

Abstract
In this study, we investigated the effect of the xanthine oxidase (XO) inhibitor, allopurinol (ALP), on cardiac dysfunction, oxidative‐nitrosative stress, apoptosis, poly(ADP‐ribose) polymerase (PARP) activity and fibrosis associated with diabetic cardiomyopathy in mice. Diabetes was induced in C57/BL6 mice by injection of streptozotocin. Control and diabetic animals were treated with ALP or placebo. Left ventricular systolic and diastolic functions were measured by pressure–volume system 10 weeks after established diabetes. Myocardial XO, p22phox, p40phox, p47phox, gp91phox, iNOS, eNOS mRNA and/or protein levels, ROS and nitrotyrosine (NT) formation, caspase3/7 and PARP activity, chromatin fragmentation and various markers of fibrosis (collagen‐1, TGF‐β, CTGF, fibronectin) were measured using molecular biology and biochemistry methods or immunohistochemistry. Diabetes was characterized by increased myocardial, liver and serum XO activity (but not expression), increased myocardial ROS generation, p22phox, p40phox, p47phox, p91phox mRNA expression, iNOS (but not eNOS) expression, NT generation, caspase 3/7 and PARP activity/expression, chromatin fragmentation and fibrosis (enhanced accumulation of collagen, TGF‐β, CTGF and fibronectin), and declined systolic and diastolic myocardial performance. ALP attenuated the diabetes‐induced increased myocardial, liver and serum XO activity, myocardial ROS, NT generation, iNOS expression, apoptosis, PARP activity and fibrosis, which were accompanied by improved systolic (measured by the evaluation of both load‐dependent and independent indices of myocardial contractility) and diastolic performance of the hearts of treated diabetic animals. Thus, XO inhibition with ALP improves type 1 diabetes‐induced cardiac dysfunction by decreasing oxidative/nitrosative stress and fibrosis, which may have important clinical implications for the treatment and prevention of diabetic cardiomyopathy and vascular dysfunction.
Keywords: oxidative stress, diabetic cardiomyopathy, iNOS, peroxynitrite, fibrosis
Introduction
Cardiovascular complications are the most common cause of morbidity and mortality in diabetic patients. The presence of myocardial left ventricular dysfunction (both diastolic and later systolic one) independent of atherosclerosis, coronary artery disease in diabetes, defined as ‘diabetic cardiomyopathy’, has been well documented in both human beings and animals [1, 2]. The mechanisms of diabetic cardiac dysfunction are multiple and may involve increased oxidative/nitrosative stress [3, 4, 5, 6], and activation of its downstream effector pathways (e.g. poly(ADP‐ribose) polymerase (PARP)) [7, 8], apoptosis [3, 9, 10], changes in the composition of extracellular matrix with enhanced cardiac fibrosis and increased inflammation [11, 12].
An increasing number of researchers during the past decade have suggested that xanthine oxidase (XO)‐derived superoxide generation plays an important role in various forms of ischaemic and other types of tissue and vascular injuries, inflammatory diseases and chronic heart failure ([13, 14, 15, 16, 17]; reviewed in [18, 19, 20]). The XO inhibitor allopurinol (ALP) and its active metabolite oxypurinol showed multitude of beneficial effects in the treatment of these conditions both in experimental animal models and in small‐scale human clinical trials [20]. In this study, we tested the effect of ALP on cardiac dysfunction, oxidative‐nitrosative stress, apoptosis, PARP activity and fibrosis associated with diabetic cardiomyopathy using a mouse model of type 1 diabetes.
| Gene | Forward primer | Reverse primer |
|---|---|---|
| iNOS | ATTCACAGCTCATCCGGTACG | GGATCTTGACCATCAGCTTGC |
| eNOS | CATTTTCGGACTCACATTGCG | TTGGTCAACCGAACGAAGTG |
| gp91phox | GACCATTGCAAGTGAACACCC | AAATGAAGTGGACTCCACGCG |
| p22phox | ATGGAGCGATGTGGACAGAAG | TAGATCACACTGGCAATGGCC |
| p40phox | TTCAAAGACCTGCTAGCGCT | TCCTTCTGTGTGACATGCAGC |
| p47phox | TTCCATCCCCAAATGCAAAG | TCAGATGCCCTAAAACCGGAG |
| XO | AAAGGACCAGACGATTGCTCC | TCACACGTTCCCCTTCAAAAC |
| Caspase3 | GGACTGTGGCATTGAGACAG | CGACCCGTCCTTTGAATTTC |
| TGFβ | TCTACAACCAACACAACCCGG | GAGCGCACAATCATGTTGGAC |
| Caspase 7 | ACGACATTGACGCTAATCCCC | TGCCATGCTCATTCAGGATG |
| Fibronectin | TGCAGTGACCAACATTGATCGC | AAAAGCTCCCGGATTCCATCC |
| CTGF | ACTATGATGCGAGCCAACTGC | TGTCCGGATGCACTTTTTGC |
| Collagen 1 | TGGCCTTGGAGGAAACTTTG | CTTGGAAACCTTGTGGACCAG |
| actin | TGCACCACCAACTGCTTAG | GGATGCAGGGATGATGTTC |
Materials and methods
Animals and treatment
All the animal protocols conformed to the National Institutes of Health (NIH) guidelines and were approved by the Institutional Animal Care and use Committee of the National Institute on Alcohol Abuse and Alcoholism. Diabetes was induced in 25 six‐ to eight‐week‐old male C57/BL6J mice (male, Jackson Laboratories, Bar Harbor, ME, USA) by intraperitoneal (i.p.) injection of streptozotocin (STZ, Sigma Chemicals, MO, USA) at the dose of 50 mg/kg dissolved in 100 mM citrate buffer pH 4.5 for 5 consecutive days. After 5 days, the blood glucose levels were measured using Ascensia Counter Glucometer (Bayer Health Care, NY, USA) by mandibular puncture blood sampling. Mice, which had blood sugar values >300 mg/dl, were used for the study. Diabetic mice were randomly segregated to two groups. One group served as diabetic control (D), whereas the other was treated with ALP (D + ALP) (Sigma) at 1 mmol/l in the drinking water for 10 weeks as described earlier [21]. The control groups (n= 25) received either vehicle/water (C) or ALP (C + ALP) alone for the same duration. After 10 weeks of treatment, animals were killed and tissues were harvested and processed for biochemical measurements.
