

Abstract
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by progressive decline in multiple cognitive domains. Its pathological hallmarks include senile plaques and neurofibrillary tangles. Mild cognitive impairment (MCI) is the earliest detectable stage of AD with limited symptomology and no dementia. The yearly conversion rate of patients from MCI to AD is 10–15%, although conversion back to normal is possible in a small percentage. Early diagnosis of AD is important in an attempt to intervene or slow the advancement of the disease. Early AD (EAD) is a stage following MCI and characterized by full‐blown dementia; however, information involving EAD is limited. Oxidative stress is well‐established in MCI and AD, including protein oxidation. Protein nitration also is an important oxidative modification observed in MCI and AD, and proteomic analysis from our laboratory identified nitrated proteins in both MCI and AD. Therefore, in the current study, a proteomics approach was used to identify nitrated brain proteins in the inferior parietal lobule from four subjects with EAD. Eight proteins were found to be significantly nitrated in EAD: peroxiredoxin 2, triose phosphate isomerase, glutamate dehydrogenase, neuropolypeptide h3, phosphoglycerate mutase1, H+– transporting ATPase, α‐enolase and fructose‐1,6‐bisphosphate aldolase. Many of these proteins are also nitrated in MCI and late‐stage AD, making this study the first to our knowledge to link nitrated proteins in all stages of AD. These results are discussed in terms of potential involvement in the progression of this dementing disorder.
Keywords: protein nitration, early Alzheimer’s disease, oxidative stress, proteomics
Introduction
Mild cognitive impairment (MCI), considered to be the earliest detectable form of Alzheimer’s disease (AD), converts to AD at the annual rate of 10–15%[1]. Post‐translational modification of brain proteins, specifically nitration, is reported in both MCI and late‐stage AD [2, 3, 4]. Protein nitration is an important oxidative modification in neurodegenerative disease [2, 3, 5, 6, 7]. Nitric oxide, produced by the conversion of L‐arginine to L‐citrulline via nitric oxide synthase, can react with superoxide anion to form the powerful oxidant, peroxynitrite (ONOO−). Peroxynitrite can then react with carbon dioxide (CO2) to form the intermediate nitrosoperoxylcarbonate, which is spontaneously rearranged and homolytically cleaved to form carbonate (CO3•−) and nitrite radical (NO2•). NO2• can react with a tyrosine residue to form 3‐nitrotyrosine (3‐NT). Upon nitration, protein inactivation occurs in several key proteins including glyceraldehyde 3‐phosphate dehydrogenase [8, 9], tyrosine hydroxylase [10], heme oxygenase, manganese superoxide dismutase [11, 12, 13, 14] and glutamine synthetase [15, 16]. Proteins are significantly nitrated in MCI and AD [2, 3, 17, 18, 19] as well as other neurodegenerative diseases including Parkinson’s disease (PD) [5] and amyotrophic lateral sclerosis [10].
Early AD (EAD) is the next step beyond MCI and thus intermediate between MCI and more advanced AD. However, information involving this disease stage is limited. Nitrosative stress is well‐documented in MCI and late AD as indexed by an increase in 3‐NT [19, 20]. Increased protein nitration also can result in detrimental cellular effects [2, 3, 21]. In the current study, a proteomics approach was used to identify nitrated proteins in EAD to determine if any proteins overlap with proteins already known to be nitrated in this early stage of the disease following MCI.
Materials and methods
Inferior parietal lobule (IPL) samples from four EAD patients and four age‐matched controls were provided by the Rapid Autopsy Program of the University of Kentucky Alzheimer’s Disease Center (UK ADC). The normal control subjects in this study were two women and two men, whose average age at death was 79 ± 2.4 years (Table 1). The EAD patients were three women and one man, whose average age at death was 86 ± 4.0 years (Table 1). All subjects underwent neuropsychological testing and neurological and physical examinations annually. All of the samples came from the UK ADC’s normal control group. Normal control subjects had no cognitive complaints, normal cognitive test scores and normal neurological examinations. EAD patients met the following criteria: progressive memory loss as reported by an informant, deficits in two or more cognitive domains, altered activities of daily living, clinical dementia rating score of 0.5 to 1.0 (mild dementia), a nonfocal neurological evaluation and no other cause for dementia [22, 23]. The post‐mortem interval (PMI) at autopsy was 2.9 hrs for control and 2.4 hrs for EAD patients (Table 1). EAD patients met the National Institute on Aging/Regan Institute [24] intermediate or high likelihood criteria for the neuropathological diagnosis of AD. Controls met the low likelihood criteria for the neuropathological diagnosis of AD and had no other significant neuropathological alterations. Braak staging (score) characterizes severity of disease based on distribution pattern of neurofibrillary tangles and neurophil threads [25]. There are six levels of Braak staging, whereby the higher the stage, the more severe the stage of AD. Although, ideally a greater number of subjects would be desirable, it is rare to obtain brain from EAD subjects because most patients live long enough to convert to late‐stage AD. Therefore, only four independent EAD samples were available through the UK ADC Brain Bank.
Table 1.
Profile of EAD subjects
| Age (year) | Gender | Brain weight (g) | PMI (hrs) | Braak Score | |
|---|---|---|---|---|---|
| Control 1 | 79 | Male | 1300 | 2.25 | II |
| Control 2 | 75 | Female | 1080 | 3.50 | I |
| Control 3 | 86 | Female | 1310 | 2.25 | II |
| Control 4 | 77 | Male | 1310 | 3.50 | I |
| Average | 79 ± 2.4 | 1250 ± 57 | 2.9 ± 0.4 | 1.5 | |
| EAD 1 | 83 | Female | 1160 | 2.25 | V |
| EAD 2 | 96 | Female | 1180 | 1.60 | V |
| EAD 3 | 88 | Male | 1340 | 2.75 | V |
| EAD 4 | 77 | Female | 1190 | 3.00 | V |
| Average | 86 ± 4.0 | 1217.5 ± 41 | 2.4 ± 0.3 | 5.0 |
Sample preparation
Brain samples were minced and suspended in Media I buffer (10 mM HEPES buffer (pH 7.4), 137 mM NaCl, 4.6 mM KCl, 1.1 mM KH2PO4, 0.1 mM EDTA, 0.6 mM MgSO4, leupeptin (0.5 mg/ml), pepstatin (0.7 μg/ml), type II S soybean trypsin inhibitor (0.5 μg/ml) and PMSF (40 μg/ml)). To remove debris, brain homogenates were centrifuged at 14,000 ×g for 10 min. Protein concentration of the supernatant was determined by the BCA protein assay. Protein samples (200 μg) were precipitated by addition of ice‐cold 100% trichloroacetic acid (TCA) to a final concentration of 15% for 10 min. on ice. Precipitates were centrifuged for 2 min. at 14,000 ×g at 4°C. The pellet was kept and washed with 1 ml of 1:1 (v/v) ethyl acetate/ethanol three times. The resultant pellet was dissolved in rehydration buffer (8 M urea, 2 M thiourea, 2% CHAPS, 0.2% (v/v) biolytes, 50 mM dithiothreitol (DTT), and bromophenol blue). Samples were sonicated in rehydration buffer three times for 15 sec. intervals.
