

Abstract
A pro-drug 8 of a synthetic analog 7 is more active in its anti-proliferative activity against human breast cancer MDA-MB-231 cells possessing high Catechol-O-methyltransferase (COMT) activity than the pro-drugs of EGCG and the analog 5. The higher activity of 8 is attributed to it not being a substrate of COMT.
Introduction
Regular drinking of green tea has been associated with reduced incidence of a variety of cancers. (-)-Epigallocatechin-3-gallate [1, (-)-EGCG] is the most abundant constituent in green tea extract and the most biologically active among the green tea polyphenols (GTPs). A number of epidemiological and biological studies have shown that (-)-EGCG can reduce or inhibit tumor growth in breast1,2,3, lung 4, GI 5 and urinary 6,7 tracts.
We previously reported that the tumor cellular proteasome is an important target of (-)-EGCG 8. The eukaryotic proteasome is a large multi-catalytic, multi-subunit protease complex. Inhibition of the chymotrypsin-like activity of the proteasome has been associated with induction of tumor cell apoptosis 9,10,11. We had previously found that (-)-EGCG inhibits the chymotrypsin-like activity of the proteasome in vitro (IC50=86-194 nM) and in intact tumor cells (1– 10 μM) (8). In silico docking studies have indicated that (-)-EGCG binds to the N-terminal threonine (Thr1) of the proteasomal chymotrypsin active site thus inhibiting the proteasomal chymotrypsin-like activity 12. Acylation of the threonine hydroxy group by the gallate ester function in EGCG accounts for the inhibition. This explains why green tea polyphenols without the ester function, such as epigallocatechin (EGC, 2), is essentially inactive (IC50=12 mM).8 Synthetic (+)-EGCG (3)13, the enantiomer of the natural (-)-EGCG, and other synthetic analogs of green tea catechins with an ester bond have also been found to inhibit the proteasomal chymotrypsin-like activity 14,15. The fact that the synthetic (+)-EGCG (3) is equally potent a proteasome inhibitor (IC50=170 nM) as the natural (-)-EGCG and that they both bind to the same active site in proteasome suggested that the active site may be nearly symmetrical and can accommodate both enantiomers equally well 12. On that basis, we designed a simple synthetic analog 5 which is symmetrical and contains the gallate ester function. Interestingly, compound 5 was found to be nearly as potent a proteasome inhibitor as either 1 or 3, with an IC50=340 nM.14
The challenge in developing (-)-EGCG for cancer prevention and therapy is its low bioavailability, partly due to its poor absorption and its instability under neutral or alkaline conditions (i.e. physiologic pH) and partly due to biologically inactivating processes such as methylation.16,17 We have previously tried to improve the bioavailability of (-)-EGCG through a pro-drug approach. 18 Acetylation of (-)-EGCG gave the peracetate of (-)-EGCG (4, pro-EGCG). Even though 4 itself is not active as a proteasome inhibitor, it exhibited enhanced growth inhibitory activity relative to EGCG in a number of cancer cell lines. 19 Improved bioavailability was observed in vivo: intragastric administration of 4 to CF-1 mice led to higher concentration of EGCG in plasma, small intestinal and colonic tissues compared with administration of equimolar doses of EGCG19. More importantly, the enhanced bioavailability also manifested in enhanced bioactivity in vivo. In animal xenograft models, Pro-EGCG (4) was found to be more effective than EGCG (1) at equivalent dosages in inhibiting tumor growth for breast tumors 20 and for androgen-independent prostate cancer.21