Hemodynamic measurements using pressure–volume conductance system in mice
Left ventricular performance was analysed in mice anaesthetized with 2% isoflurane as previously described [22, 23]. All pressure–volume loop data were analysed using a cardiac pressure–volume analysis program (PVAN3.5; Millar Instruments, Houston, TX, USA), and the heart rate (HR), maximal left ventricular systolic pressure (LVSP), left ventricular end‐diastolic pressure (LVEDP), stroke volume (SV), maximal slope of systolic pressure increment (+dP/dt), diastolic decrement (–dP/dt), ejection fraction (EF), cardiac output (CO) and stroke work (SW) were computed. The relaxation time constant (τ), an index of diastolic function, was also calculated by two different methods (Weiss method: regression of log[pressure]versus time; Glantz method: regression of dP/dt versus pressure). All hemodynamic parameters were also determined under conditions of changing preload, elicited by transiently compressing the inferior vena cava (IVC) in ventilated anaesthetized animals following thoracotomy. Since +dP/dt may be preload‐dependent, in these animals pressure–volume (PV) loops recorded at different preloads were used to derive other useful systolic function indices that are not influenced by loading conditions and cardiac mass. These measures include the dP/dt‐end‐diastolic volume (EDV) relation (dP/d–EDV), the preload‐recruitable stroke work (PRSW), which represents the slope of the relation between stroke work and EDV and is independent of chamber size and mass, and the end‐systolic pressure–volume relation (ESPVR, E max) [23]. After the hemodynamic measurements were made, animals were killed and tissue samples collected.
Reverse transcription and real‐time PCR
Heart tissues were homogenized and total RNA was isolated using Trizol LS reagent (Invitrogen, CA, USA) according to manufacturer’s instruction. The RNA was treated with RNase‐free DNase (Ambion, TX, USA) to remove traces of genomic DNA contamination. Total RNA was then reverse‐transcribed to cDNA using the Super‐Script II (Invitrogen) and the target genes were amplified using the standard real‐time PCR kit (Applied Biosystems, Foster City, CA, USA). The amplification was performed in real‐time PCR system (Applied Biosystems) using the following conditions: initial denaturation at 95°C for 2 min., followed by 35 cycles were performed at 95°C for 30 sec. and 60°C for 30 sec. The fold induction/repression in gene expression by real‐time RT‐PCR was calculated after adjusting for actin using the formula 2−ΔΔCt as described earlier [22]. Primers used for amplification of respective genes are described below:
Determination of XO activity
Xanthine oxidase (XO) levels in the myocardial tissues were determined using Amplex‐Red assay kit obtained from Invitrogen.
Determination of PARP activity
PARP activities in the heart homogenates were performed using the universal colorimetric assay kit (Trevigen Inc., Gaithersburg, MD, USA).
Caspase 3/7 assay
Caspase 3/7 activities in the heart samples were performed using the Apo‐ONE caspase 3/7 kit (Promega, Madison, WI, USA). All samples were run in duplicate and the values were expressed as fluorescence units.
Cell death ELISA
The quantitative determination of cytoplasmic histone‐associated‐DNA‐fragments (mono and oligonucleosomes) due to in vivo cell death was measured using the sandwich ELISA kit according to the protocol supplied by the vendor (Roche Diagnostics, GmbH, Indianapolis, IN, USA).
Determination of myocardial 3‐nitrotyrosine (3‐NT) content
Quantification of 3‐NT levels in the heart tissue extracts were performed using the sandwich ELISA kit according to the manufacturer’s protocol (Hycult Biotechnology, Uden, The Netherlands).
Western immunoblot analysis
Heart tissues were homogenized in mammalian tissue protein extraction reagent (TPER, Pierce Biotechnology, IL, USA) supplemented with protease and phosphatase inhibitors (Roche, GmbH). Then the samples were kept on ice for 1 hr, followed by centrifugation at 13,000 rpm for 30 min. at 4°C. The supernatants were carefully collected and protein content was determined using Lowry assay kit (Bio‐Rad, CA, USA). Thirty μg of protein was resolved in 12% SDS‐PAGE and transferred to nitrocellulose membranes (GE Healthcare, Piscataway, NJ, USA). Blocking was performed for 2 hrs at room temperature with 5% non‐fat skimmed milk powder prepared in PBS containing 0.1% tween 20 (Sigma). After washing with PBST, membranes were probed with either mouse monoclonal iNOS (BD‐Biosciences, CA, USA), eNOS rabbit monoclonal (Cell Signaling Technology, MA, USA), cleaved caspase ‐3 antibody (Asp175) (Cell Signaling Technology, MA, USA) or XO monoclonal (AbCam, Cambridge, MA, USA) at 1:1000 dilution for 12 hrs at 4°C. After subsequent washing with PBST, the secondary antibody‐goat anti‐rabbit HRP or goat anti‐mouse HRP (Pierce Biotechnology) was incubated at RT for 1 hr. Then the membranes were developed using chemiluminescence detection kit (Super signal ‐west pico substrate, Pierce). To confirm uniform loading, membranes were stripped and re‐probed with β‐actin (Chemicon, CA, USA).
Immunohistochemistry
Hearts were fixed in 4% buffered formalin. After paraffin embedding, 5 μm sections were stained for 3‐NT antibody (mouse monoclonal, Cayman Chemicals, MI, USA) at 1:100 dilution for 12 hrs at 4°C. Then the sections were developed with Vectastain ABC – DAB kit (Vector Laboratories, Burlingame, CA, USA). Subsequently, the sections were counterstained with nuclear fast red for 3 min. Finally, the sections were dehydrated in ethanol, cleared in xylene, mounted and observed in the light microscope.