Measurement of 3‐nitrotyrosine
Levels of total 3‐NT were determined immunochemically [3]. Samples (5 μl) were incubated with 5 μl Laemmli buffer without reducing agents (0.125 M Tris base pH 6.8, 4% (v/v) sodium dodecyl sulfate (SDS) and 20% (v/v) glycerol). The resulting sample (250 ng) was loaded per well in the slot blot apparatus. Samples were loaded onto a nitrocellulose membrane under vacuum pressure. The membrane was blocked with 3% (w/v) bovine serum albumin (BSA) in wash blot for 2 hrs and incubated with a 1:2000 dilution of anti‐3‐NT polyclonal antibody (Sigma Aldrich, St. Louis, MO, USA) in wash blot for 2 hrs. Following completion of the primary antibody incubation, the membranes were washed three times in wash blot for 5 min. each. An anti‐rabbit IgG alkaline phosphatase secondary antibody (Sigma Aldrich) was diluted 1:3000 in wash blot and added to the membrane for 1 hr. The membrane was washed in wash blot three times for 5 min. and developed using Sigmafast BCIP/NBT (5‐bromo‐4‐chloro‐3‐indolyl phosphate/nitro blue tetrazolium) tablets (Sigma). Blots were dried, scanned into TIFF files with Adobe Photoshop (San Jose, CA, USA), and quantitated with Scion Image. The specificity of anti‐3‐NT antibody was confirmed by preincubation of the antibody with free 3‐NT (10 mM) that revealed no non‐specific binding of the antibody, confirming our previous result [26].
Two‐dimensional gel electrophoresis
Two‐dimensional polyacrylamide gel electrophoresis was performed with a Bio‐Rad IEF Cell system using 110‐mm pH 3–10 immobilized pH gradients (IPG) strips and Criterion 8–16% resolving gels. IPG strips were actively rehydrated at 50 V 20°C followed by isoelectric focusing: 300 V for 2 hrs linear gradient, 1200 V for 4 hrs slow gradient, 8000 V for 8 hrs linear gradient and 8000 V for 10 hrs rapid gradient. Gel strips were equilibrated for 10 min. prior to second‐dimension separation in solution A (0.375M Tris‐HCl, pH 8.8 containing 6M urea (Bio‐Rad, Hercules, CA, USA), 2% (w/v) SDS, 20% (v/v) glycerol and 0.5% DTT (Bio‐Rad), followed by re‐equilibration for 10 min. in solution B (0.375M Tris‐HCl pH 8.8 containing 6 M urea, 2% (w/v) SDS, 20% (v/v glycerol, and 4.5% iodoacetamide (IA) (Bio‐Rad)). Control and EAD strips were placed on the 8–16% Criterion gels, unstained molecular standards were applied, and electrophoresis was performed at 200 V for 65 min. Gels were fixed in a solution containing 10% (v/v) methanol, 7% (v/v) acetic acid for 20 min. and stained overnight at room temperature with agitation in 50 ml of SYPRO Ruby gel stain (Bio‐Rad). Gels were then destained with 50 ml deionized water overnight.
Immunochemical detection
For immunoblotting analysis, electrophoresis was performed as stated previously, and gels were transferred to a nitrocellulose membrane. The membranes were blocked with 3% BSA in phosphate‐buffered saline containing 0.01% (w/v) sodium azide and 0.2% (v/v) Tween 20 (PBST) overnight at 4°C. The membranes were incubated with anti‐nitrotyrosine polyclonal antibody (3‐NT) (Sigma‐Aldrich), diluted 1:100 in wash blot for 2 hrs at room temperature with rocking. Following completion of the primary antibody incubation, the membranes were washed three times in wash blot for 5 min. each. An anti‐rabbit IgG alkaline phosphatase secondary antibody (Sigma) was diluted 1:3000 in wash blot and incubated with the membranes for 1 hr at room temperature. The membranes were washed in wash blot three times for 5 min. and developed using Sigmafast Tablets (BCIP/NBT substrate) (Sigma) to yield 2D Western blots.
Image analysis
Analysis of gel maps and membranes comparing protein levels and tyrosine nitration content between control and EAD IPL samples was performed with PDQuest image analysis software (Bio‐Rad). The immunoreactivity of the Western blot was normalized to the actual protein content as measured by the intensity of a SYPRO Ruby (Bio‐Rad) protein stain. Images from SYPRO Ruby‐stained gels, used to measure protein content, were obtained performed with a UV transilluminator (λex= 470 nm, λem= 618 nm, Molecular Dynamics, Sunnyvale, CA, USA). Nitrocellulose membranes were scanned with Adobe Photoshop on a Microtek Scanmaker 4900.
In‐gel trypsin digestion
Samples were prepared according to the method described by Thongboonkerd et al.[27]. Briefly, the protein spots were cut and removed from the gel with a clean razor blade. The gel pieces were placed into individual, clean 1.5 ml microcentrifuge tubes and kept overnight at –20°C. The gel pieces were thawed and washed with 0.1 M ammonium bicarbonate (NH4HCO3) (Sigma) for 15 min. at room temperature. Acetonitrile (Sigma) was added to the gel pieces and incubated for an additional 15 min. The liquid was removed and the gel pieces were allowed to dry. The gel pieces were rehydrated with 20 mM DTT (Bio‐Rad) in 0.1 M NH4HCO3 and incubated for 45 min. at 56°C. The DTT was removed and replaced with 55 mM IA (Bio‐Rad) in 0.1 M NH4HCO3 for 30 min. in the dark at room temperature. The liquid was drawn off and the gel pieces were incubated with 50 mM NH4HCO3 at room temperature for 15 min. Acetonitrile was added to the gel pieces for 15 min. at room temperature. All solvents were removed and the gel pieces were allowed to dry for 30 min. The gel pieces were rehydrated with addition of a minimal volume of 20 ng/μl modified trypsin (Promega, Madison, WI, USA) in 50 mM NH4HCO3. The gel pieces were chopped and incubated with shaking overnight (∼18 hrs) at 37°C.
