-2 secretion, and NF-κB-mediated transcription in the Jurkat T cell line.

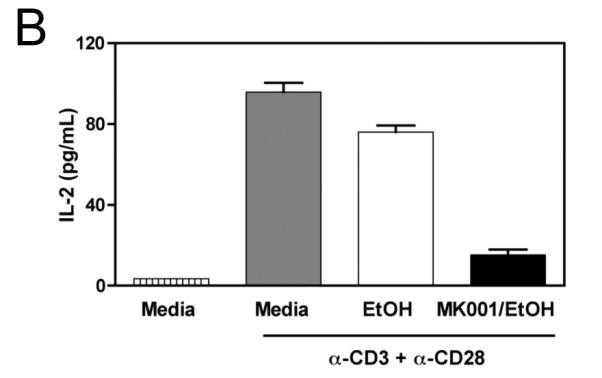

(A) Spontaneous proliferation of the human Jurkat tumor cell line was detected using 3H-thymidine incorporation (mean cpm of quaduplicate wells). Results from parallel cultures treated with MK001 (20 μg/mL) dissolved in either DMSO or 95% ethanol are shown in this representative experiment. (B) Jurkat T cells were stimulated to produce IL-2 using plate-bound anti-CD3 and soluble anti-CD28 stimulation, and compared to the negative control (95% ethanol) and MK001 (20 μg/mL) treatment. IL-2 was quantified in supernatants collected 24 hours after stimulation using ELISA. (C) Jurkat T cells were transfected with 5 μg of a plasmid encoding a luciferase reporter gene under the control of an NF-κB responsive promoter. Cells were treated with MK001 or DMSO for 30 minutes prior to stimulation. (No cellular toxicity has been noted with the MK001 concentrations used at 30 minutes, data not shown). NF-κB transcription was activated via the TCR using plate-bound anti-CD3, anti-CD3 + anti-CD28 antibodies, or with 15 ng/mL TNF-α, and luciferase activity was measured 4 hours after TCR activation or TNF-α treatment. Treatment with MK001 resulted in a dose-dependent inhibition of TCR-induced NF-κB activation. Error bars in all panels indicate one standard deviation among 4 replicates per condition.
DISCUSSION
Hepatic inflammation in chronic liver disease is histologically evaluated by the extent of infiltration by lymphocytes and the degree of hepatocyte cell death or necrosis, usually near lymphocyte aggregates.21 Clinical studies have demonstrated a clear association between greater hepatic inflammation and subsequent fibrosis progression.14, 15 Thus, control of inflammation is likely to be a useful strategy for ameliorating the sequelae of chronic liver disease. Here we demonstrate that a standardized preparation of silymarin, MK001, as well as two components of silymarin, silybin A and silybin B (individually and combined as silibinin), have the ability to dose-dependently inhibit T cell proliferation and pro-inflammatory cytokine secretion in vitro. Furthermore, MK001 was able to inhibit NF-κB-dependent transcription in Jurkat T cells stimulated through T cell receptor engagement, a property that is likely associated with its ability to inhibit T cell proliferation and cytokine secretion. These effects suggest that silymarin, at sufficiently high oral doses in humans, could potentially result in control of hepatic inflammation in chronic liver disease.