Methylation of (-)-EGCG occurs by catechol-O-methyltransferase (COMT), an enzyme widely distributed throughout the body.22 In humans, a single gene for COMT encodes both a soluble COMT (S-COMT) and a membrane-bound COMT (MB-COMT). A single nucleotide polymorphism (G to A) in codon 108 (S-COMT) or 158 (MB-COMT) results in a valine to methionine (Val to Met) substitution, leading to a high- (Val/Val [H/H]), intermediate- (Val/Met [H/L]), or low-activity (Met/Met [L/L]) form of COMT.23 There is a three-to-four-fold difference in enzyme activity between the high- and low-activity expressed genes.24 A recent case-control study of breast cancer in Asian-American women revealed that women who consumed green tea and who also carried the low activity COMT polymorphism had a reduced risk of breast cancer.25 In contrast, among those who were homozygous for the high activity COMT allele, breast cancer risk did not differ between tea drinkers and non-tea drinkers. These data suggest that EGCG and other tea polyphenols may be less cancer-protective upon methylation. In support of this possibility, catechins were known to be substrates of human COMT 26, 27. Recently, we found that, using synthetic methylated catechins which are metabolites or potential metabolites of tea EGCG in biomethylation 28 , the proteasome inhibition potency decreased as the number of methyl groups in methylated EGCG increased29. Metabolic O-methylation of EGCG may therefore reduce the effectiveness of EGCG in its anti-cancer activity29 in support of the human study.25
In this study, we wish to build on the observation that the simple analog 5 is almost as effective a proteasome inhibitor as the natural EGCG. We want to know if acetylation of 5 would enhance the cytotoxic activity of 5 just as pro-EGCG exhibited enhanced growth inhibitory activity relative to EGCG. We also want to see if the problem of metabolic O-methylation can be circumvented by using analogs of EGCG. We hypothesize that an analog such as 7, being devoid of the o-catechol structure, should not be a substrate of COMT. We report here the proteasome inhibition and cytotoxic activity of 7 as well as its peracetate 8 against breast cancer MDA-MB-231 cells possessing high COMT activity. Their activities are compared with the natural EGCG (1) and the analogs 5 as well as their peracetates 4 and 6 respectively.
Results
Chemical Synthesis
The synthesis of compounds 5 and 7 as well as their peracetates 6 and 8 is outlined in Scheme 1. 1,4-Dihydronaphthalene (11) was dihydroxylated with osmium tetraoxide to give the cis-diol 12. Acylation of 12 with DCC/DMAP and an equivalent of benzyl-protected gallic acid 13 gave the corresponding monobenzoate 14. When more than 2 equivalents of 13 were used, the dibenzoate 15 was obtained. Removal of the benzyl protection of 14 and 15 by palladium catalyzed hydrogenolysis gave compounds 16 and 5 respectively. Acetylation of 16 and 5 gave the corresponding acetates 17 and 6. Similar sequence of reactions of 12 with 3,5-dibenzyloxybenzoic acid (18) gave the series 7 and 21 as well as their acetates 8 and 22.
Scheme 1.
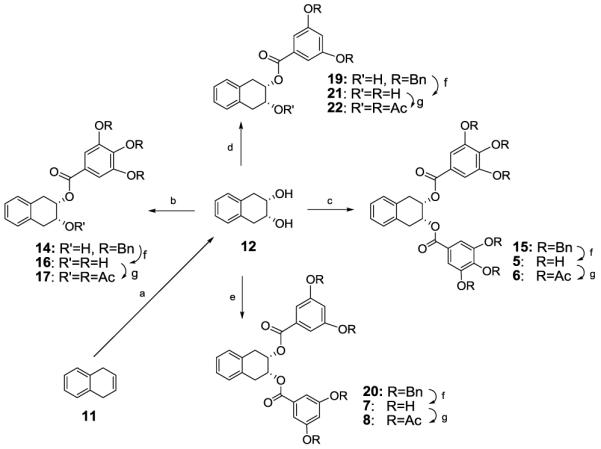
(a) OsO4, NMO, Acetone/H2O; (b) 3,4,5-tris(benzyloxy)benzoic acid (1.1 eq), DCC, DMAP, CH2Cl2; (c) 3,4,5-tris(benzyloxy)benzoic acid (2.1 eq), DCC, DMAP, CH2Cl2; (d) 3,5-bis(benzyloxy)benzoic acid (1.1 eq), DCC, DMAP, CH2Cl2; (e) 3,5-bis(benzyloxy)benzoic acid (2.1 eq), DCC, DMAP, CH2Cl2; (f) Pd/C, H2, THF/MeOH; (g) Ac2O, Py.