Mass spectrometry
All mass spectra were obtained at the University of Louisville Mass Spectrometry Facility on a Bruker Autoflex MALDI TOF (matrix assisted laser desorption‐time of flight) mass spectrometer (Bruker Daltonic, Billerica, MA, USA) operated in the reflectron mode to generate peptide mass fingerprints. Peptides resulting from in‐gel digestion were analysed on a 384 position, 600 μm Anchor‐Chip™ Target (Bruker Daltonics, Bremen, Germany) and prepared according to AnchorChip recommendations (AnchorChip Technology, Rev. 2, Bruker Daltonics). Briefly, 1 μl of tryptic digest was mixed with 1 ml of alpha‐cyano‐4‐hydroxycinnamic acid (0.3 mg/ml in ethanol: acetone, 2:1 ratio) directly on the target and allowed to dry at room temperature. The sample spot was washed with 1 μl of 1% TFA solution for approximately 60 sec. The TFA droplet was gently blown off the sample spot with compressed air. The resulting diffuse sample spot was recrystallized (refocused) performed with 1 μl of a solution of ethanol:acetone: 0.1% TFA (6:3:1 ratio). Reported spectra are a summation of 100 laser shots. External calibration of the mass axis was used for acquisition and internal calibration performed with either trypsin autolysis ions or matrix clusters and was applied after acquisition for accurate mass determination.
Protein identification
Peptide mass fingerprinting was used to identify proteins from tryptic peptide fragments by utilizing the MASCOT search engine (http://www.matrixscience.com) based on the entire NCBI protein database. Database searches were conducted allowing for up to one missed trypsin cleavage and using the assumption that the peptides were monoisotopic, oxidized at methionione residues and carbamiodomethylated at cysteine residues. Mass tolerance of 100 ppm/g was the window of error allowed for matching the peptide mass values. Statistical comparisons of 3‐NT protein levels on 2D gels and corresponding matched anti‐3NT positive spots on 2D‐Western blots from EAD IPL samples and age‐matched control samples were performed using Student’s t‐test.
Enzyme assays (ATP synthase and glutamate dehydrogenase)
Mitochondrial ATP synthase activity was measured spectrophotometrically at 340 nm by coupling the production of ADP to the oxidation of NADPH via the pyruvate kinase and lactate dehydrogenase reaction (coupled assay) as described [28]. The reaction mixture (0.2 ml final volume) contained: 100 mM Tris (pH 8.0), 4 mM Mg‐ATP (substrate), 2 mM MgCl2, 50 mM KCl, 0.2 mM EDTA, 0.23 mM NADH, 1 mM phosphoenolpyruvate, 1.4 unit pyruvate kinase, 1.4 unit lactate dehydrogenase and about 25–50 μg proteins (brain homogenates), and was assayed at 30°C. The assay was carried out in a microtitre plate reader (Bio‐Tek Instrument Inc., Winooski, VT, USA).
Glutamate dehydrogenase catalyses the reversible conversion of α‐ketoglutarate to L‐glutamate, and its activity was determined by spectrophotometric methods to measure the oxidation of NADH to NAD+ at 340 nm in the presence of the ammonium ion. The reaction mixture consists of 90 mM triethanolamine HCl, 13 mM α‐ketoglutarate, 53 mM ammonium acetate, 0.06 mM β‐ NADH (substrate), 0.25 mM EDTA and 5 μl sample. The reaction was assayed at 25°C.
Statistical analysis
The data were analysed by Student’s t‐tests. According to Maurer et al.[29], generalized statistical tests applicable to proteomics data are unavailable, in contrast to microarray data. Because of the small number of proteins identified in this study, compared to the large quantities of genes analysed by microarrays, a value of p < 0.05 was considered significant. Similarly, after protein identification using the NCBI database, a MOWSE score of over 61 was considered significant. Probability‐based MOWSE scores were estimated by comparing search results against estimated random match population and were reported as –10 * Log10 (P), where P is the probability that the identification of the protein is not correct. All protein identifications were in the expected size and pI range based on position in the gel.
Results
The total level of nitrated proteins in the IPL of EAD patients yielded a significant 18% increase compared with age‐matched controls (Fig. 1). Using proteomics analysis, 8 proteins were significantly nitrated (p < 0.05) in EAD IPL according to PDQuest software analysis of 2D gels (Fig. 2A and B) and 2D Western blots (Fig. 3A and B) probed appropriately with anti‐3‐NT antibody. These excessively nitrated brain proteins compared with control IPL include peroxiredoxin 2 (Prx2), triose phosphate isomerase (TPI), glutamate dehydrogenase (GDH), neuropolypeptide h3, phosphoglycerate mutase 1 (PGM1), H+ transporting ATPase, alpha enolase, and fructose 1,6‐bisphosphate aldolase (ALDO1). Table 2 provides the proteomics results from which these proteins were identified.
Figure 1.
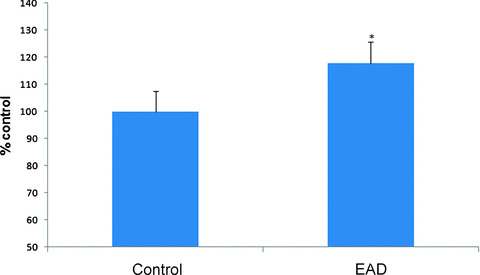
3‐Nitrotyrosine levels in inferior parietal lobule indexed in early Alzheimer’s disease (EAD) and age‐matched control. Mean ± S.E.M. N = 4, P < 0.0.5.
Figure 2.
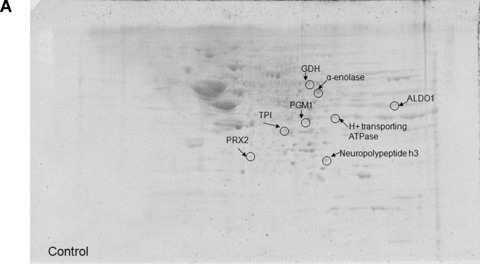
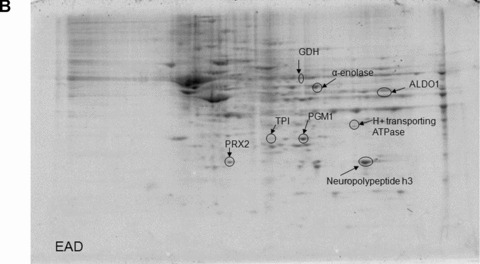
Representative 2D gel images of IPL proteins from age‐matched control (A) and early Alzheimer’s disease (EAD) subjects (B).