The data described herein are in agreement with the limited number of earlier reports regarding the effect of silymarin on lymphocyte function and support the notion that silymarin and its components can have immunosuppressive/anti-inflammatory effects. Expansion of T cells is important step in the amplification of an immune response. Our finding that silymarin inhibited antigen-specific and non-specific T cell proliferation extends our knowledge of the inhibitory effect of silymarin on cellular proliferation beyond that shown with tumor cell lines7 to freshly isolated T cells from HCV-infected and uninfected individuals. With regard to effects on cytokine secretion, Schumann et al. described immunosuppressive/anti-inflammatory effects upon silibinin pre-treatment in a murine ConA-induced T cell dependent hepatitis model.16 Consistent with our in vitro data, elevated intrahepatic mRNA levels of IL-2, IL-4, IFN-γ and TNF-α and splenocyte secretion of the same cytokines were significantly inhibited by silibinin. Other effects included increases in intrahepatic and splenocyte IL-10 levels (which differed from our in vitro results), but reinforced the overall immunosuppressive profile of silibinin’s effects in their model, as well as reduction in ALT and AST levels and inhibition of hepatocyte apoptosis. In a separate report, Lee et al. described silibinin’s ability to inhibit human dendritic cell maturation in vitro, which also impacts immune responsiveness.17
Silymarin has a short half-life, with rapid conjugation in the liver and primary excretion in bile.18, 19 However, achieving control of hepatic inflammation in vivo will likely require high and/or frequent oral dosing of silymarin in its currently available forms. Recently published pharmacokinetic analyses of oral Legalon® silymarin in HCV-infected patients described steady-state maximum plasma concentrations for free and total (free and conjugated) flavonolignans at the 700 mg dose between 0.4-2.7 μg/mL, and 4.1-18.9 μg/mL, respectively,20 where higher levels of silymarin were found with higher degrees of liver fibrosis.19 It is unclear whether free or total levels are relevant for the described bioactive effects; note that the latter concentrations are within the range of the anti-inflammatory effects described here and the anti-viral activity described previously.8 In addition, these plasma values are likely to underestimate silymarin levels in the liver. Results from rat21 and human studies18 indicate that the highest concentrations are found in liver, suggesting a localization of silymarin effects to this site. As a consequence, the likelihood of adverse effects on immune responses generated in lymph nodes or at distant sites of infection appears to be relatively low. Whether such effects occur in vivo will need to be evaluated directly through trials of silymarin including both pharmacokinetic and immunological analyses. Fortunately, silymarin has been described to be very well tolerated, even at high doses, making it possible to test whether the dosages required to induce the desired anti-inflammatory effects will be safely tolerated. Ideally, such studies to assess the therapeutic window for silymarin administration would identify a silymarin dose that would sufficiently dampen injurious hepatic immune responses to reduce liver disease progression without inducing generalized immunosuppression. The challenge and key to unlocking the promise of silymarin will likely be related to the ability to attain optimal concentrations in vivo.
Specifically in the case of HCV infection, however, it is important to consider the possibility that the inflammatory lymphocyte response is also likely to be controlling viral replication to some degree, such that inhibition of lymphocyte responses could theoretically unleash HCV replication and lead to deleterious sequelae. Arguing against this possibility, however, our group previously demonstrated that the same preparation of silymarin (MK001), used at the same concentration as in this report, inhibited HCV replication in vitro.8 Furthermore, Ferenci et al. have since demonstrated that intravenous silibinin decreased circulating HCV RNA levels in humans.22 On the basis of these findings taken in toto, we propose a model for potential silymarin efficacy in HCV infection which includes both anti-viral and anti-inflammatory effects. We hypothesize that decreased hepatic inflammation--due to direct inhibition by silymarin, or due indirectly to decreased viral replication, or both--has the potential to induce long-term benefit to the infected liver. Future studies will be needed to determine if one effect dominates over the other in vivo, although this would not be an issue in situations where hepatic inflammation is not due to viral infection. In light of the clearly demonstrated anti-viral effect of silymarin and its components, we predict that its immunosuppressive/anti-inflammatory effects are unlikely to lead to substantial increases in direct liver injury due to uncontrolled viral replication in hepatitis C.
In summary, we have demonstrated that a standardized preparation of silymarin, MK001, and two subcomponents, silybin A and silybin B, were able to dose-dependently inhibit proliferation and pro-inflammatory cytokine secretion by freshly isolated PBMC from HCV-infected and uninfected subjects. These findings are important, because patients and clinicians have taken a keen interest in silymarin as a potential alternative therapy for chronic liver disease, and its mechanism of action is incompletely understood. Whether inhibition of T cell proliferation and cytokine secretion in vitro might translate into clinical benefit will be determined by randomized controlled trials evaluating silymarin efficacy. It is important to note that the potential for clinical benefit has already been demonstrated by the recent report of intravenous silibinin which led to substantial reduction of HCV RNA levels.22 We speculate that effects on immune function would most likely have been detected in this setting, had they been tested. Additional studies are needed to better understand the mechanism of action and pharmacokinetics of silymarin both in vitro and in vivo, as these data may reveal new indications for the use of this promising intervention and provide new benchmarks for effective dosing.
Supplementary Material
ACKNOWLEDGEMENTS
The authors wish to thank Jee-Hang Tse for his help in preparation of the manuscript, and Terri Mathisen, RN for help in recruitment of subjects for this study. We are also grateful to Michael Houghton, PhD and Kevin Crawford (Chiron Corp.) for the gift of HCV protein antigens.
FINANCIAL SUPPORT: S.J.P. is partially supported by grant R21 AT002895 from the National Center for Complementary and Alternative Medicines/National Institutes of Health. The isolation of pure compounds from milk thistle was supported by grant R01 CA104286 to T.N.G./N.H.O. from the National Cancer Institute/NIH.
