

Abstract
Cholestasis results when excretion of bile acids from the liver is interrupted. Liver injury occurs during cholestasis, and recent studies showed that inflammation is required for injury. Our previous studies demonstrated that early growth response factor-1 (Egr-1) is required for development of inflammation in liver during cholestasis, and that bile acids upregulate Egr-1 in hepatocytes. What remains unclear is the mechanism by which bile acids upregulate Egr-1. Bile acids modulate gene expression in hepatocytes by activating the farnesoid X receptor (FXR) and through activation of mitogen-activated protein kinase (MAPK) signaling. Accordingly, the hypothesis was tested that bile acids upregulate Egr-1 in hepatocytes by FXR and/or MAPK-dependent mechanisms. Deoxycholic acid (DCA) and chenodeoxycholic acid (CDCA) stimulated upregulation of Egr-1 to the same extent in hepatocytes isolated from wild-type mice and FXR knockout mice. Similarly, upregulation of Egr-1 in the livers of bile duct-ligated (BDL) wild-type and FXR knockout mice was not different. Upregulation of Egr-1 in hepatocytes by DCA and CDCA was prevented by the MEK inhibitors U0126 and SL-327. Furthermore, pretreatment of mice with U0126 prevented upregulation of Egr-1 in the liver after BDL. Results from these studies demonstrate that activation of MAPK signaling is required for upregulation of Egr-1 by bile acids in hepatocytes and for upregulation of Egr-1 in the liver during cholestasis. These studies suggest that inhibition of MAPK signaling may be a novel therapy to prevent upregulation of Egr-1 in liver during cholestasis.
Keywords: Early growth response factor-1, Bile acids, Mitogen-activated protein kinases, Farnesoid X receptor, Inflammation, Cholestasis
Introduction
Cholestasis is a condition that occurs when bile flow from the liver is disrupted (Li and Crawford, 2004). This results in increased concentrations of toxic bile acids in the liver and blood (Lindblad et al., 1977; Setchell et al., 1997). If cholestasis is left untreated, this disease results in hepatocellular injury, bile duct proliferation, and activation of hepatic stellate cells and portal fibroblasts, leading to fibrosis (Gujral et al., 2003; Ramadori and Saile, 2004). Over time as this disease progresses, the liver becomes cirrhotic and the patient will be in danger of liver failure (Paumgartner, 2006).
Inflammation, consisting primarily of neutrophils, has been shown to play an integral role in the development of hepatocellular injury during cholestasis (Gujral et al., 2003; Gujral et al., 2004) Neutrophil accumulation has been shown to occur in animals and humans with obstructive jaundice (Gulubova, 1998; Yamashiki et al., 1998; Neuman et al., 2002). Studies from our laboratory have shown that early growth response factor-1 (Egr-1), a transcription factor, is important for inflammation and injury during cholestasis (Kim et al., 2006). In these studies, Egr-1 was upregulated in hepatocytes of mice subjected to bile duct ligation (BDL), a model of cholestatic liver disease. Furthermore, Egr-1 knockout mice subjected to bile duct ligation had reduced liver injury, fewer neutrophils in the liver, and reduced expression of proinflammatory mediators compared to wild-type mice (Kim et al., 2006). These studies suggested that Egr-1 is a key mediator of inflammation in the liver during cholestasis. Our results demonstrated further that the bile acid, deoxycholic acid (DCA), increased Egr-1 protein levels in primary mouse hepatocytes (Kim et al., 2006). This suggested that the stimulus for upregulation of Egr-1 in liver during cholestasis may be bile acids. What remains unknown, however, is the molecular mechanism by which bile acids increase Egr-1 levels in hepatocytes. Identification of the signaling pathways that stimulate upregulation of Egr-1 could lead to the development of therapeutics that attenuate the inflammatory response in the liver during cholestasis.
Bile acids modulate gene expression in hepatocytes by several mechanisms. For example, bile acids activate the nuclear receptor, farnesoid X receptor (FXR). Upon bile acid binding, FXR heterodimerizes with RXR (Makishima et al., 1999) and binds to response elements in the promoters of genes, and modulates gene transcription by suppressing or enhancing gene expression (Claudel et al., 2005). In addition to FXR, bile acids modulate gene expression by activating mitogen-activated protein kinase (MAPK) signaling (Rao et al., 2002). Bile acids stimulate ligand-independent activation of the epidermal growth factor receptor which stimulates phosphorylation and activation of raf kinase, MEK, and Erk1/2 (Rao et al., 2002). Whether activation of FXR and/or MAPK signaling is required for upregulation of Egr-1 by bile acids is not known. Accordingly, the hypothesis was tested that upregulation of Egr-1 in hepatocytes in vitro by bile acids and in vivo during cholestasis requires FXR and/or MAPK signaling.