Figure 3.

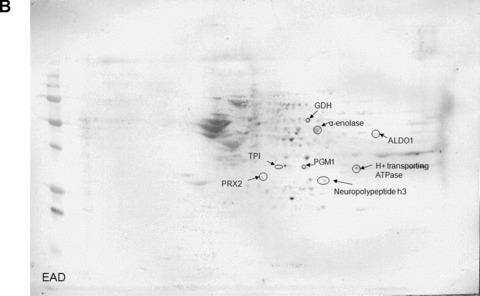
Representative 2D Western blot of inferior parietal lobule probed with anti‐3‐nitrotyrosine for control (A) and early Alzheimer’s disease (EAD) (B).
Table 2.
Proteomic identification of nitrated proteins found in EAD IPL
| gI accessioning number | Name | Mowse score | pI | Apparent molecular weight (Mr) | # peptides matched | Protein nitration (% control) | Probability |
|---|---|---|---|---|---|---|---|
| gi|32189392 | Peroxiredoxin 2 (Prx2) | 135 | 5.67 | 21,918 | 11/34 | 187 ± 0.15 | <0.05 |
| gi|4507645 | Triose phosphate isomerase (TPI) | 74 | 6.51 | 26,807 | 6/18 | 463 ± 63.5 | <0.02 |
| gi|4885281 | Glutamate dehydrogenase (GDH) | 112 | 7.66 | 61,701 | 14/40 | 376 ± 29.1 | <0.05 |
| gi|913159 | Neuropolypeptide h3 | 104 | 7.42 | 21,027 | 9/36 | 23.5 ± 0.04* | <0.02 |
| gi|4505753 | Phosphoglycerate mutase 1 (PGM) | 91 | 6.75 | 28,769 | 9/32 | 230 ± 4.20 | <0.05 |
| gi|4502317 | H+ transporting ATPase | 83 | 7.71 | 26,186 | 7/13 | 84.8 ± 0.05* | <0.04 |
| gi|4503571 | α‐Enolase | 191 | 6.99 | 47,350 | 20/48 | 516 ± 25.1 | <0.01 |
| gi|4930167 | Fructose 1,6‐bisphosphate aldolase (ALDO1) | 80 | 8.39 | 39,720 | 7/18 | 199 ± 21.5 | <0.04 |
*With an n= 4, if one blot has a particularly less intense protein spot for a statistically significant protein spot, this can cause an overall lower level of protein nitration. The overall % protein nitration is based on the average of protein nitration levels of all statistically significant protein spots. Ideally, one should have a greater n to avoid this potential problem, but samples from EAD are extremely rare, so we are fortunate just to have n= 4.
Because our earlier research demonstrated that oxidatively modified proteins generally are dysfunctional [19, 30, 31, 32, 33, 34, 35], the activities of a subset of identified nitrated proteins for IPL of EAD subjects were determined. ATP synthase activity was measured in EAD IPL and was 30% lower in EAD IPL compared with age‐matched controls (Fig. 4). ATP synthase also is lower in brains from subjects with MCI, and these results are consistent with the notion of loss of enzymatic activity of oxidatively modified proteins [30, 32]. Glutamate dehydrogenase activity also was reduced by 40% in EAD IPL compared with age‐matched controls (Fig. 5). Iwatsuji et al. reported lowered GDH activity in lymphocytes from AD patients compared with control [36].
Figure 4.
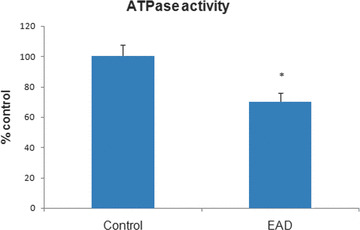
Activity of ATP synthase in early Alzheimer’s disease (EAD) inferior parietal lobule compared with age‐matched control. Bars represent mean ± S.E.M, *P < 0.05; n= 4 for each group. ATP synthase activity is significantly decreased in EAD samples.
Figure 5.
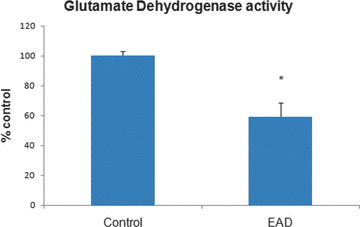
Reduced glutamate dehydrogenase activity in early Alzheimer’s disease (EAD) inferior parietal lobule compared with control. Bars represent mean ± S.E.M, *P < 0.05; n= 4 for each group.
Discussion
Several proteins were found to be significantly nitrated in EAD IPL. They are involved in antioxidant defence, energy metabolism, lipid asymmetry, and ATP production. This discussion has been written by categorizing these oxidatively modified proteins by function. In this manner, it is easier to envisage how oxidative modification can greatly impair protein function and how this can correlate with AD pathology.
Antioxidant defence
Peroxiredoxins are peroxidases that serve in an antioxidant capacity by reducing hydrogen peroxide. There are six forms of peroxiredoxin. Prx 1 ‐2, ‐3, ‐4 and ‐5 use thioredoxin as an electron donor, whereas Prx‐6 uses glutathione. Prx2 (or thio‐specific antioxidant), only expressed in neurons, blocks apoptosis by preventing the opening of the mitochondrial permeability pore [37] and thereby promotes neuronal cell survival [38, 39]. Overexpression of Prx2 in cell lines ameliorates apoptotic effects induced by oxidants [40]. Prx2, which can be neuroprotective, is increased in PD [41] as well as AD and Down syndrome [42, 43]. Prx2 also reduces peroxynitrite to detoxify RNS [44]. Loss of protein function hampers peroxidase activity and would be predicted to yield an increase in cytotoxic RNS.
We hypothesize that these findings have relevance to the emerging concepts of redox regulation of cellular stress response (vitagenes) in response to oxidative and nitrosative stress [45, 46]. In particular, the family of peroxiredoxins, coupled to thiol homeostasis, could exert a neuroprotective role in age‐related neurodegenerative disorders such as AD, and may be a promising, novel therapeutic target for this and other age‐related neurodegenerative disorders.