Determination of DHE fluorescence in the frozen heart sections
Myocardial ROS/superoxide generation was determined using dihydroethidium (DHE) staining using fluorescence microscope (Olympus IX‐81, PA, USA) in 5‐μm frozen myocardial sections as described earlier [24]. The relative DHE fluorescence was measured using the Slidebook software (SLIDEBOOK Inc, CO, USA).
Sirius red staining for collagen
Paraffin tissue sections were stained with picro‐sirius red satin solution for 1 hr at RT. Then slides were washed in two changes of acidified water (0.5% acetic acid) for 2 min. and excess of water was removed by blotting. Finally, the sections were dehydrated in 100% alcohol and cleared in xylene and mounted with cover glass.
Statistical analysis
Values were represented as mean ± S.E.M. Statistical evaluation of the data was determined by performing the ANOVA followed by Tukey’s post hoc test for multiple comparison. The analysis was performed using the statistical software package (GraphPad‐Prism‐4, CA, USA) and P < 0.05 was regarded statistically significant.
Results
Animals
Induction of diabetes in male C57BL/6 mice by STZ lead to reduction in the body weight with concomitant increase in the blood glucose levels in mice (body weight (g): C = 26.5 ± 0.3 versus D = 22.1 ± 0.2; P < 0.01); blood glucose (mg/dl): C = 107.9 ± 2.8 versus D = 415.8 ± 16.1; P < 0.01). ALP treatment for 10 weeks did not significantly alter either the body weight or blood glucose levels in diabetic animals (D = 22.1 ± 0.2 g versus D + ALP = 21.6 ± 0.2 g; D = 415.8 ± 16.1 mg/dl versus D + ALP = 413.0 ± 17.7 mg/dl, respectively). There were no significant changes in body weight or plasma glucose levels in mice treated with ALP alone or vehicle (C = 26.5 ± 0.3 g versus C + ALP = 26.2 ± 0.2 g; C = 107.9 ± 2.8 mg/dl versus C + ALP = 104.4 ± 2.2 mg/dl, respectively).
Cardiac function
Diabetes was characterized by impaired systolic left ventricular function evidenced by decreases in both load‐dependent (cardiac output, stroke work, LVSP, + dP/dt and EF), and load‐independent (Emax, PRSW and dP/dt‐EDV) indices of myocardial contractility (Fig. 1). Diabetes also impaired various indices of diastolic function (LVEDP, –dP/dt, tau and the slope of the EDPVR; Fig. 1), consistent with enhanced myocardial fibrosis observed in these animals (see later below and Fig. 8). ALP treatment significantly improved the diabetes‐induced systolic and diastolic dysfunctions (Fig. 1).
Figure 1.
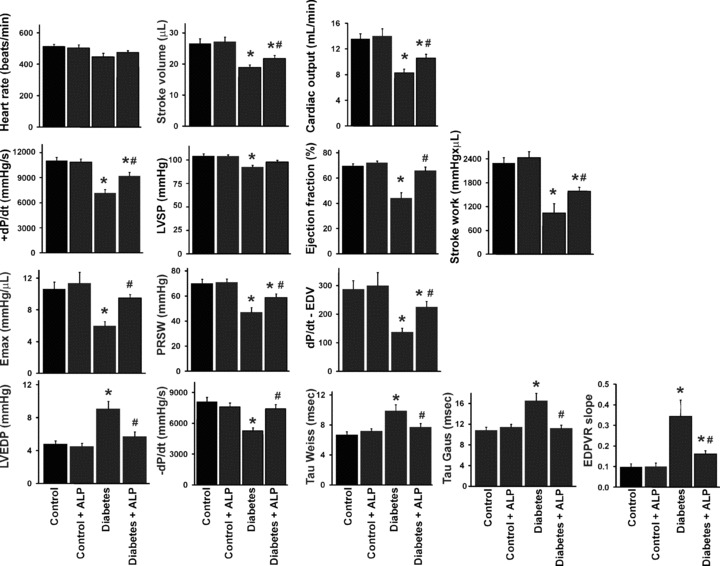
Effects of diabetes and allopurinol (ALP) on left ventricular function. Effect of diabetes and ALP on load‐dependent (cardiac output, stroke work, LVSP, + dP/dt and EF) and load‐ and/or heart rate‐independent (Emax, PRSW and dP/dt‐EDV) indices of myocardial contractility, diastolic function (LVEDP, –dP/dt, tau and the slope of the EDPVR), heart rate and stroke volume (n= 10–13/group; *P < 0.05 versus control; #P < 0.05 versus diabetes).
Figure 8.
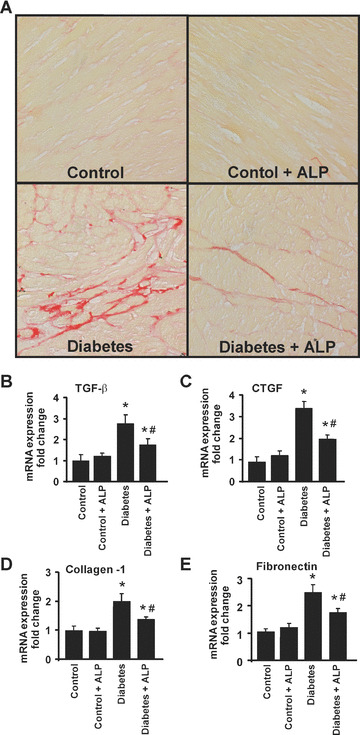
Effects of diabetes and allopurinol (ALP) on myocardial fibrosis and expression of pro‐fibrotic genes. (A) Shown are the representative light microscope images from three independent experiments for fibrosis when the collagen deposition was revealed by staining using Sirius red of paraffin embedded sections (400× magnification). Note the increased fibrosis in the diabetic myocardium (red), which is attenuated by ALP treatment. Real‐time quantitative RT‐PCR was performed in myocardium of the respective groups as indicated. (B) TGF‐β; (C) CTGF; (D) Collagen‐1; (E) Fibronectin. Note up‐regulation of pro‐fibrotic genes in hearts of diabetic mice, which attenuated by ALP treatment (n= 6; *P < 0.05 versus control; #P < 0.05 versus diabetes).