Materials and Methods
Animals care
C57BL/6 mice (Harlan, Madison, WI) and FXR knockout mice with a congenic C57BL/6 background were used for all studies. Generation of the FXR knockout mice was described previously (Sinal et al., 2000). Mice were maintained on a 12-h light/dark cycle under controlled temperature (18-21°C) and humidity. Food (Rodent Chow; Harlan-Teklad, Madison, WI) and tap water were allowed ad libitum. All procedures on animals were carried out in accordance with the Guide for the Care and Use of Laboratory Animals promulgated by the National Institutes of Health and were approved by the institutional IACUC committee at the University of Kansas Medical Center.
Hepatocyte Isolation
Hepatocytes were isolated from the livers of mice by collagenase perfusion as described in detail by us previously (Kim et al., 2006). The hepatocytes were cultured in Williams' medium E containing 10% FBS and Penicillin-Streptomycin. After a 3 hour attachment period, the medium with unattached cells was removed, and fresh medium added. The cells were cultured overnight before addition inhibitors and bile acids.
Bile Duct Ligation
Male, C57BL/6 and FXR knockout mice, 8-12 weeks of age, were anesthetized with isoflurane. A midline laparotomy was performed and the bile duct ligated with 3-0 surgical silk. The abdominal incision was closed with sutures, and the mice received 0.2 mg/kg Buprenex by subcutaneous injection. For studies with U0126, mice received 100 mg/kg U0126 dissolved in DMSO or DMSO alone by intraperitoneal injection, 1 hour before surgery.
Real-time PCR
RNA was isolated from livers and hepatocytes using TRI reagent (Sigma Chemical Company, St. Louis, MO), and reverse transcribed into cDNA as described by us previously (Kim et al., 2006). Real-time PCR was used to quantify the mRNA levels of Egr-1 and 18S on an Applied Biosystems Prism 7300 Real-time PCR Instrument (Applied Biosystems, Foster City, CA) using the SYBR green DNA PCR kit (Applied Biosystems) as described (Kim et al., 2006). The sequences of the primers were as follows: 18S Forward: 5′-TTG ACG GAA GGG CAC CAC CAG-3′; 18S Reverse: 5′-GCA CCA CCA CCC ACG GAA TCG-3′; Egr-1 Forward: 5′-GGC AGA GGA AGA CGA TGA AG-3′; and Egr-1 Reverse: 5′-GAC GAGTTATCC CAG CCA AA-3′.
Western Blot
Total protein was isolated from hepatocytes and liver. In brief, hepatocytes and liver were lysed in RIPA buffer (1×PBS, 1% IGPAL, 0.5% Sodium Deoxycholate and 0.5% SDS) containing Complete protease inhibitor (Roche Applied Science Indianapolis, IN) and PhosSTOP phosphatase inhibitor (Roche Applied Science Indianapolis, IN). The lysates were sonicated, and total protein concentrations measured using the BCA Protein Assay Kit (Pierce Rockford, IL). Phospho-p44/42 MAPK (Erk1/2), total p44/42 MAPK, Egr-1 and β-actin levels were measured using western blotting. Equal amounts of protein were separated on a 4.5-15% gradient Criterion polyacrylamide gel (Bio Rad Hercule, CA) and transferred to a PVDF membrane (Immoblion-P Bedford, MA). The membrane was incubated with anti-phospho-p44/42 MAPK antibody, anti-p44/p42 MAPK antibody, anti-Egr-1 antibody (all from Cell Signaling Technology, Inc.) or anti-β-actin antibody (Sigma Chemical Co., St. Louis, MO) followed by incubation with the appropriate horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology Santa Cruz, CA). The bands were visualized using the ECL detection kit (Bio Rad Hercules, CA).
Statistical Analysis
Results are presented as the mean ± SEM. Data were analyzed by Analysis of Variance (ANOVA). ANOVAs were performed on log X-transformed data in instances in which variances were not homogenous. Comparisons among group means were made using the Student-Newman-Keuls test. The criterion for significance was p < 0.05 for all studies.
Results
Deoxycholic Acid and Chenodeoxycholic Acid Increase Egr-1 mRNA and Protein Levels in Primary Mouse Hepatocytes
Exposure of primary mouse hepatocytes to deoxycholic acid (DCA) or chenodeoxycholic acid (CDCA) caused a dose-dependent increase in Egr-1 mRNA and protein in primary mouse hepatocytes (Fig. 1).
Fig. 1.