Bioassays
We first examined the ability of the analogs to inhibit the chymotrypsin-like activity of purified 20S proteasome (Figure 2). EGCG potently inhibited the proteasomal chymotryptic activity consistent with our previous observation.8 As shown in Figure 2, compound 5 which can be considered as an analog of EGCG, inhibited the proteasomal chymotrypsin-like activity with an IC50 value of 19 μM (Fig. 2A). Compound 16, which can be considered as the analog of (-)-EGC, is not active even at a concentration of 50 μM. On the other hand, compound 7, with an IC50 of 29 μM, is less active than EGCG or 5 (Fig. 2A). This is consistent with our previous observation that for EGCG, when the number of hydroxyl groups in the B ring or D ring is reduced, their proteasomal inhibition activity is also reduced.15,30 Not surprisingly, compound 21 is not active in proteasome inhibition even at 50 μM. As expected, the peracetates 4, 6, 8, 17 and 22 are all not active in proteasome inhibition under in vitro conditions (Fig. 2B).
Figure 2.
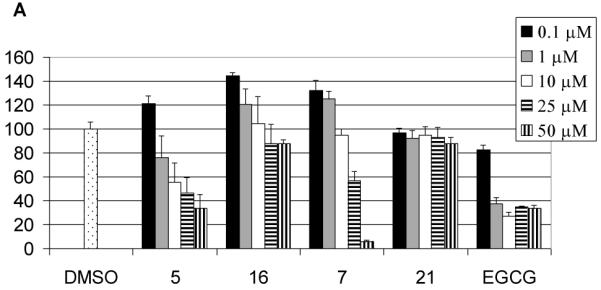
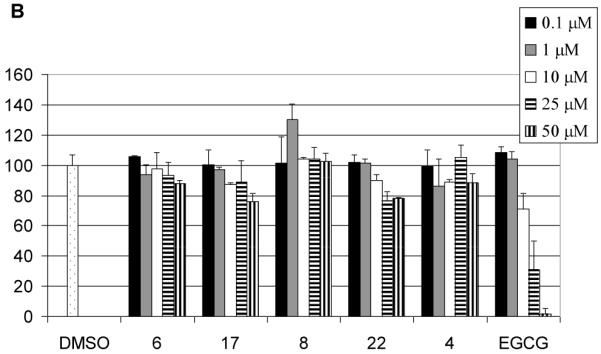
Proteasome inhibition by EGCG analogs. Purified 20S proteasome (35 ng) was incubated with compound 5, 7, 16, or 21 (A) or MDA-MB-231 cell extract (5.7 μg) was incubated with compound 6, 8, 17, or 22 (B) at indicated concentrations for 2 hours, followed by measuring the proteasomal chymotrypsin-like activity. EGCG was used as a comparison.
To test whether COMT could affect the proteasome inhibitory effects of the two EGCG analogs 5 and 7, we tested proteasome inhibition by compounds 5 and 7 in cells containing COMT. Human breast cancer MDA-MB-231 cell lysates that contains high COMT activity were treated with different concentrations of compound 5 or 7. Compound 7 inhibited 18-51% proteasomal activity at 1-10 μM while compound 5 only inhibited 10-16% proteasomal activity at the same conditions (Fig. 3A). Since compound 5 was more potent in inhibiting purified 20S proteasome than compound 7 (Fig. 2) and compound 5 but not 7 contains COMT-recognizable structure (Figure 1), these results suggested that compound 5 may be more susceptible to COMT modification than compound 7. As a control, EGCG at 10 μM only inhibited ~22% of the chymotrypsin-like activity in these cells, also not as active as compound 7 (Fig. 3A). Presumably, EGCG is also susceptible to COMT from the lysate. Indeed, compound 5 or EGCG activity was improved when the lysate was pre-treated with a COMT inhibitor, 3,5-dinitrocatechol (DNC). In contrast, compound 7’s activity was only slightly increased at the same condition (Fig. 3B), further confirming that compound 7 is resistant to COMT modification.