Effects of diabetes and ALP on myocardial, liver and serum XO expression and/or activity
Western blot analysis of the myocardial and liver tissue extracts revealed no change in the protein expression of XO in the diabetic mice compared with controls with or without ALP (Fig. 2A). Similar change was observed with the mRNA expression of XO in the myocardium (Fig. 1B). However, XO activity was markedly elevated in the liver, heart and serum of diabetic animals when compared with controls; and this increase was attenuated by ALP (Fig. 2C).
Figure 2.
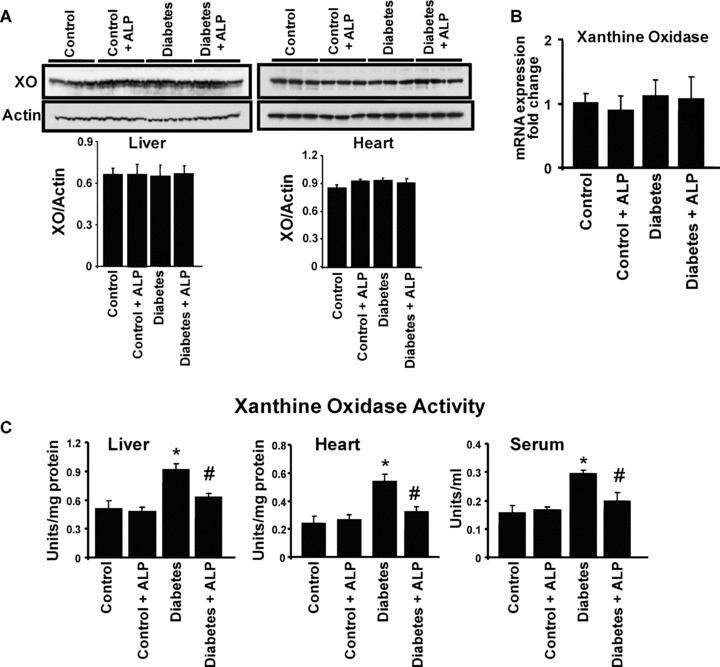
Effects of diabetes and allopurinol (ALP) on myocardial, liver and serum xanthine oxidase (XO) expression and/or activity. Mice were treated as described in the Methods section and 10 weeks later, animals were killed and hearts were excised. Then protein extracts were subjected to Western blot analysis for the determination of XO expression in the heart and liver tissues. (A) Western blot for XO expression in the liver and heart tissues. The bottom panel denotes the quantification of XO expression. The XO expression was unaltered in untreated or ALP treated diabetic mice (n= 3 in each group). (B) Denotes the mRNA expression of XO in the myocardial tissues in the respective groups as indicated (n= 6–8 in each group). (C) XO activity in liver, heart tissue homogenates and serum of control and diabetic animals with or without ALP treatment. Note the increased activity of XO in diabetic tissues and serum, which was suppressed upon ALP treatment (n= 5/group, *P < 0.05 versus control; #P < 0.05 versus diabetes).
Effects of diabetes and ALP on superoxide/ROS production in the myocardium of diabetic mice
XO‐induced superoxide/ROS generation was determined in the frozen section of myocardial tissues using the DHE fluorescence as described in the Methods section. Our observation revealed markedly increased DHE oxidation fluorescence in the diabetic myocardial tissues (Fig. 3A and B) when compared with myocardium from control mice (Fig. 3A and B). ALP attenuated increased superoxide/ROS generation in hearts of diabetic animals, but not in controls. (Fig. 3A and B).
Figure 3.
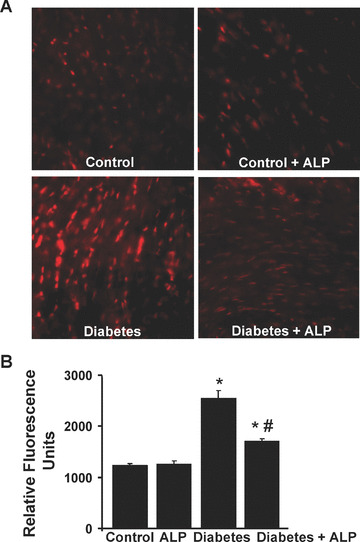
Effects of diabetes and allopurinol (ALP) on myocardial superoxide/ROS generation. (A) Shown are the representative images of DHE fluorescence in the respective myocardial sections (400× magnification). (B) Denotes the quantification of the DHE fluorescence using the Slidebook software (n= 6/group, *P < 0.05 versus control; #P < 0.05 versus diabetes).
Effects of diabetes and ALP on the mRNA expression of NADP(H) oxidase subunits
It has been demonstrated earlier that in diabetic vascular complications there is significant increase in the superoxide production attributed to the NADPH oxidase activation [25]. Therefore, we determined the mRNA expression of the NADPH oxidase subunits in the heart tissues and studied the effects of ALP treatment on these variables. The mRNA expression of p22phox (Fig. 4A), p40phox (Fig. 4B), p47phox (Fig. 4C) and gp91phox (Fig. 4D) were significantly elevated in the myocardial tissues of diabetic mice, compared with controls and were not affected by ALP treatment (Fig. 4).
Figure 4.
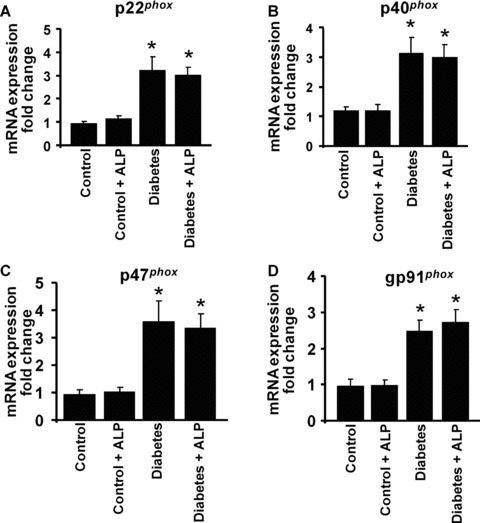
Effects of diabetes and allopurinol (ALP) on NADPH oxidase mRNA expression in the myocardial tissues. Total RNA was extracted from the myocardial tissues and real‐time quantitative RT‐PCR was performed to determine the expression of NADPH oxidase subunits. (A) mRNA expression of p22phox, (B) p40phox, (C) p47phox, (D) gp91phox, respectively, in the myocardial tissues as indicated. Note the enhanced expression of NADPH oxidase subunits in the diabetic mice, which is not affected by ALP treatment (n= 6–8/group; *P < 0.05 versus control; #P < 0.05 versus diabetes).