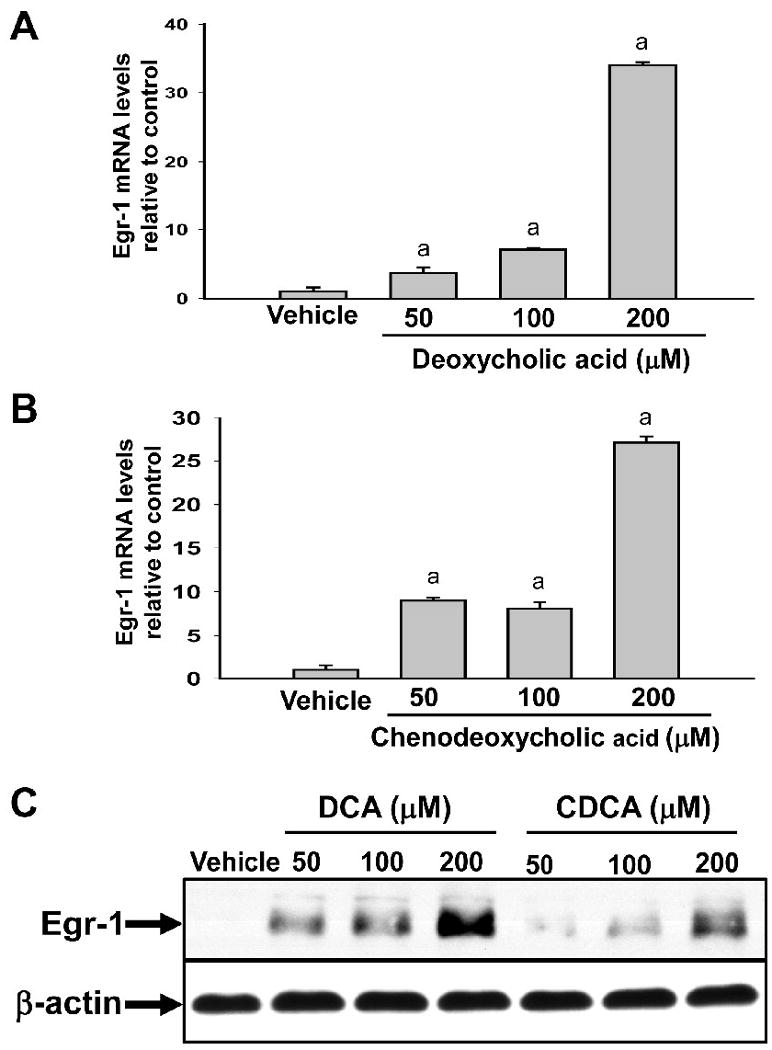
Upregulation of Egr-1 mRNA and protein in primary mouse hepatocytes treated with bile acids. Hepatocytes were isolated from mice and treated with either DCA or CDCA. (A and B) Two hours later, Egr-1 mRNA levels were quantified by real-time PCR. Data are expressed as mean +/- SEM; n=3. aSignificantly different (p < 0.05) from vehicle-treated hepatocytes. (C) Egr-1 and β-actin proteins were detected by western blot. Representative of an n=3.
Upregulation of Egr-1 by Bile Acids does not require FXR
FXR is a nuclear receptor activated by bile acids and represents a possible signaling mechanism by which bile acids could affect Egr-1 expression (Makishima et al., 1999). To determine whether FXR is required for upregulation of Egr-1 by bile acids, hepatocytes were isolated from wild-type and FXR knockout mice and treated with either DCA or CDCA. Treatment of hepatocytes from wild-type mice with DCA or CDCA increased Egr-1 mRNA levels (Fig. 2). Bile acids increased Egr-1 mRNA levels in hepatocytes isolated FXR knockout mice to the same extent as hepatocytes from wild-type mice (Fig. 2). Next it was determined whether FXR is required for upregulation of Egr-1 in the livers of bile duct-ligated mice. For these studies, wild-type and FXR knockout mice were subjected to either sham operation or bile duct ligation, and Egr-1 mRNA levels were quantified in the liver. Egr-1 mRNA was increased to the same extent in the livers of wild-type and FXR knockout mice subjected to bile duct ligation (Fig. 2).
Fig. 2.
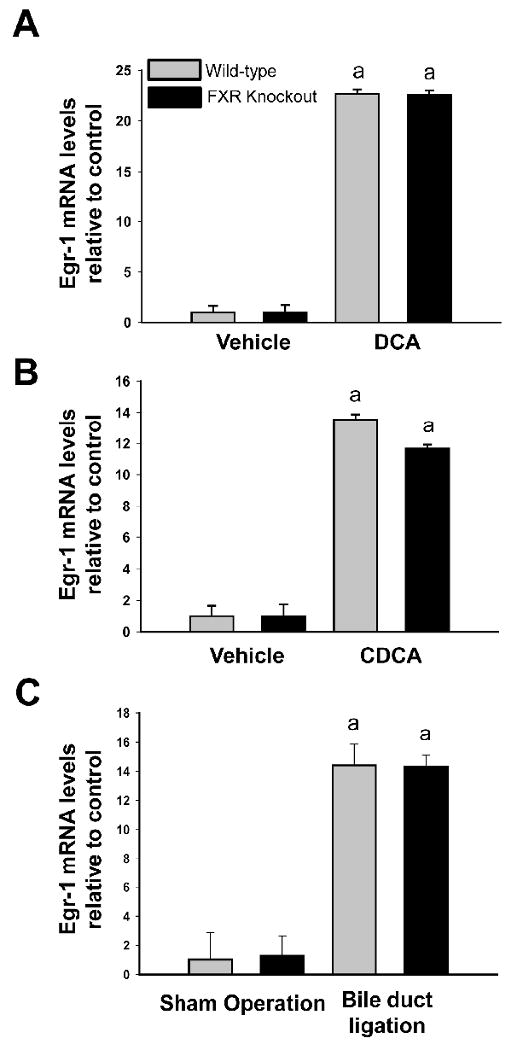
Upregulation of Egr-1 in primary mouse hepatocytes by bile acids and in liver during cholestasis does not require FXR. Primary mouse hepatocytes were isolated from wild-type and FXR knockout mice and treated with either (A) DCA or (B) CDCA. Two hours later, Egr-1 mRNA levels were quantified by real-time PCR. Data are expressed as mean +/- SEM; n=3. aSignificantly different (p < 0.05) from vehicle-treated hepatocytes. (C) Wild-type and FXR knockout mice were subjected to bile duct ligation or sham surgery. Three days later, Egr-1 mRNA levels were quantified in the liver by real-time PCR. Data are expressed as mean +/- SEM; n=3. aSignificantly different (p < 0.05) from sham-operated mice.