Figure 3.
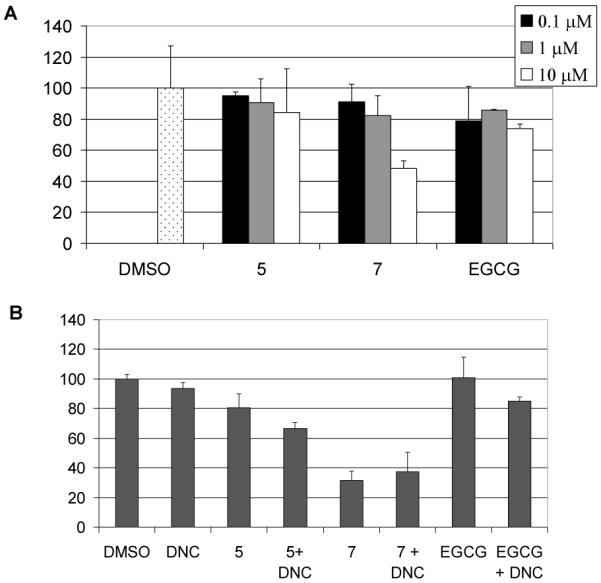
Inhibition on cellular proteasome by EGCG and EGCG analogs. (A) MDA-MB-231 cell extracts (5.7 μg) were incubated with different concentrations of compound 5 or 7 or EGCG for 2 hours, followed by performance of proteasomal chymotrypsin-like activity assay. EGCG was used as a control. (B) MDA-MB-231 cell extracts (5.7 μg) were pre-treated with 10 μM DNC for 20 minutes, followed by co-incubation with 5, or 7 or EGCG for 2 h. The proteasomal chymotrypsin-like activity was measured.
Figure 1.
Structures of EGCG and analogs
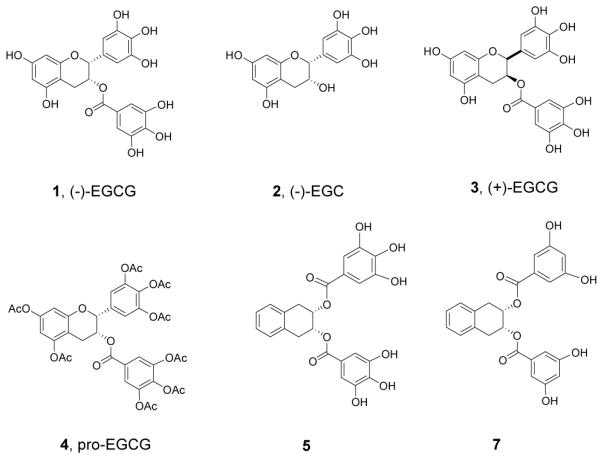
It was previously reported that pro-EGCG (4) exhibited enhanced growth inhibitory activity relative to EGCG (1) in a number of cancer cell lines.18, 20, 21, 30 We now found that the peracetates 6 and 8 were also more potent in inhibiting cell growth compared to their non-acetylated precursors 5 and 7 (Fig. 4). It was reasonable to assume that the peracetates 6 and 8 function similarly as prodrugs to release 5 and 7 intracellularly after absorption. We next compared the growth inhibitory activity of 4 with the peracetates 6 and 8 on human breast cancer MDA-MB-231 cells. The results are summarized in Figure 4. Among these pro-drugs, compound 8, the pro-drug of compound 7, was the most potent analog, showing 70-79% inhibition in MDA-MB-231 cells (Fig. 4) at 25 to 50 μM. Next to compound 8, compound 6, the pro-drug of compound 5 induced about 50% inhibition in MDA-MB-231 cells (Fig. 4). Both are more potent than pro-EGCG (4) itself which showed 0 to 32% inhibition at these concentrations. It is tempting therefore to suggest that the higher potency of compound 8 in inhibition of MDA-MB-231 proliferation is due to the resistance of compound 7 to COMT methylation. By contrast, EGCG, though more potent an inhibitor for purified 20S proteasome, is less potent in the inhibition of MDA-MB-231 proliferation because it is readily subject to biomethylation by the COMT in MDA-MB-231 cells29.