THE STUDY SPONSOR HAD NO ROLE IN THE COLLECTION, ANALYSIS OR INTERPRETATION OF THE DATA IN THIS MANUSCRIPT
ABBREVIATIONS
- HCV
hepatitis C virus
- RNA
ribonucleic acid
- TNF-α
Tumor Necrosis Factor-alpha
- FN-γ
Interferon-gamma
- ELISA
Enzyme Linked Immuno Sorbent Assay
- PHA
Phytohemagglutinin
- B-LCL
B-Lymphoblastoid Cell Line
- PBMC
Peripheral Blood Mononuclear Cells
- HLA
Human Leukocyte Antigen
- ALT
alanine aminotransferase
- DMSO
Dimethyl Sulfoxide
- EtOH
Ethanol
- CFSE
Carboxy Fluoroscein Succinimidyl Ester
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
FINANCIAL DISCLOSURES: NONE
NO CONFLICTS OF INTEREST EXIST
REFERENCES
- 1.Alter HJ, Seeff LB. Recovery, persistence, and sequelae in hepatitis C virus infection: a perspective on long-term outcome. Seminars in Liver Disease. 2000;20:17–35. doi: 10.1055/s-2000-9505. [DOI] [PubMed] [Google Scholar]
- 2.Manns MP, McHutchison JG, Gordon SC, Rustgi VK, Shiffman M, Reindollar R, Goodman ZD, Koury K, Ling M, Albrecht JK. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet. 2001;358:958–65. doi: 10.1016/s0140-6736(01)06102-5. [DOI] [PubMed] [Google Scholar]
- 3.Fried MW, Shiffman ML, Reddy KR, Smith C, Marinos G, Goncales FL, Haussinger D, M D, G C, Dhumeaux D, Craxi A, Lin A, Hoffmann J, Yu J. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. New England Journal of Medicine. 2002;347:975–982. doi: 10.1056/NEJMoa020047. [DOI] [PubMed] [Google Scholar]
- 4.Davis GL, Albright JE, Cook SF, Rosenberg DM. Projecting future complications of chronic hepatitis C in the United States. Liver Transpl. 2003;9:331–8. doi: 10.1053/jlts.2003.50073. [DOI] [PubMed] [Google Scholar]
- 5.Strader DB, Bacon BR, Lindsay KL, La Brecque DR, Morgan T, Wright EC, Allen J, Khokar MF, Hoofnagle JH, Seeff LB. Use of complementary and alternative medicine in patients with liver disease. Am J Gastroenterol. 2002;97:2391–7. doi: 10.1111/j.1572-0241.2002.05993.x. [DOI] [PubMed] [Google Scholar]
- 6.Seeff LB, Curto TM, Szabo G, Everson GT, Bonkovsky HL, Dienstag JL, Shiffman ML, Lindsay KL, Lok AS, Di Bisceglie AM, Lee WM, Ghany MG. Herbal product use by persons enrolled in the hepatitis C Antiviral Long-Term Treatment Against Cirrhosis (HALT-C) Trial. Hepatology. 2008;47:605–12. doi: 10.1002/hep.22044. [DOI] [PubMed] [Google Scholar]
- 7.Agarwal R, Agarwal C, Ichikawa H, Singh RP, Aggarwal BB. Anticancer potential of silymarin: from bench to bed side. Anticancer Res. 2006;26:4457–98. [PubMed] [Google Scholar]
- 8.Polyak SJ, Morishima C, Shuhart MC, Wang CC, Liu Y, Lee DYW. Inhibition of T cell inflammatory cytokines, hepatocyte NF-κB signaling and HCV infection by standardized silymarin. Gastroenterology. 2007;132:1925–36. doi: 10.1053/j.gastro.2007.02.038. [DOI] [PubMed] [Google Scholar]
- 9.Saller R, Meier R, Brignoli R. The use of silymarin in the treatment of liver diseases. Drugs. 2001;61:2035–63. doi: 10.2165/00003495-200161140-00003. [DOI] [PubMed] [Google Scholar]
- 10.Jacobs BP, Dennehy C, Ramirez G, Sapp J, Lawrence VA. Milk thistle for the treatment of liver disease: a systematic review and meta-analysis. Am J Med. 2002;113:506–15. doi: 10.1016/s0002-9343(02)01244-5. [DOI] [PubMed] [Google Scholar]
- 11.Spengler U, Nattermann J. Immunopathogenesis in hepatitis C virus cirrhosis. Clin Sci (Lond) 2007;112:141–55. doi: 10.1042/CS20060171. [DOI] [PubMed] [Google Scholar]