Upregulation of Egr-1 by Bile Acids Requires MAPK Activation
Bile acids have been shown activate the MAPK cascade in hepatocytes (Rao et al., 2002). Accordingly, we next determined whether MAPK activation is required from upregulation of Egr-1 in hepatocytes. Treatment with either DCA or CDCA for 15 minutes stimulated phosphorylation of ERK1/2 in hepatocytes. Pretreatment with U0126, a MEK inhibitor, prevented phosphorylation of ERK1/2 after bile acid treatment (Fig. 3A). Treatment of hepatocytes with either DCA or CDCA increased Egr-1 mRNA levels (Fig. 3B and 3C) and protein levels (Fig. 4). Pretreatment with U0126 completely prevented upregulation of Egr-1 mRNA and protein by both DCA and CDCA (Fig. 3 and 4). To confirm these results, hepatocytes were pretreated with SL-327, another MEK inhibitor. Pretreatment of hepatocytes with SL-327 attenuated the increase in Erk1/2 phosphorylation and the increase in Egr-1 mRNA and protein in hepatocytes treated with DCA and CDCA (Fig. 5 and 6).
Fig. 3.
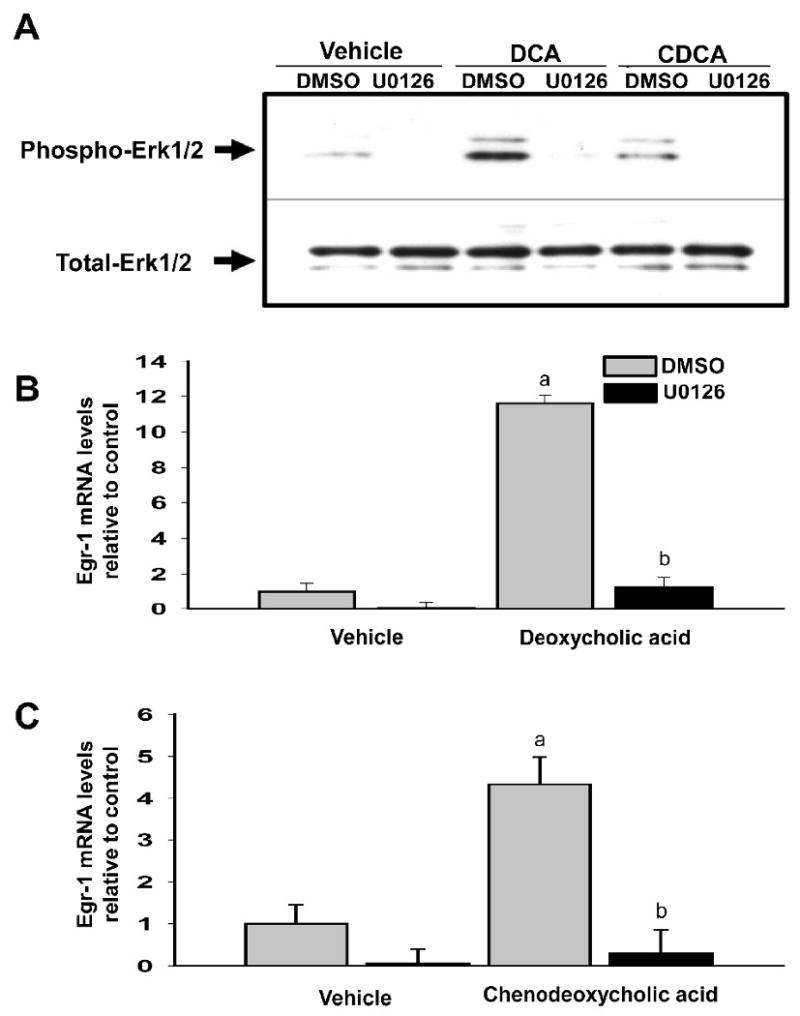
U0126 prevents upregulation of Egr-1 mRNA in bile acid-treated hepatocytes. Primary mouse hepatocytes were treated with 10μM UO126 or vehicle followed by either 200μM DCA or 200μM CDCA 30 minutes later. (A) After a 15 minute incubation, phosphorylated-Erk1/2 and total-Erk1/2 were detected by western blot. Representative of an n=3. (B and C) After 2 hours, Egr-1 mRNA levels were quantified by real-time PCR. Data are expressed as mean +/- SEM; n=3. aSignificantly different (p < 0.05) from control-treated hepatocytes. bSignificantly different (p < 0.05) from hepatocytes treated with vehicle and bile acid.