Figure 4.
Inhibition on cell proliferation by EGCG analogs. Human breast cancer MDA-MB-231 cells were treated with 25 or 50 μM compound 5 or its pro-drug 6, or compound 7 or its pro-drug 8 for 24 hours, followed by MTT assay. Pro-EGCG (4) was used as a comparison.
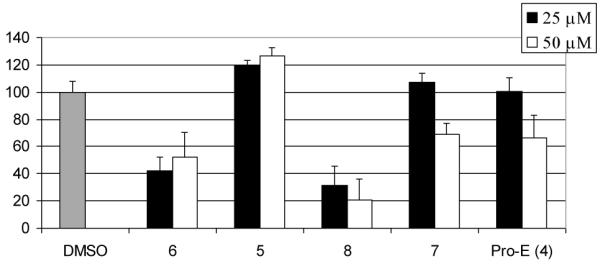
To test the validity of the above argument, we examined whether the inhibitory activity of compounds 8 (as prodrug of 7) or 6 (as prodrug of 5) as well as that of pro-EGCG (4) is affected by the addition of 3,5-dinitrocatechol (DNC), a tight-binding inhibitor of COMT.37 We hypothesize that if DNC inhibits COMT-mediated methylation of compound 5 or EGCG, we would observe increased growth-inhibitory activity of compound 6 or pro-EGCG (4) on the addition of DNC. On the other hand, the growth inhibitory activity of compound 8 would not be significantly affected by the addition of DNC as compound 7 presumably should not be a substrate of COMT. To test this idea, MDA-MB-231 cells were treated with compounds 8 or 6, the prodrugs of compounds 7 or 5, in the presence or absence of DNC. Compound 6 alone at 50 μM inhibited 48% cell proliferation. With the co-addition of 10 μM DNC, compound 6 efficacy was increased, showing 88% inhibition on cell proliferation (Fig. 5). Similarly, Pro-EGCG (4) efficacy was greatly improved from 42% inhibition to 89% inhibition when combined with DNC. However, compound 8 efficacy was not greatly improved by co-treatment with DNC, showing 69% and 84% inhibition on cell proliferation without or with the presence of DNC. These results are consistent with the hypothesis and suggest that for the compounds which could be methylated by COMT in cells, inhibition of COMT could increase their activity by reducing biomethylation. Compared with compound 7, compound 5 and Pro-E are more susceptible towards COMT modification.
Figure 5.
Effects of DNC on EGCG analogs efficacy against cell proliferation. Human breast cancer MDA-MB-231 cells with high COMT activity were treated with 50 μM EGCG analogs for 24 hours in the absence or presence of 10 μM DNC, followed by MTT assay. Pro-EGCG was used as a comparison.