Metabolic enzymes
As noted above, TPI is a glycolytic enzyme that catalyses production of glyceraldehyde‐3‐phosphate (G3P), which is required to continue glycolysis to produce ATP. In AD brain, TPI is oxidatively modified [2, 3], which can affect ATPases, ion motive pumps and potential gradients.
Alpha enolase hydrolyses 2‐phosphoglycerate to phosphoenolpyruvate in glycolysis and impairment of this enzyme can greatly affect ATP production. Reduced enzyme activity of α‐enolase has been previously established in brain from subjects with MCI [47] and late AD [2, 48]. Altered energy metabolism is a common motif in neurodegenerative diseases as α‐enolase also is oxidatively modified in MCI [47], late AD [31, 49], PD [50], Huntington’s disease [33] and amyotrophic lateral sclerosis [51]. Notably, α‐enolase is the only nitrated protein to overlap MCI, EAD and late AD [2, 31, 47]. This observation supports the notion that changes in energy metabolism need to be investigated further to gain more knowledge about AD pathogenesis and potentially offer targets for therapeutic intervention of AD.
PGM1 catalyses the interconversion of 3‐phosphoglycerate to 2‐phosphoglycerate, leading to a second equivalent of ATP produced in glycolysis. Enzyme activity is decreased in AD [52], which could result in protein dysfunction and increased glycolytic intermediates, reduced pyruvate production and decreased ATP production. PGM1 is oxidatively modified in rat brain treated with Aβ(1–42) and AD hippocampus. In the gracile axonal dystrophy mouse brain in which Aβ is deposited, PGM1 is significantly increased, which contributes to the notion of reduced energy metabolism in neurodegenerative diseases [53, 54].
ALDO1 cleaves fructose1,6 ‐bisphosphate into dihydroxyacetone phosphate (DHAP) and G3P in glycolysis. Levels of ALDO1 are significantly decreased in AD hippocampus [4] and this enzyme is oxidatively modified in the olfactory bulb of aged mice [55]. Getchell et al. reported increased 3‐NT immunoreactivity in olfactory bulbs of late AD patients compared with age‐matched controls [56]. A diminished sense of smell (olfaction) is observed in some AD patients, as in the case of aging and several age‐related neurodegenerative diseases [57, 58]. ALDO1 and TPI deficiencies are associated with hemolytic anaemia resulting in mitochondrial myopathy [59, 60, 61]. Enzyme activity is reduced and impairment can cause increased levels of fructose 1,6‐ bisphosphate, inhibition of complete glycolysis, and ATP depletion.
Lipid asymmetry
Neuropolypeptide h3, also known as hippocampal cholinergic neurostimulating peptide, phosphatidylethanolamine binding protein (PEBP), or Raf kinase inhibitor protein, plays a role in cholinergic process by upregulating the enzyme, choline acetyltransferase. As a phosphatidylethanolamine binding protein, PEBP is important in phospholipid asymmetry. A signal for initiation of apoptosis is phophatidylserine exposure on the outer leaflet of the membrane lipid bilayer. Neuropolypeptide h3 is nitrated in late AD [2] and modified by the lipid peroxidation product, HNE, in synaptosomes treated with Aβ(1–42) [62]. Loss of PEBP may affect lipid asymmetry as loss of activity is observed in AD [63].
ATP production
Glutamate dehydrogenase (GDH), located in the mitochondrial matrix, catalyses the reversible deamination of glutamate to α‐ketoglutarate, ammonium and NAD.+ This α‐ketoglurate can be channeled into the Kreb’s cycle to stimulate ATP production. GDH is important in preventing accumulation of extracellular glutamate. Indeed, loss of function of GDH can cause excess glutamate, which in the brain leads to excitotoxicity [64]. NMDA receptors are stimulated by excess glutamate causing an increase in Ca2+ influx. Disruption of Ca2+ homeostasis can lead to changes in long‐term potentiation and consequently, diminished learning and memory. Altered homeostasis of Ca2+ also leads to cytochrome c release, as well as activation of calpain, a calcium‐sensitive protease. Such alterations would initiate neuronal death and may be important in AD [65, 66, 67].
As complex V in the mitochondrial electron transport chain, H+ transporting two‐sector ATPase (ATP synthase) catalyses ADP phosphorylation in ATP generation. Nitration of this protein can cause mitochondrial complex disturbance. As noted above, ATP synthase expression [68] and activity are decreased in AD [69, 70, 71, 72]. H+ transporting two‐sector ATPase is oxidized in synaptosomes treated with the peptide Aβ(1–42) [62]. Reduction in enzyme activity in brain of subjects with EAD (Fig. 4), coupled with changes in the mitochondrial complex, can lead to electron leakage and reactive oxygen species production, potentiating oxidative stress in AD via mitochondrial dysfunction [73, 74, 75].
To our knowledge, this is the first report of elevated 3‐NT in brain of subjects with EAD. In conclusion, the identified nitrated proteins in EAD IPL play major roles in antioxidant defence and energy metabolism directly or indirectly linked to AD pathology. Comparative analysis of nitrated proteins between MCI, EAD, and late AD showed α‐enolase was the only protein nitrated in all three disease stages [2, 3, 31, 35, 47]. Aldolase, phosphoglycerate mutase 1, neuropolypeptide h3 and TPI are oxidatively modified in EAD and late AD brain [2, 31, 35, 47]. Our data suggest that protein nitration of these specific brain proteins could be a mechanism to trigger the further conversion of EAD to more advanced AD. A Venn diagram (Fig. 6) shows the relationship between nitrated proteins in MCI, EAD and late AD. Future studies using animal models of AD at different ages and different Aβ pathologies should help to further delineate mechanisms of AD pathogenesis and to perhaps lead to development of effective therapies to combat disease progression.
Figure 6.
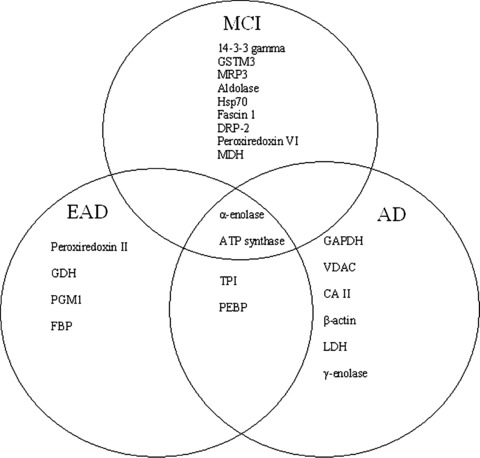
Venn diagram of functional relationship between nitrated proteins in mild cognitive impairment (MCI) early Alzheimer’s disease (EAD) and Alzheimer’s disease (AD).