Fig. 4.
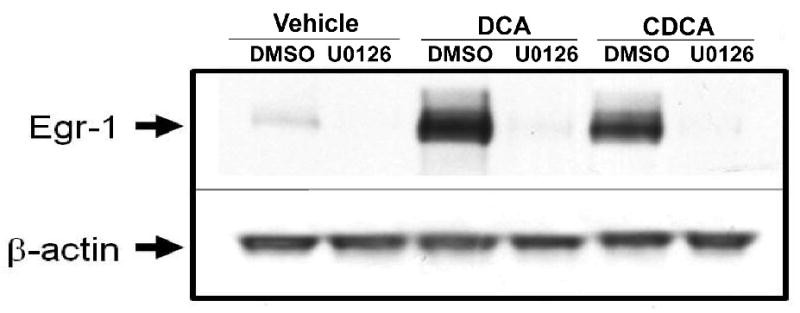
U0126 prevents upregulation of Egr-1 protein in bile acid-treated hepatocytes. Primary mouse hepatocytes were treated with 10μM U0126 or vehicle followed by either DCA or CDCA 30 minutes later. Egr-1 and β-actin proteins were detected by western blot. Representative of an n=3.
Fig. 5.
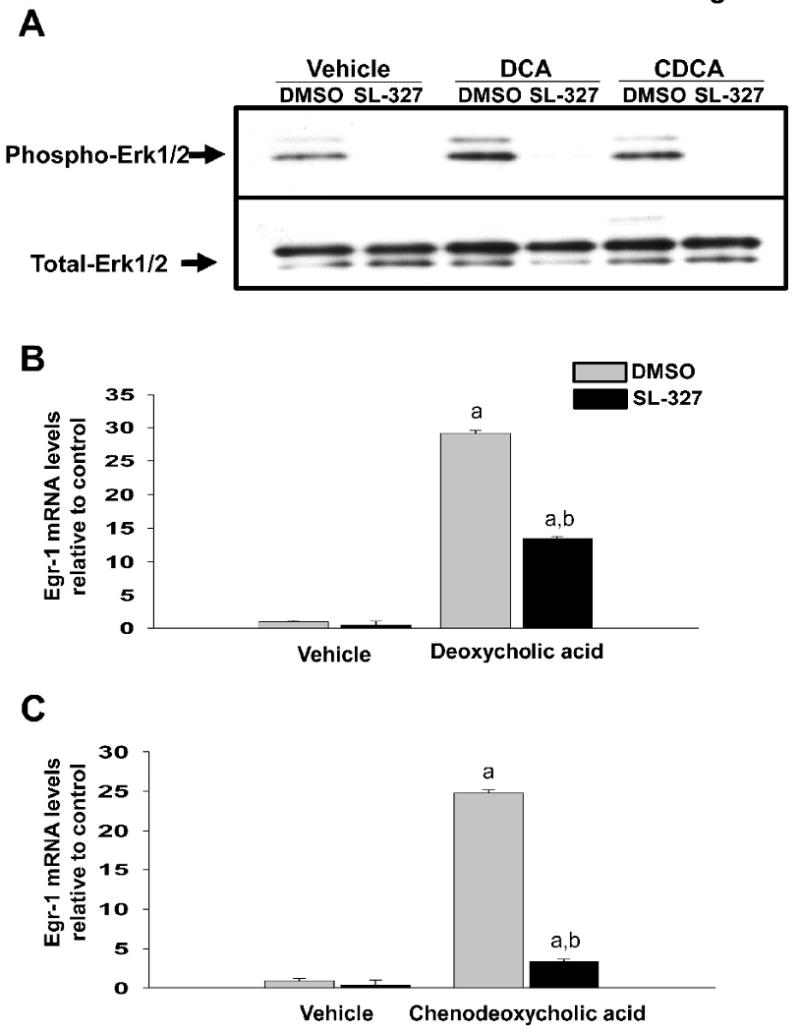
SL-327 prevents upregulation of Egr-1 mRNA in bile acid-treated hepatocytes. Primary mouse hepatocytes were treated with 20μM SL-327 or vehicle followed by either 200μM DCA or 200μM CDCA 30 minutes later. (A) After a 15 minute incubation, phosphorylated-Erk1/2 and total-Erk1/2 were detected by western blot. Representative of an n=3. (B and C) After 2 hours, Egr-1 mRNA levels were quantified by real-time PCR. Data are expressed as mean +/- SEM; n=3. aSignificantly different (p < 0.05) from control-treated hepatocytes. bSignificantly different (p < 0.05) from hepatocytes treated with vehicle and bile acid.