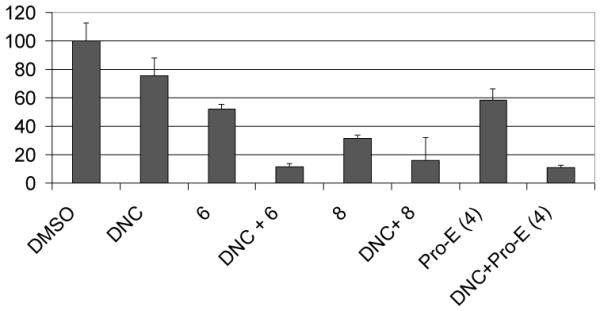
Next, we would like to see if these prodrugs indeed function by proteasome inhibition and if compound 8 is more potent to hit the target, the proteasome. Indeed, higher levels of ubiquitinated proteins were accumulated by compound 8 compared with compound 6 and Pro-EGCG (4) in MDA-MB-231 cells (Fig. 6), indicating that more proteasome activity was inhibited by compound 8.
Figure 6.
Accumulation of proteasome substrates. MDA-MB-231 cells were treated with 50 μM EGCG analogs for 22 hours. Extracted proteins were subject to Western blotting analysis using antibodies against ubiquitinated proteins and actin.
Discussion
The health benefits of green tea and its main constituent (-)-EGCG have been widely supported by results from epidemiological, cell culture, and animal studies (31). Since there are no toxic effects associated with tea drinking, the attraction of using green tea extract or EGCG as therapeutic agents is considerable. Yet, the U. S. Food and Drug Administration (FDA), after reviewing the human data, concluded in 2005 that “it is highly unlikely that green tea reduces the risk of breast cancer or prostate cancer”.32 A major challenge in extrapolating the biological activities of green tea polyphenols in vitro to possible benefits in vivo is its bioavailability,33 The poor bioavailability of EGCG can be attributed to the following factors: (a) the ease of degradation of EGCG in alkaline or neutral conditions,34 (b) poor cellular uptake because of the high aqueous solubility of EGCG and poor hydrophobicity to be absorbed by cells and (c) metabolic transformations such as methylation, glucuronidation and sulfation after absorption.16
In the present study, we demonstrated that a prodrug of a simple synthetic analog 5, the acetate 6, enhanced the cytotoxic activity of 5 just as pro-EGCG (4) enhanced the growth inhibitory activity relative to EGCG (Figure 4). An even simplier analog 7, though not as potent an inhibitor of purified proteasome as EGCG or 5 (Figure 2), was found to be a better proteasome inhibitor when incubated with MDA-MB-231 cell extracts (Figure 3). Furthermore, the prodrug of 7, the acetate 8, was found to be more potent in growth inhibition than either 6 or pro-EGCG (Figure 4). In order to explain the enhanced cytotoxicity of 7 (from 8) towards MDA-MB-231cells, we postulated that this is due to the biomethylation of 5 or EGCG by COMT in MDA-MB-231cells. We have recently assayed the COMT genotype of various breast cancer cell lines via DNA sequencing and MDA-MB-231 was found to express the COMT-H allele.37 Biomethylation of 5 or EGCG by COMT would lead to methylated metabolites. We have previously showed that the proteasome inhibition potency decreased as the number of methyl groups in methylated EGCG increased.30 Compound 7, in contrast, does not have the o-catechol structure and should not be a substrate of COMT and biomethylated. Supporting evidence for the role of COMT in reducing the proteasome inhibition activity of EGCG and 5 is provided by the addition of DNC, a known COMT inhibitor37 (Figure 3B). The activity of either EGCG or 5 was enhanced in the presence of DNC. By contrast, the proteasome inhibition activity of 7 was not affected by the addition of DNC. Similarly, the antiproliferative activities of pro-EGCG and 6 but not 8 is affected by co-treatment with DNC. Figure 5 showed that addition of DNC potently enhanced the antiproliferative activities of both pro-EGCG and 6 but not 8. Inhibition of COMT by DNC restored the potencies of EGCG and 5 because they are better proteasome inhibitors than 7.
These results suggest that COMT does play an important role in affecting the antiproliferative activities of EGCG and its analogs. This is especially critical in cells having high COMT activities, such as the MDA-MB-231 breast cancer cells. The results are also in agreement with the epidemiological study which showed that for Asian-American women who were homozygous for the high activity COMT allele, breast cancer risk did not differ between tea drinkers and non-tea drinkers.25 Such information will have to be taken into consideration for any effort to develop green tea polyphenols or analogs into therapeutics for cancer treatment.