- 12.Graf TN, Wani MC, Agarwal R, Kroll DJ, Oberlies NH. Gram-scale purification of flavonolignan diastereoisomers from Silybum marianum (Milk Thistle) extract in support of preclinical in vivo studies for prostate cancer chemoprevention. Planta Med. 2007;73:1495–501. doi: 10.1055/s-2007-990239. [DOI] [PubMed] [Google Scholar]
- 13.Morishima C, Polyak SJ, Ray R, Doherty MC, Di Bisceglie AM, Malet PF, Bonkovsky HL, Sullivan DG, Gretch DR, Rothman AL, Koziel MJ, Lindsay KL. Hepatitis C Virus-Specific Immune Responses and Quasi-Species Variability at Baseline Are Associated with Nonresponse to Antiviral Therapy during Advanced Hepatitis C. J Infect Dis. 2006;193:931–40. doi: 10.1086/500952. [DOI] [PubMed] [Google Scholar]
- 14.Yano M, Kumada H, Kage M, Ikeda K, Shimamatsu K, Inoue O, Hashimoto E, Lefkowitch JH, Ludwig J, Okuda K. The long-term pathological evolution of chronic hepatitis C. Hepatology. 1996;23:1334–40. doi: 10.1002/hep.510230607. [DOI] [PubMed] [Google Scholar]
- 15.Ghany MG, Kleiner DE, Alter H, Doo E, Khokar F, Promrat K, Herion D, Park Y, Liang TJ, Hoofnagle JH. Progression of fibrosis in chronic hepatitis C. Gastroenterology. 2003;124:97–104. doi: 10.1053/gast.2003.50018. [DOI] [PubMed] [Google Scholar]
- 16.Schumann J, Prockl J, Kiemer AK, Vollmar AM, Bang R, Tiegs G. Silibinin protects mice from T cell-dependent liver injury. J Hepatol. 2003;39:333–40. doi: 10.1016/s0168-8278(03)00239-3. [DOI] [PubMed] [Google Scholar]
- 17.Lee JS, Kim SG, Kim HK, Lee TH, Jeong YI, Lee CM, Yoon MS, Na YJ, Suh DS, Park NC, Choi IH, Kim GY, Choi YH, Chung HY, Park YM. Silibinin polarizes Th1/Th2 immune responses through the inhibition of immunostimulatory function of dendritic cells. J Cell Physiol. 2007;210:385–97. doi: 10.1002/jcp.20852. [DOI] [PubMed] [Google Scholar]
- 18.Lorenz D, Lucker PW, Mennicke WH, Wetzelsberger N. Pharmacokinetic studies with silymarin in human serum and bile. Methods Find Exp Clin Pharmacol. 1984;6:655–61. [PubMed] [Google Scholar]
- 19.Schrieber SJ, Wen Z, Vourvahis M, Smith PC, Fried MW, Kashuba AD, Hawke RL. The pharmacokinetics of silymarin is altered in patients with hepatitis C virus and nonalcoholic Fatty liver disease and correlates with plasma caspase-3/7 activity. Drug Metab Dispos. 2008;36:1909–16. doi: 10.1124/dmd.107.019604. [DOI] [PubMed] [Google Scholar]
- 20.Schrieber SJ, Soule TA, Wen Z, Wahed AS, Smith PC, Fried MW, Reddy KR, Navarro VJ, Afdhal NH, Belle SH, Berman J, Liu QY, Doo E, Hawke RL. Phase I study to evaluate the safety, tolerability, and pharmacokinetics of silymarin following chronic dosing in patients with chronic hepatitis C. Gastroenterology. 2008;134(Supplement):P435396. [Google Scholar]
- 21.Wu JW, Lin LC, Hung SC, Chi CW, Tsai TH. Analysis of silibinin in rat plasma and bile for hepatobiliary excretion and oral bioavailability application. J Pharm Biomed Anal. 2007;45:635–41. doi: 10.1016/j.jpba.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 22.Ferenci P, Scherzer TM, Kerschner H, Rutter K, Beinhardt S, Hofer H, Schoniger-Hekele M, Holzmann H, Steindl-Munda P. Silibinin is a potent antiviral agent in patients with chronic hepatitis C not responding to pegylated interferon/ribavirin therapy. Gastroenterology. 2008;135:1561–7. doi: 10.1053/j.gastro.2008.07.072. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.