Fig. 6.

SL-327 prevents upregulation of Egr-1 protein in bile acid-treated hepatocytes. Primary mouse hepatocytes were treated with 20μM SL-327or vehicle followed by either DCA or CDCA 30 minutes later. Egr-1 and β-actin proteins were detected by western blot. Representative of an n=3.
Increased Egr-1 expression prevented with U0126 pretreatment in vivo
Since U0126 prevented upregulation of Egr-1 by bile acids in vitro, we next determined whether U0126 would prevent upregulation of Egr-1 in the livers of mice after bile duct ligation, a model of cholestasis. Bile duct ligation in mice stimulated phosphorylation of ERK (Fig. 7). Pretreatment of the mice with U0126 completely prevented phosphorylation of the ERK (Fig. 7). Bile duct ligation in mice increased Egr-1 protein levels (Fig. 7). Pretreatment of mice with U0126 prevented the increase in Egr-1 protein after bile duct ligation (Fig. 7).
Fig. 7.
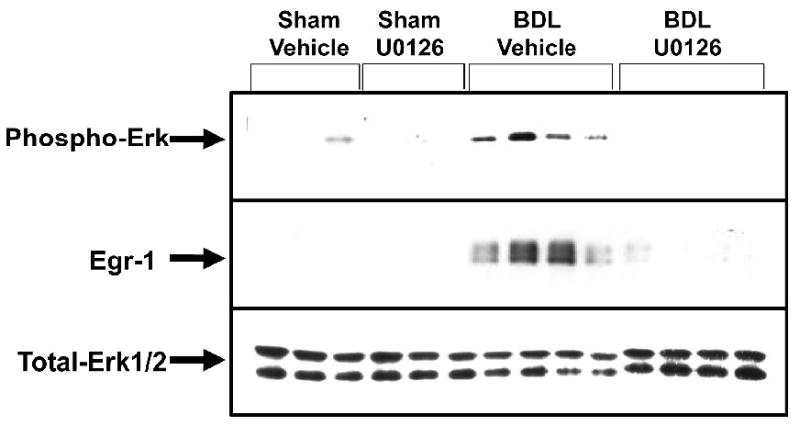
U0126 prevents upregulation of Egr-1 in the liver after bile duct ligation. Mice were treated with 100 mg/kg U0126 or vehicle. One hour later, the mice were subjected to bile duct ligation or sham operation. After 6 hours, phosphorylated, total Erk1/2 and Egr-1 protein were detected in the liver by western blot. n=3 for sham-operated mice and n=4 for bile duct-ligated mice.
Discussion
Inflammatory cell infiltration into the liver during cholestasis has been shown to exacerbate liver injury (Gujral et al., 2004). Studies have shown that upregulation of inflammatory mediators is important for inflammatory injury during cholestasis (Gujral et al., 2004; Wintermeyer et al., 2009). Our studies have identified Egr-1 as a key regulator of these proinflammatory molecules in hepatocytes in vivo during cholestasis (Kim et al., 2006). Furthermore, our studies demonstrated that bile acids stimulate upregulation of Egr-1 in hepatocytes (Fig. 1), suggesting that bile acids may be the stimulus for upregulation of Egr-1 in liver during cholestasis (Kim et al., 2006). What remained unknown from these studies, however, is the molecular mechanism by which bile acids upregulate Egr-1 in hepatocytes. The present studies demonstrated that MAPK signaling is important for upregulation of Egr-1 by bile acids in vitro and in vivo.
As discussed, bile acids activate many different signaling pathways in hepatocytes, including FXR and the MAPK cascade. FXR is the endogenous bile acid receptor, and has been shown to be activated during cholestasis and regulate expression of numerous genes (Makishima et al., 1999; Wagner et al., 2003). Our studies indicate that FXR is not required for upregulation of Egr-1 by bile acids in vitro or in vivo (Figs. 2). Consistent with these results, previous studies demonstrated that bile duct ligation in FXR knockout mice does not reduce liver injury or increase survival in these mice when compared to wild-type mice (Wagner et al., 2003). We saw similar results in our bile duct ligation studies (data not shown). If FXR were a key regulator of Egr-1, it would be anticipated that liver injury would be reduced in FXR knockout mice, since Egr-1 is required for inflammation and injury in the liver during cholestasis.