Effects of diabetes and ALP on myocardial eNOS and iNOS expressions
There was no change in the eNOS expression the hearts of either diabetic mice or diabetic mice treated with ALP (Fig. 5A). On the contrary, iNOS expression was markedly induced ∼2.5‐fold in diabetic hearts when compared with control mice (Fig. 5B), which was significantly attenuated by the ALP treatment.
Figure 5.
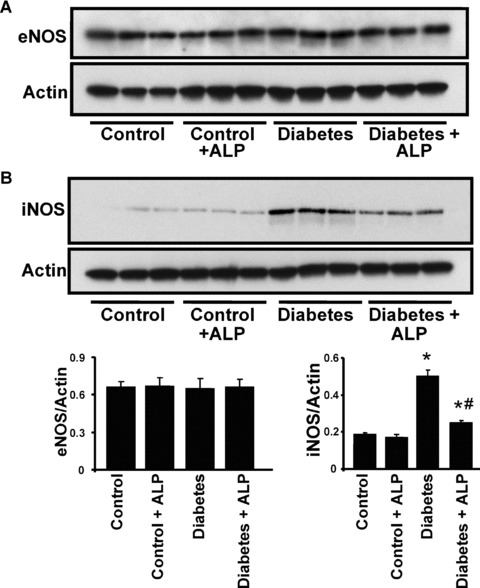
Effects of diabetes and allopurinol (ALP) on myocardial eNOS and iNOS expression. (A) Representative immunoblot revealing the eNOS protein expression in the myocardial tissues. The adjacent panel shows the quantification of the eNOS expression. (B) Western immunoblot demonstrating the iNOS expression in the myocardial tissues. The adjacent panel denotes the quantification of iNOS expression (n= 6; *P < 0.05 versus control; #P < 0.05 versus diabetes).
Effect of diabetes and ALP on myocardial 3‐NT accumulation
The myocardial 3‐NT accumulation (a foot‐print of peroxynitrite formation or more broadly nitrosative stress [26, 27, 28]) in the heart tissues was determined by immunohistochemical staining and quantitative ELISA. As shown in Fig. 6A, diffuse 3‐NT accumulation was significantly higher in the myocardium of diabetic mice compared with controls. ALP treatment of diabetic mice attenuated the 3‐NT accumulation in heart sections (Fig. 6A). Similar results were obtained using quantitative ELISA (Fig. 6B).
Figure 6.
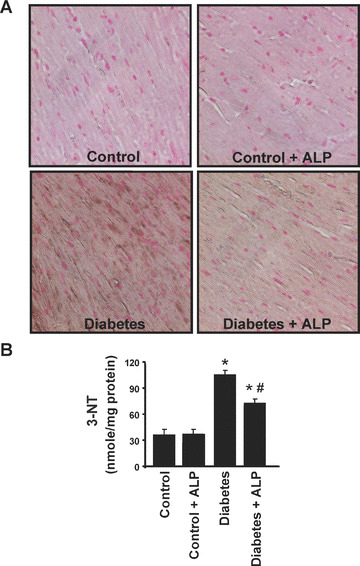
Effects of diabetes and allopurinol (ALP) on myocardial 3‐nitrotyrosine (NT) accumulation. (A) Shown are the representative images from three independent experiments for 3‐NT immunohistochemial staining in the paraffin sections of the respective myocardial tissues (400× magnification). Note the increased accumulation of 3‐NT in the diabetic myocardial sections, which is attenuated by ALP treatment. (B) Denotes the quantification of 3‐NT levels in the myocardial tissues by ELISA as described in the Methods section (n= 5/group; *P < 0.05 versus control; #P < 0.05 versus diabetes).
Effect of diabetes and ALP on myocardial apoptosis and PARP activity
Western blot analysis (Fig. 7A) and quantitative determination of caspase 3/7 activity by ELISA (Fig. 7B) revealed increased apoptosis in diabetic hearts compared with controls. ALP treatment of diabetic mice, but not controls, attenuated the caspase 3/7 activation (Fig. 7A and B). There were also markedly increased mRNA expressions of caspase 3 and 7 in diabetic hearts, which were attenuated by ALP treatment (Fig. 7C.) Myocardial chromatin fragmentation and PARP activity were also elevated in hearts from diabetic mice (Fig. 7D and E), which were attenuated by ALP treatment.
Figure 7.
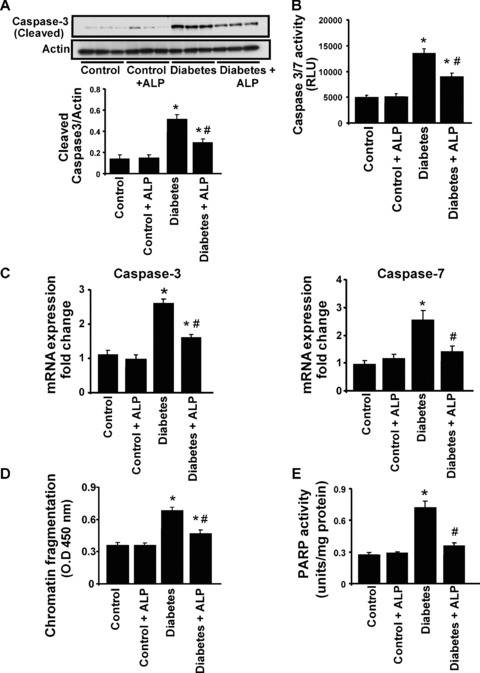
Effects of diabetes and allopurinol (ALP) on myocardial apoptosis and PARP activity. (A) Shown is the representative immunoblot depicting the cleaved caspase 3 expression in the myocardial tissues. The adjacent panel denotes the quantification of cleaved caspase 3 expression in the heart tissues (n= 6; *P < 0.05 versus control; #P < 0.05 versus diabetes. (B) Represents the quantification of caspase 3/7 activity in the myocardial tissues as described in the Methods section (n= 5/group; *P < 0.05 versus control; #P < 0.05 versus diabetes). (C) Shows the mRNA expression of caspase 3/7, respectively, in the myocardial tissues (n= 6; *P < 0.05 versus control; #P < 0.05 versus diabetes). (D) Quantitative determination of apoptosis by determination of chromatin condensation by ELISA (n= 5/group, *P < 0.05 versus control; #P < 0.05 versus diabetes). (E) PARP activity was determined in the myocardial tissues as described in the Methods section (n= 5/group, *P < 0.05 versus control; #P < 0.05 versus diabetes).