Acknowledgements
This research was supported in part by grants from NIH to D.A.B. [AG‐10836; AG‐05119] and W.R.M. [AG‐05119 and 3P30 AG‐028383].
References
- 1. Petersen RC. Mild cognitive impairment: transition between aging and Alzheimer’s disease. Neurologia . 2000; 15: 93–101. [PubMed] [Google Scholar]
- 2. Castegna A, Thongboonkerd V, Klein JB, et al . Proteomic identification of nitrated proteins in Alzheimer’s disease brain. J Neurochem . 2003; 85: 1394–401. [DOI] [PubMed] [Google Scholar]
- 3. Sultana R, Poon HF, Cai J, et al . Identification of nitrated proteins in Alzheimer’s disease brain using a redox proteomics approach. Neurobiol Dis . 2006; 22: 76–87. [DOI] [PubMed] [Google Scholar]
- 4. Sultana R, Boyd‐Kimball D, Cai J, et al . Proteomics analysis of the Alzheimer’s disease hippocampal proteome. J Alzheimers Dis . 2007; 11: 153–64. [DOI] [PubMed] [Google Scholar]
- 5. Good PF, Hsu A, Werner P, et al . Protein nitration in Parkinson’s disease. J Neuropathol Exp Neurol . 1998; 57: 338–42. [DOI] [PubMed] [Google Scholar]
- 6. Casoni F, Basso M, Massignan T, et al . Protein nitration in a mouse model of familial amyotrophic lateral sclerosis: possible multifunctional role in the pathogenesis. J Biol Chem . 2005; 280: 16295–304. [DOI] [PubMed] [Google Scholar]
- 7. Urushitani M, Shimohama S. The role of nitric oxide in amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord . 2001; 2:71–81. [DOI] [PubMed] [Google Scholar]
- 8. Kanski J, Alterman MA, Schoneich C. Proteomic identification of age‐dependent protein nitration in rat skeletal muscle. Free Radic Biol Med . 2003; 35: 1229–39. [DOI] [PubMed] [Google Scholar]
- 9. Kanski J, Behring A, Pelling J, et al . Proteomic identification of 3‐nitrotyrosine‐containing rat cardiac proteins: effects of biological aging. Am J Physiol Heart Circ Physiol . 2005; 288:H371–81. [DOI] [PubMed] [Google Scholar]
- 10. Reynolds MR, Berry RW, Binder LI. Nitration in neurodegeneration: deciphering the “Hows”“nYs”. Biochemistry . 2007; 46: 7325–36. [DOI] [PubMed] [Google Scholar]
- 11. Anantharaman M, Tangpong J, Keller JN, et al . Beta‐amyloid mediated nitration of manganese superoxide dismutase: implication for oxidative stress in a APPNLH/NLH X PS‐1P264L/P264L double knock‐in mouse model of Alzheimer’s disease. Am J Pathol . 2006; 168: 1608–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jung O, Marklund SL, Xia N, et al . Inactivation of extracellular superoxide dismutase contributes to the development of high‐volume hypertension. Arterioscler Thromb Vasc Biol . 2007; 27: 470–7. [DOI] [PubMed] [Google Scholar]
- 13. MacMillan‐Crow LA, Thompson JA. Tyrosine modifications and inactivation of active site manganese superoxide dismutase mutant (Y34F) by peroxynitrite. Arch Biochem Biophys . 1999; 366: 82–8. [DOI] [PubMed] [Google Scholar]
- 14. Tangpong J, Cole MP, Sultana R, et al . Adriamycin‐mediated nitration of manganese superoxide dismutase in the central nervous system: insight into the mechanism of chemobrain. J Neurochem . 2007; 100: 191–201. [DOI] [PubMed] [Google Scholar]
- 15. Berlett BS, Friguet B, Yim MB, et al . Peroxynitrite‐mediated nitration of tyrosine residues in Escherichia coli glutamine synthetase mimics adenylylation: relevance to signal transduction. Proc Natl Acad Sci USA . 1996; 93: 1776–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gorg B, Qvartskhava N, Voss P, et al . Reversible inhibition of mammalian glutamine synthetase by tyrosine nitration. FEBS Lett . 2007; 581: 84–90. [DOI] [PubMed] [Google Scholar]
- 17. Good PF, Werner P, Hsu A, et al . Evidence of neuronal oxidative damage in Alzheimer’s disease. Am J Pathol . 1996; 149: 21–8. [PMC free article] [PubMed] [Google Scholar]
- 18. Reynolds MR, Reyes JF, Fu Y, et al . Tau nitration occurs at tyrosine 29 in the fibrillar lesions of Alzheimer’s disease and other tauopathies. J Neurosci . 2006; 26: 10636–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Butterfield DA, Reed TT, Perluigi M, et al . Elevated levels of 3‐nitrotyrosine in brain from subjects with amnestic mild cognitive impairment: implications for the role of nitration in the progression of Alzheimer’s disease. Brain Res . 2007; 1148: 243–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tohgi H, Abe T, Yamazaki K, et al . Alterations of 3‐nitrotyrosine concentration in the cerebrospinal fluid during aging and in patients with Alzheimer’s disease. Neurosci Lett . 1999; 269: 52–4. [DOI] [PubMed] [Google Scholar]
- 21. Sultana R, Reed T, Perluigi M, et al . Proteomic identification of nitrated brain proteins in amnestic mild cognitive impairment: a regional study. J Cell Mol Med . 2007; 11: 839–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grundman M, Petersen RC, Ferris SH, et al . Mild cognitive impairment can be distinguished from Alzheimer disease and normal aging for clinical trials. Arch Neurol . 2004; 61: 59–66. [DOI] [PubMed] [Google Scholar]
- 23. Markesbery WR, Schmitt FA, Kryscio RJ, et al . Neuropathologic substrate of mild cognitive impairment. Arch Neurol . 2006; 63: 38–46. [DOI] [PubMed] [Google Scholar]
- 24. Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease. Neurobiol Aging . 1997; 18: S1–2. [PubMed] [Google Scholar]
- 25. Braak H, Braak E. Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol . 1991; 82: 239–59. [DOI] [PubMed] [Google Scholar]