Our studies demonstrated that MAPK activation is required for upregulation of Egr-1 in vitro after bile acid treatment (Figs. 3, 4, 5 and 6) and in vivo during cholestasis (Fig. 7). The MAPK cascade has been shown to be activated in primary rat hepatocytes treated with bile acids in vitro (Rao et al., 2002). Studies have shown that MAPK activation results in phosphorylation and activation of the transcription factor Elk-1 (Mayer and Thiel, 2009). Mayer et al. demonstrated that phosphorylated Elk-1 binds to the Egr-1 promoter in response to cellular Ca2+ changes in some cell types (Mayer et al., 2008; Mayer and Thiel, 2009). This suggests that Elk-1 may be the transcription factor responsible for upregulation of Egr-1 in bile acid-treated hepatocytes. Further studies are needed, however, to test this possibility. Our results also demonstrated that MAPK signaling is activated in the liver during cholestasis, and that inhibition of Erk phosphorylation by a MEK inhibitor prevents upregulation of Egr-1 (Fig. 7). Interestingly, it was recently demonstrated that the kinase inhibitor, sorafenib, prevents Erk activation in the livers of bile duct-ligated rats and that this inihibitor reduces portal hypertension (Hennenberg et al., 2009). Whether sorafenib also affected inflammation in the liver was not investigated in these studies. These results in combination with ours suggest that Erk inhibition may not only be effective at reducing portal hypertension in patients with cholestasis, but may be effective at alleviating inflammatory liver injury in these patients.
Rao et. al. demonstrated previously that most conjugated and unconjugated bile acids stimulate Erk1/2 phosphorylation in hepatocytes (Rao et al., 2002). Therefore, it is likely that most bile acids will also promote upregulation of Egr-1. Activation of MAPK signaling in hepatocytes by bile acids was shown to be dependent upon ligand-independent activation of the epidermal growth factor receptor (Rao et al., 2002). Consistent with a role for this pathway in regulation of Egr-1 in hepatocytes, it was shown that epidermal growth factor upregulates Egr-1 in hepatocytes both in vitro and in vivo (Tsai et al., 2001). Therefore, therapeutic inhibition of the epidermal growth factor receptor may also be useful at preventing bile acid-induced inflammation in the liver during cholestasis by preventing upregulation of the proinflammatory transcription factor, Egr-1.
Collectively, our studies indicate that during cholestasis, elevated concentrations of bile acids activate MAPK signaling in hepatocytes. MAPK signaling then stimulates upregulation of Egr-1 which regulates expression of proinflammatory mediators that stimulate neutrophil accumulation and activation in the liver. Our studies also suggest that therapeutic targeting of the MAPK pathway in patients with cholestasis may be effective at inhibiting Egr-1 expression.
Acknowledgments
The authors thank Callie Wentling for her excellent technical assistance. In addition, the authors thank Dr. Grace Guo, Ph.D. for providing FXR knockout mice for these studies.
Financial Support: This study was supported by NIH grants DK073566 (B.L.C.) and COBRE (Center of Biomedical Research Excellence) P20 RR021940 as well as the Molecular Biology Core and the Histology Core supported by the COBRE grant. In addition, this work was supported by grant number P20 RR016475 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH).
Abbreviations
- Egr-1
early growth response factor-1
- DCA
deoxycholic acid
- CDCA
chenodeoxycholic acid
- MAPK
mitogen-activated protein kinases
- FXR
farnesoid X receptor
- Erk
extracellular signal-regulated kinase
- BDL
bile duct ligation
Footnotes
Conflict of Interest Statement: The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Claudel T, Staels B, Kuipers F. The Farnesoid X receptor: a molecular link between bile acid and lipid and glucose metabolism. Arteriosclerosis, thrombosis, and vascular biology. 2005;25:2020–2030. doi: 10.1161/01.ATV.0000178994.21828.a7. [DOI] [PubMed] [Google Scholar]
- Gujral JS, Farhood A, Bajt ML, Jaeschke H. Neutrophils aggravate acute liver injury during obstructive cholestasis in bile duct-ligated mice. Hepatology (Baltimore, Md. 2003;38:355–363. doi: 10.1053/jhep.2003.50341. [DOI] [PubMed] [Google Scholar]
- Gujral JS, Liu J, Farhood A, Hinson JA, Jaeschke H. Functional importance of ICAM-1 in the mechanism of neutrophil-induced liver injury in bile duct-ligated mice. Am J Physiol Gastrointest Liver Physiol. 2004;286:G499–507. doi: 10.1152/ajpgi.00318.2003. [DOI] [PubMed] [Google Scholar]