Effect of diabetes and ALP on myocardial fibrosis
Sirius red staining of the formalin fixed heart sections revealed marked interstitial fibrosis in the diabetic myocardium characterized by increased collagen accumulation, which was attenuated by ALP treatment (Fig. 8A). As shown in Fig. 8B–E, mRNA expression of transforming growth factor‐beta (TGF‐β; Fig. 8B), connective tissue growth factor (CTGF; Fig. 8C), collagen‐1 (Fig. 8D) and fibronectin (Fig. 8E) were all elevated in the diabetic heart tissues, when compared with the controls and were attenuated by ALP treatment.
Discussions
Complex changes in the mechanical, biochemical, structural and electrical properties of the heart, which may be responsible for the development of an early diastolic dysfunction and increased incidence of cardiac arrhythmias in diabetic patients, are characteristic features of diabetic cardiomyopathy. In spite of the accumulating knowledge obtained from different models of diabetes, the mechanism of diabetic cardiac dysfunction still remains elusive [1, 2].
The current study demonstrates that STZ‐induced diabetes in mice is associated with a marked depression of both systolic and diastolic function of the left ventricle. These results are consistent with earlier reports showing depressed cardiac function in different mouse [3, 7, 11, 29, 30] models of type 1 diabetes and compromised cardiac conductivity and/or performance reported in diabetic patients. The results presented here document that in STZ‐induced mouse model of type 1 insulin‐dependent diabetes, the impaired cardiac function is associated with increased myocardial XO activity, oxidative/nitrosative stress and fibrosis, which can be attenuated by pharmacological inhibition of the XO by ALP.
Multitude of experimental and clinical studies have suggested that increased sympathetic activity, activated cardiac renin‐angiotensin system, myocardial ischaemia/functional hypoxia and elevated circulating levels of glucose result in oxidative/nitrosative stress and inflammation, eventually culminating in cellular dysfunction and death in cardiomyocytes and endothelial cells (both apoptotic and necrotic) promoting increased remodelling and fibrosis, processes which play a critical role in the development of subsequent diabetic cardiomyopathy [3, 4, 5, 6, 7, 8, 9, 10, 11, 12]. Furthermore, increased oxidative/nitrosative stress may also lead to inactivation of key cardiac ion channels by oxidation, nitration and/or nitrosylation, thereby contributing to impaired repolarization reported in both diabetic animals as well as in human beings with type 1 diabetes.
Hyperglycaemia triggers oxidative stress via numerous mechanisms involving activation of the polyol pathway, glucose auto‐oxidation, alterations of cellular redox state, increased formation of diacylglycerol and the subsequent activation of protein kinase C, and accelerated nonenzymatic formation of advanced glycation end products. Superoxide anion appears to play a particularly important role in the pathogenesis of diabetic cardiovascular dysfunction, and this reactive oxidant was reported to activate many of the above‐mentioned pathways [6, 31]. The cellular sources of superoxide anion in diabetes are multiple and may include NAD(P)H and XO, the mitochondrial respiratory chain among many others [19, 20, 31]. Recent studies suggest that XO may play an important role in the generation of free radicals in diabetes. There is an elevation the plasma and liver XO levels in type 1 diabetic patients [32] and increased endothelial superoxide formation in aorta from alloxan‐induced diabetic rabbits can be blocked by the XO inhibitor ALP [32]. Diabetes also causes an increase of XO activity in the liver of rats, and XO is released from the liver of these animals [32]. Increased plasma XO activity in diabetic mice correlates with the degree of superoxide generation 2 weeks after the onset of diabetes, and can be normalized by pretreatment with XO inhibitors ALP or oxypurinol [33]. In type 1 diabetic patients, ALP treatment attenuated the degree of oxidative stress (haemoglobin glycation, glutathione oxidation and lipid peroxidation) [32], whereas in type 2 diabetic patients with mild hypertension, prolonged treatment with ALP resulted in significant improvements in peripheral endothelium‐dependent vasorelaxant function [34]. Consistently with these results, we found increased XO activity in the liver, myocardium and serum of diabetic mice, which could be normalized by ALP treatment. ALP treatment also attenuated the myocardial ROS generation in our mouse model of diabetes. In contrast, ALP had no effects on diabetes‐induced increased expression of various isoforms of NAD(P)H oxidases.