- 26. Sultana R, Butterfield DA. Oxidatively modified GST and MRP1 in Alzheimer’s disease brain: implications for accumulation of reactive lipid peroxidation products. Neurochem Res . 2004; 29: 2215–20. [DOI] [PubMed] [Google Scholar]
- 27. Thongboonkerd V, Luengpailin J, Cao J, et al . Fluoride exposure attenuates expression of Streptococcus pyogenes virulence factors. J Biol Chem . 2002; 277: 16599–605. [DOI] [PubMed] [Google Scholar]
- 28. Zheng J, Ramirez VD. Piceatannol, a stilbene phytochemical, inhibits mitochondrial F0F1‐ATPase activity by targeting the F1 complex. Biochem Biophys Res Commun . 1999; 261: 499–503. [DOI] [PubMed] [Google Scholar]
- 29. Maurer HH, Peters FT. Toward high‐throughput drug screening using mass spectrometry. Ther Drug Monit . 2005; 27: 686–8. [DOI] [PubMed] [Google Scholar]
- 30. Butterfield DA, Perluigi M, Sultana R. Oxidative stress in Alzheimer’s disease brain: new insights from redox proteomics. Eur J Pharmacol . 2006; 545: 39–50. [DOI] [PubMed] [Google Scholar]
- 31. Castegna A, Aksenov M, Thongboonkerd V, et al . Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part II: dihydropyrimidinase‐related protein 2, alpha‐enolase and heat shock cognate 71. J Neurochem . 2002; 82: 1524–32. [DOI] [PubMed] [Google Scholar]
- 32. Hensley K, Hall N, Subramaniam R, et al . Brain regional correspondence between Alzheimer’s disease histopathology and biomarkers of protein oxidation. J Neurochem . 1995; 65: 2146–56. [DOI] [PubMed] [Google Scholar]
- 33. Perluigi M, Poon HF, Maragos W, et al . Proteomic analysis of protein expression and oxidative modification in r6/2 transgenic mice: a model of Huntington disease. Mol Cell Proteomics. 2005; 4: 1849–61. [DOI] [PubMed] [Google Scholar]
- 34. Poon HF, Vaishnav RA, Getchell TV, et al . Quantitative proteomics analysis of differential protein expression and oxidative modification of specific proteins in the brains of old mice. Neurobiol Aging 2006; 27: 1010–19. [DOI] [PubMed] [Google Scholar]
- 35. Sultana R, Perluigi M, Butterfield DA. Redox proteomics identification of oxidatively modified proteins in Alzheimer’s disease brain and in vivo and in vitro models of AD centered around Abeta(1–42). J Chromatogr B Analyt Technol Biomed Life Sci . 2006; 833: 3–11. [DOI] [PubMed] [Google Scholar]
- 36. Iwatsuji K, Nakamura S, Kameyama M. Lymphocyte glutamate dehydrogenase activity in normal aging and neurological diseases. Gerontology . 1989; 35: 218–24. [DOI] [PubMed] [Google Scholar]
- 37. Kowaltowski AJ, Vercesi AE, Rhee SG, et al . Catalases and thioredoxin peroxidase protect Saccharomyces cerevisiae against Ca(2+)‐induced mitochondrial membrane permeabilization and cell death. FEBS Lett . 2000; 473: 177–82. [DOI] [PubMed] [Google Scholar]
- 38. Zhang P, Liu B, Kang SW, et al . Thioredoxin peroxidase is a novel inhibitor of apoptosis with a mechanism distinct from that of Bcl‐2. J Biol Chem . 1997; 272: 30615–8. [DOI] [PubMed] [Google Scholar]
- 39. Ichimiya S, Davis JG, O’Rourke DM, et al . Murine thioredoxin peroxidase delays neuronal apoptosis and is expressed in areas of the brain most susceptible to hypoxic and ischemic injury. DNA Cell Biol . 1997; 16: 311–21. [DOI] [PubMed] [Google Scholar]
- 40. Bae JY, Ahn SJ, Han W, et al . Peroxiredoxin I and II inhibit H(2)O(2)‐induced cell death in MCF‐7 cell lines. J Cell Biochem . 2007; 101: 1038–45. [DOI] [PubMed] [Google Scholar]
- 41. Basso M, Giraudo S, Corpillo D, et al . Proteome analysis of human substantia nigra in Parkinson’s disease. Proteomics . 2004; 4: 3943–52. [DOI] [PubMed] [Google Scholar]
- 42. Kim SH, Fountoulakis M, Cairns N, et al . Protein levels of human peroxiredoxin subtypes in brains of patients with Alzheimer’s disease and Down syndrome. J Neural Transm Suppl . 2001; 61: 223–35. [DOI] [PubMed] [Google Scholar]
- 43. Krapfenbauer K, Engidawork E, Cairns N, et al . Aberrant expression of peroxiredoxin subtypes in neurodegenerative disorders. Brain Res . 2003; 967: 152–60. [DOI] [PubMed] [Google Scholar]
- 44. Rhee SG, Yang KS, Kang SW, et al . Controlled elimination of intracellular H(2)O(2): regulation of peroxiredoxin, catalase, and glutathione peroxidase via post‐translational modification. Antioxid Redox Signal . 2005; 7: 619–26. [DOI] [PubMed] [Google Scholar]
- 45. Calabrese V, Guagliano E, Sapienza M, et al . Redox regulation of cellular stress response in aging and neurodegenerative disorders: role of vitagenes. Neurochem Res . 2007; 32: 757–73. [DOI] [PubMed] [Google Scholar]
- 46. Calabrese V, Sultana R, Scapagnini G, et al . Nitrosative stress, cellular stress response, and thiol homeostasis in patients with Alzheimer’s disease. Antioxid Redox Signal . 2006; 8: 1975–86. [DOI] [PubMed] [Google Scholar]
- 47. Butterfield DA, Poon HF, St Clair D, et al . Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: insights into the development of Alzheimer’s disease. Neurobiol Dis . 2006; 22: 223–32. [DOI] [PubMed] [Google Scholar]
- 48. Newman SF, Sultana R, Perluigi M, et al . An increase in S‐glutathionylated proteins in the Alzheimer’s disease inferior parietal lobule, a proteomics approach. J Neurosci Res . 2007; 85: 1506–14. [DOI] [PubMed] [Google Scholar]
- 49. Sultana R, Perluigi M, Butterfield DA. Protein oxidation and lipid peroxidation in brain of subjects with Alzheimer’s disease: insights into mechanism of neurodegeneration from redox proteomics. Antioxid Redox Signal . 2006; 8: 2021–37. [DOI] [PubMed] [Google Scholar]