- Gulubova MV. Intercellular adhesion molecule-1 (ICAM-1) expression in the liver of patients with extrahepatic cholestasis. Acta histochemica. 1998;100:59–74. doi: 10.1016/s0065-1281(98)80006-8. [DOI] [PubMed] [Google Scholar]
- Hennenberg M, Trebicka J, Stark C, Kohistani AZ, Heller J, Sauerbruch T. Sorafenib targets dysregulated Rho kinase expression and portal hypertension in rats with secondary biliary cirrhosis. Br J Pharmacol. 2009;157:258–270. doi: 10.1111/j.1476-5381.2009.00158.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim ND, Moon JO, Slitt AL, Copple BL. Early growth response factor-1 is critical for cholestatic liver injury. Toxicol Sci. 2006;90:586–595. doi: 10.1093/toxsci/kfj111. [DOI] [PubMed] [Google Scholar]
- Li MK, Crawford JM. The pathology of cholestasis. Seminars in liver disease. 2004;24:21–42. doi: 10.1055/s-2004-823099. [DOI] [PubMed] [Google Scholar]
- Lindblad L, Lundholm K, Schersten T. Bile acid concentrations in systemic and portal serum in presumably normal man and in cholestatic and cirrhotic conditions. Scandinavian journal of gastroenterology. 1977;12:395–400. doi: 10.3109/00365527709181679. [DOI] [PubMed] [Google Scholar]
- Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, Hull MV, Lustig KD, Mangelsdorf DJ, Shan B. Identification of a nuclear receptor for bile acids. Science. 1999;284:1362–1365. doi: 10.1126/science.284.5418.1362. [DOI] [PubMed] [Google Scholar]
- Mayer SI, Thiel G. Calcium influx into MIN6 insulinoma cells induces expression of Egr-1 involving extracellular signal-regulated protein kinase and the transcription factors Elk-1 and CREB. Eur J Cell Biol. 2009;88:19–33. doi: 10.1016/j.ejcb.2008.07.002. [DOI] [PubMed] [Google Scholar]
- Mayer SI, Willars GB, Nishida E, Thiel G. Elk-1, CREB, and MKP-1 regulate Egr-1 expression in gonadotropin-releasing hormone stimulated gonadotrophs. J Cell Biochem. 2008;105:1267–1278. doi: 10.1002/jcb.21927. [DOI] [PubMed] [Google Scholar]
- Neuman M, Angulo P, Malkiewicz I, Jorgensen R, Shear N, Dickson ER, Haber J, Katz G, Lindor K. Tumor necrosis factor-alpha and transforming growth factor-beta reflect severity of liver damage in primary biliary cirrhosis. J Gastroenterol Hepatol. 2002;17:196–202. doi: 10.1046/j.1440-1746.2002.02672.x. [DOI] [PubMed] [Google Scholar]
- Paumgartner G. Medical treatment of cholestatic liver diseases: From pathobiology to pharmacological targets. World J Gastroenterol. 2006;12:4445–4451. doi: 10.3748/wjg.v12.i28.4445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramadori G, Saile B. Portal tract fibrogenesis in the liver. Lab Invest. 2004;84:153–159. doi: 10.1038/labinvest.3700030. [DOI] [PubMed] [Google Scholar]
- Rao YP, Studer EJ, Stravitz RT, Gupta S, Qiao L, Dent P, Hylemon PB. Activation of the Raf-1/MEK/ERK cascade by bile acids occurs via the epidermal growth factor receptor in primary rat hepatocytes. Hepatology. 2002;35:307–314. doi: 10.1053/jhep.2002.31104. [DOI] [PubMed] [Google Scholar]
- Setchell KD, Rodrigues CM, Clerici C, Solinas A, Morelli A, Gartung C, Boyer J. Bile acid concentrations in human and rat liver tissue and in hepatocyte nuclei. Gastroenterology. 1997;112:226–235. doi: 10.1016/s0016-5085(97)70239-7. [DOI] [PubMed] [Google Scholar]
- Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102:731–744. doi: 10.1016/s0092-8674(00)00062-3. [DOI] [PubMed] [Google Scholar]
- Tsai JC, Liu L, Zhang J, Spokes KC, Topper JN, Aird WC. Epidermal growth factor induces Egr-1 promoter activity in hepatocytes in vitro and in vivo. Am J Physiol Gastrointest Liver Physiol. 2001;281:G1271–1278. doi: 10.1152/ajpgi.2001.281.5.G1271. [DOI] [PubMed] [Google Scholar]
- Wagner M, Fickert P, Zollner G, Fuchsbichler A, Silbert D, Tsybrovskyy O, Zatloukal K, Guo GL, Schuetz JD, Gonzalez FJ, Marschall HU, Denk H, Trauner M. Role of farnesoid X receptor in determining hepatic ABC transporter expression and liver injury in bile duct-ligated mice. Gastroenterology. 2003;125:825–838. doi: 10.1016/s0016-5085(03)01068-0. [DOI] [PubMed] [Google Scholar]
- Wintermeyer P, Cheng CW, Gehring S, Hoffman BL, Holub M, Brossay L, Gregory SH. Invariant natural killer T cells suppress the neutrophil inflammatory response in a mouse model of cholestatic liver damage. Gastroenterology. 2009;136:1048–1059. doi: 10.1053/j.gastro.2008.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashiki M, Kosaka Y, Nishimura A, Watanabe S, Nomoto M, Ichida F. Analysis of serum cytokine levels in primary biliary cirrhosis patients and healthy adults. Journal of clinical laboratory analysis. 1998;12:77–82. doi: 10.1002/(SICI)1098-2825(1998)12:2<77::AID-JCLA1>3.0.CO;2-G. [DOI] [PMC free article] [PubMed] [Google Scholar]