Hyperglycemia, through the activation of NF‐κB induces the increased ROS generation, which in turn favors the induction of iNOS, which can increase the generation of NO in the diabetic hearts [28]. Superoxide anion interacts with nitric oxide, forming the oxidant peroxynitrite (ONOO−), which attacks various biomolecules, leading to—among other things—the production of a modified amino acid, nitrotyrosine [28, 35]. Small amount of peroxynitrite may be generated in normal cells and may play various physiological regulatory functions (e.g. in signalling processes; for reviews see [36, 37, 38]). However, several lines of evidence support the pathogenetic role of excessive endogenous peroxynitrite formation in diabetic cardiovascular [6, 39, 40] and other complications [41] both in experimental animals and in human beings. For example, the degree of cell death and/or dysfunction correlates with levels of NT in endothelial cells, cardiomyocytes and fibroblasts from myocardial biopsies of diabetic patients [9], hearts of experimental diabetic rats or mice [3, 9, 29] and hearts perfused with high glucose concentrations [5]. The NT immunoreactivity is increased in the microvasculature of type 2 diabetic patients and correlates with fasting blood glucose, HbA1c, intracellular adhesion molecule, vascular cellular adhesion molecule and endothelial dysfunction [42]. Peroxynitrite has been reported to attack various biomolecules, leading to compromised cardiovascular function and cell death (both apoptotic and necrotic) in hearts and other tissues via multiple complex mechanisms [6, 27, 28]. One of these pathways involves DNA strand breakage and consequent activation of the nuclear enzyme PARP, which emerges as a major effector pathway in the development of various interrelated diabetic complications [8, 43], including cardiomyopathy [7]. Consistently with the importance of these pathways in the development of diabetic cardiomyopathy, we found increased iNOS (but not eNOS) expression, NT generation, apoptosis and PARP activity in diabetic heart accompanied by increased fibrosis. Remarkably, ALP treatment of diabetic mice attenuated not only the oxidative/nitrosative stress, PARP activation and apoptosis, but also fibrosis in diabetic hearts.
Based on the results of the current study, we conclude that the XO‐derived superoxide production contributes to the development of diabetic cardiomyopathy, and XO inhibition with ALP improves diabetes‐induced cardiac dysfunction by decreasing oxidative/nitrosative stress and the activation of its downstream effector pathways (e.g. PARP) and fibrosis. This coupled with the beneficial effects of ALP in various animal models of heart failure, also observed in small‐scale human clinical trials (reviewed in [20, 44]), is particularly encouraging from the therapeutic point of view forecasting multiple possible benefits of ALP treatment in diabetic patients.
Acknowledgements
This study was supported by Intramural Research Program of the NIH/NIAAA to P.P.
References
- 1. Fein FS. Diabetic cardiomyopathy. Diabetes Care . 1990; 13: 1169–79. [DOI] [PubMed] [Google Scholar]
- 2. Regan TJ, Ahmed S, Haider B, et al . Diabetic cardiomyopathy: experimental and clinical observations. N Engl J Med . 1994; 91: 776–8. [PubMed] [Google Scholar]
- 3. Kajstura J, Fiordaliso F, Andreoli AM, et al . IGF‐1 overexpression inhibits the development of diabetic cardiomyopathy and angiotensin II‐mediated oxidative stress. Diabetes . 2001; 50: 1414–24. [DOI] [PubMed] [Google Scholar]
- 4. Ceriello A. New insights on oxidative stress and diabetic complications may lead to a “causal” antioxidant therapy. Diabetes Care . 2003; 26: 1589–96. [DOI] [PubMed] [Google Scholar]
- 5. Ceriello A, Quagliaro L, D’Amico M, et al . Acute hyperglycemia induces nitrotyrosine formation and apoptosis in perfused heart from rat. Diabetes . 2002; 51: 1076–82. [DOI] [PubMed] [Google Scholar]
- 6. Pacher P, Szabo C. Role of peroxynitrite in the pathogenesis of cardiovascular complications of diabetes. Curr Opin Pharmacol . 2006; 6: 136–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pacher P, Liaudet L, Soriano FG, et al . The role of poly(ADP‐ribose) polymerase activation in the development of myocardial and endothelial dysfunction in diabetes. Diabetes . 2002; 51: 514–21. [DOI] [PubMed] [Google Scholar]
- 8. Pacher P, Szabo C. Role of poly(ADP‐ribose) polymerase‐1 activation in the pathogenesis of diabetic complications: endothelial dysfunction, as a common underlying theme. Antioxid Redox Signal . 2005; 7: 1568–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Frustaci A, Kajstura J, Chimenti C, et al . Myocardial cell death in human diabetes. Circ Res . 2000; 87: 1123–32. [DOI] [PubMed] [Google Scholar]
- 10. Fiordaliso F, Li B, Latini R, et al . Myocyte death in streptozotocin‐induced diabetes in rats in angiotensin II‐ dependent. Lab Invest . 2000; 80: 513–27. [DOI] [PubMed] [Google Scholar]
- 11. Westermann D, Rutschow S, Jager S, et al . Contributions of inflammation and cardiac matrix metalloproteinase activity to cardiac failure in diabetic cardiomyopathy: the role of angiotensin type 1 receptor antagonism. Diabetes . 2007; 56: 641–6. [DOI] [PubMed] [Google Scholar]
- 12. Westermann D, Van Linthout S, Dhayat S, et al . Cardioprotective and anti‐ inflammatory effects of interleukin converting enzyme inhibition in experimental diabetic cardiomyopathy. Diabetes. 2007; 56: 1834–41. [DOI] [PubMed] [Google Scholar]
- 13. Ekelund UE, Harrison RW, Shokek O, et al . Intravenous allopurinol decreases myocardial oxygen consumption and increases mechanical efficiency in dogs with pacing‐induced heart failure. Circ Res . 1999; 85: 437–45. [DOI] [PubMed] [Google Scholar]
- 14. Cappola TP, Kass DA, Nelson GS, et al . Allopurinol improves myocardial efficiency in patients with idiopathic dilated cardiomyopathy. Circulation . 2001; 104: 2407–11. [DOI] [PubMed] [Google Scholar]
- 15. Amado LC, Saliaris AP, Raju SV, et al . Xanthine oxidase inhibition ameliorates cardiovascular dysfunction in dogs with pacing‐induced heart failure. J Mol Cell Cardiol . 2005; 39: 531–6. [DOI] [PubMed] [Google Scholar]