- 50. Poon HF, Frasier M, Shreve N, et al . Mitochondrial associated metabolic proteins are selectively oxidized in A30P alpha‐synuclein transgenic mice–a model of familial Parkinson’s disease. Neurobiol Dis . 2005; 18: 492–8. [DOI] [PubMed] [Google Scholar]
- 51. Ekegren T, Hanrieder J, Aquilonius SM, et al . Focused proteomics in post‐mortem human spinal cord. J Proteome Res . 2006; 5: 2364–71. [DOI] [PubMed] [Google Scholar]
- 52. Iwangoff P, Armbruster R, Enz A, et al . Glycolytic enzymes from human autoptic brain cortex: normal aged and demented cases. Mech Ageing Dev . 1980; 14: 203–9. [DOI] [PubMed] [Google Scholar]
- 53. Bowling AC, Beal MF. Bioenergetic and oxidative stress in neurodegenerative diseases. Life Sci . 1995; 56: 1151–71. [DOI] [PubMed] [Google Scholar]
- 54. Castegna A, Thongboonkerd V, Klein J, et al . Proteomic analysis of brain proteins in the gracile axonal dystrophy (gad) mouse, a syndrome that emanates from dysfunctional ubiquitin carboxyl‐terminal hydrolase L‐1, reveals oxidation of key proteins. J Neurochem . 2004; 88: 1540–6. [DOI] [PubMed] [Google Scholar]
- 55. Vaishnav RA, Getchell ML, Poon HF, et al . Oxidative stress in the aging murine olfactory bulb: redox proteomics and cellular localization. J Neurosci Res . 2007; 85: 373–85. [DOI] [PubMed] [Google Scholar]
- 56. Getchell TV, Krishna NS, Dhooper N, et al . Human olfactory receptor neurons express heat shock protein 70: age‐related trends. Ann Otol Rhinol Laryngol . 1995; 104: 47–56. [DOI] [PubMed] [Google Scholar]
- 57. Doty RL, Perl DP, Steele JC, et al . Olfactory dysfunction in three neurodegenerative diseases. Geriatrics . 1991; 46: 47–51. [PubMed] [Google Scholar]
- 58. Royall DR, Chiodo LK, Polk MS, et al . Severe dysosmia is specifically associated with Alzheimer‐like memory deficits in nondemented elderly retirees. Neuroepidemiology . 2002; 21: 68–73. [DOI] [PubMed] [Google Scholar]
- 59. Eber SW, Pekrun A, Bardosi A, et al . Triosephosphate isomerase deficiency: haemolytic anaemia, myopathy with altered mitochondria and mental retardation due to a new variant with accelerated enzyme catabolism and diminished specific activity. Eur J Pediatr .1991; 150: 761–6. [DOI] [PubMed] [Google Scholar]
- 60. Zanella A, Mariani M, Colombo MB, et al . Triosephosphate isomerase deficiency: 2 new cases. Scand J Haematol . 1985; 34: 417–24. [DOI] [PubMed] [Google Scholar]
- 61. Kishi H, Mukai T, Hirono A, et al . Human aldolase A deficiency associated with a hemolytic anemia: thermolabile aldolase due to a single base mutation. Proc Natl Acad Sci USA . 1987; 84: 8623–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Boyd‐Kimball D, Castegna A, Sultana R, et al . Proteomic identification of proteins oxidized by Abeta(1–42) in synaptosomes: implications for Alzheimer’s disease. Brain Res . 2005; 1044: 206–15. [DOI] [PubMed] [Google Scholar]
- 63. George AJ, Holsinger RM, McLean CA, et al . Decreased phosphatidylethanolamine binding protein expression correlates with Abeta accumulation in the Tg2576 mouse model of Alzheimer’s disease. Neurobiol Aging . 2006; 27: 614–23. [DOI] [PubMed] [Google Scholar]
- 64. Facheris M, Beretta S, Ferrarese C. Peripheral markers of oxidative stress and excitotoxicity in neurodegenerative disorders: tools for diagnosis and therapy? J Alzheimers Dis . 2004; 6: 177–84. [DOI] [PubMed] [Google Scholar]
- 65. Beal MF. Role of excitotoxicity in human neurological disease. Curr Opin Neurobiol . 1992; 2: 657–62. [DOI] [PubMed] [Google Scholar]
- 66. Browne SE, Beal MF. Oxidative damage and mitochondrial dysfunction in neurodegenerative diseases. Biochem Soc Trans . 1994; 22:1002–6. [DOI] [PubMed] [Google Scholar]
- 67. Tritschler HJ, Packer L, Medori R. Oxidative stress and mitochondrial dysfunction in neurodegeneration. Biochem Mol Biol Int . 1994; 34: 169–81. [PubMed] [Google Scholar]
- 68. Sergeant N, Wattez A, Galvan‐valencia M, et al . Association of ATP synthase alpha‐chain with neurofibrillary degeneration in Alzheimer’s disease. Neuroscience . 2003; 117: 293–303. [DOI] [PubMed] [Google Scholar]
- 69. Chen X, Stern D, Yan SD. Mitochondrial dysfunction and Alzheimer’s disease. Curr Alz Res . 2006; 3: 515–20. [DOI] [PubMed] [Google Scholar]
- 70. Hauptmann S, Keil U, Scherping I, et al . Mitochondrial dysfunction in sporadic and genetic Alzheimer’s disease. Exp Gerontol . 2006; 41: 668–73. [DOI] [PubMed] [Google Scholar]
- 71. Kim SH, Vlkolinsky R, Cairns N, et al . Decreased levels of complex III core protein 1 and complex V beta chain in brains from patients with Alzheimer’s disease and Down syndrome. Cell Mol Life Sci . 2000; 57: 1810–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Reix S, Mechawar N, Susin SA, et al . Expression of cortical and hippocampal apoptosis‐inducing factor (AIF) in aging and Alzheimer’s disease. Neurobiol Aging . 2007; 28: 351–6. [DOI] [PubMed] [Google Scholar]
- 73. Mancuso M, Coppede F, Migliore L, et al . Mitochondrial dysfunction, oxidative stress and neurodegeneration. J Alzheimers Dis . 2006; 10: 59–73. [DOI] [PubMed] [Google Scholar]
- 74. Petrozzi L, Ricci G, Giglioli NJ, et al . Mitochondria and neurodegeneration. Biosci Rep . 2007; 27: 87–104. [DOI] [PubMed] [Google Scholar]
- 75. Schapira AH. Oxidative stress and mitochondrial dysfunction in neurodegeneration. Curr Opin Neurol . 1996; 9: 260–4. [DOI] [PubMed] [Google Scholar]