- 16. Minhas KM, Saraiva RM, Schuleri KH, et al . Xanthine oxidoreductase inhibition causes reverse remodeling in rats with dilated cardiomyopathy. Circ Res . 2006; 98: 271–9. [DOI] [PubMed] [Google Scholar]
- 17. Saliaris AP, Amado LC, Minhas KM, et al . Chronic allopurinol administration ameliorates maladaptive alterations in Ca2 + cycling proteins and beta‐adrenergic hyporesponsiveness in heart failure. Am J Physiol Heart Circ Physiol . 2007; 292: H1328–35. [DOI] [PubMed] [Google Scholar]
- 18. Berry CE, Hare JM. Xanthine oxidoreductase and cardiovascular disease: molecular mechanisms and pathophysiological implications. J Physiol . 2004; 555: 589–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ungvari Z, Gupte SA, Recchia FA, et al . Role of oxidative‐nitrosative stress and downstream pathways in various forms of cardiomyopathy and heart failure. Curr Vasc Pharmacol . 2005; 3: 221–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pacher P, Nivorozhkin A, Szabo C. Therapeutic effects of xanthine oxidase inhibitors: renaissance half a century after the discovery of allopurinol. Pharmacol Rev . 2006; 58: 87–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stull LB, Leppo MK, Szweda L, et al . Chronic treatment with allopurinol boosts survival and cardiac contractility in murine postischemic cardiomyopathy. Circ Res . 2004; 95: 1005–11. [DOI] [PubMed] [Google Scholar]
- 22. Mukhopadhyay P, Batkai S, Rajesh M, et al . Pharmacological inhibition of CB1 cannabinoid receptor protects against doxorubicin‐induced cardiotoxicity. J Am Coll Cardiol . 2007; 50: 528–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pacher P, Nagayama T, Mukhopadhyay P, et al . Measurement of cardiac function using pressure–volume conductance catheter technique in mice and rats. Nat Protoc . 2008; 3: 1422–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Laurindo FR, Fernandes DC, Santos CX. Assessment of superoxide production and NADPH oxidase activity by HPLC analysis of dihydroethidium oxidation products. Methods Enzymol . 2008; 441: 237–60. [DOI] [PubMed] [Google Scholar]
- 25. Griendling KK, Sorescu D, Ushio‐Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res . 2000; 86: 494–501. [DOI] [PubMed] [Google Scholar]
- 26. Ferdinandy P, Schulz R. Nitric oxide, superoxide, and peroxynitrite in myocardial ischaemia‐reperfusion injury and preconditioning. Br J Pharmacol . 2003; 138: 532–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pacher P, Schulz R, Liaudet L, et al . Nitrosative stress and pharmacological modulation of heart failure. Trends Pharmacol Sci . 2005; 26: 302–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev . 2007; 87: 315–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Szabo C, Mabley JG, Moeller SM, et al . Part I: pathogenetic role of peroxynitrite in the development of diabetes and diabetic vascular complications: studies with FP15, a novel potent peroxynitrite decomposition catalyst. Mol Med . 2002; 8: 571–80. [PMC free article] [PubMed] [Google Scholar]
- 30. Westermann D, Rutschow S, Van Linthout S, et al . Inhibition of p38 mitogen‐activated protein kinase attenuates left ventricular dysfunction by mediating pro‐inflammatory cardiac cytokine levels in a mouse model of diabetes mellitus. Diabetologia . 2006; 49: 2507–13. [DOI] [PubMed] [Google Scholar]
- 31. Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature . 2001; 414: 813–20. [DOI] [PubMed] [Google Scholar]
- 32. Desco MC, Asensi M, Marquez R, et al . Xanthine oxidase is involved in free radical production in type 1 diabetes: protection by allopurinol. Diabetes . 2002; 51: 1118–24. [DOI] [PubMed] [Google Scholar]
- 33. Matsumoto S, Koshiishi I, Inoguchi T, et al . Confirmation of superoxide generation via xanthine oxidase in streptozotocin‐induced diabetic mice. Free Radic Res . 2003; 37: 767–72. [DOI] [PubMed] [Google Scholar]
- 34. Butler R, Morris AD, Belch JJ, et al . Allopurinol normalizes endothelial dysfunction in type 2 diabetics with mild hypertension. Hypertension . 2000; 35: 746–51. [DOI] [PubMed] [Google Scholar]
- 35. Szabo C, Ischiropoulos H, Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov . 2007; 6: 662–80. [DOI] [PubMed] [Google Scholar]
- 36. Ferdinandy P. Peroxynitrite: just an oxidative/nitrosative stressor or a physiological regulator as well? Br J Pharmacol . 2006; 148: 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ferdinandy P, Schulz R, Baxter GF. Interaction of cardiovascular risk factors with myocardial ischemia/reperfusion injury, preconditioning, and postconditioning. Pharmacol Rev . 2007; 59: 418–58. [DOI] [PubMed] [Google Scholar]
- 38. Liaudet L, Vassalli G, Pacher P. Role of peroxynitrite in the redox regulation of cell signal transduction pathways. Front Biosci . in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pacher P, Obrosova IG, Mabley JG, et al . Role of nitrosative stress and peroxynitrite in the pathogenesis of diabetic complications. Emerging new therapeutical strategies. Curr Med Chem . 2005; 12: 267–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pacher P, Szabo C. Role of the peroxynitrite‐poly(ADP‐ribose) polymerase pathway in human disease. Am J Pathol . 2008; 173: 2–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Obrosova IG, Mabley JG, Zsengeller Z, et al . Role for nitrosative stress in diabetic neuropathy: evidence from studies with a peroxynitrite decomposition catalyst. FASEB J . 2005; 19: 401–3. [DOI] [PubMed] [Google Scholar]
- 42. Szabo C, Zanchi A, Komjati K, et al . Poly(ADP‐Ribose) polymerase is activated in subjects at risk of developing type 2 diabetes and is associated with impaired vascular reactivity. Circulation . 2002; 106: 2680–6. [DOI] [PubMed] [Google Scholar]
- 43. Obrosova IG, Julius UA. Role for poly(ADP‐ribose) polymerase activation in diabetic nephropathy, neuropathy and retinopathy. Curr Vasc Pharmacol . 2005; 3: 267–83. [DOI] [PubMed] [Google Scholar]
- 44. Kittleson MM, Hare JM. Xanthine oxidase inhibitors: an emerging class of drugs for heart failure. Eur Heart J . 2005; 26: 1458–60. [DOI] [PubMed] [Google Scholar]