Experimental
General Experimentals for chemical synthesis
The starting materials and reagents, purchased from commercial suppliers, were used without further purification. Anhydrous methylene chloride was distilled under nitrogen from CaH2. Anhydrous DMF was distilled under vacuum from CaH2. Reaction flasks were flame-dried under a stream of N2. All moisture-sensitive reactions were conducted under a nitrogen atmosphere. Flash chromatography was carried out using silica-gel 60 (70-230 mesh). The melting points were uncorrected. 1H-NMR and 13C NMR (300 MHz) spectra were measured with TMS as an internal standard when CDCl3, CD3OD and acetone-d6 were used as solvent. High-resolution (ESI) MS spectra were recorded using a QTOF-2 Micromass spectrometer.
Preparation of cis-1,2,3,4-tetrahydro-naphthalene-2,3-diol (12)
To a solution of 1,4-dihydronaphthalene (500 mg, 3.84 mmol) in acetone/H2O (3.0/1.0 mL) was added a solution of NMO in H2O (1.43 mL, 50% wt, 6.90 mmol) and a solution of OsO4 in 2-methyl-2-propanol (313 μL, 2.5% wt., 25 μmol). The mixture was stirred at room temperature for 16 h. Saturated Na2SO3 aqueous solution (10 mL) was added and stirred for an additional 15 min. H2O (10 mL) and EtOAc (30 mL) was added and stirred for 5 min. The aqueous phase was extracted with EtOAc (4×30 mL). The combined organic phase was washed with brine (20 mL) and dried over anhydrous Na2SO4. The solution was concentrated by rotary evaporator and vacuum drying to give the crude product which was purified by silica gel chromatography (Hexane/EtOAc/CH2Cl2=5/1/1) to afford 521.7 mg (83%) of the title compound as a white solid. 1H NMR (acetone-d6, 300 MHz) δ7.13 (m, 2 H), 7.09 (m, 2 H), 4.11 (t, J = 5.4, 2 H), 3.01 (m, 4 H), 2.36 (s, 2 H); 13C NMR (acetone-d6, 75 MHz) δ132.87, 129.11, 126.25, 69.24, 34.32.
Preparation of monobenzoates 14 and 19
To a solution of the corresponding benzoic acid (0.22 mmol) in dry CH2Cl2 (20 mL), dicyclohexylcarbodiimide (DCC, 45 mg, 0.22mmol) was added. The resulting mixture was stirred at room temperature for 4h, 4-Dimethylaminopyridine (DMAP, 3 mg, 0.025 mmol.) was added, then a solution of diol 12 (33 mg, 0.2 mol) in CH2Cl2 (5 mL) was added dropwise. The mixture was stirred at room temperature overnight. Then the mixture was concentrated, EA (1 mL) was added and cooled in fridge, the urea byproduct was filtered and the filtrate was evaporated. The resulting residue was purified by column chromatography (EA/n-Hex= 1: 3) to afford the desired compound as a pale yellow amorphous solid.
Compound 14
white solid (61 % yield). 1H NMR (CDCl3, 300 MHz) δ 7.43-7.13 (m, 22H), 5.48 (brs, 1H), 5.15 (s, 2H), 5.11 (s, 4H), 4.33 (s, 1H), 3.28-3.03 (m, 4H); 13C NMR (CDCl3, 75 MHz) δ 166.4, 152.7, 142.7, 137.6, 136.9, 133.3, 132.7, 129.5, 129.3, 128.9, 128.8, 128.5, 128.3, 127.8, 126.7, 126.6, 125.3, 109.4, 75.4, 73.4, 71.4, 69.8, 68.1, 35.0, 32.2.
Compound 19
white solid (65 % yield). 1H NMR (CDCl3, 300 MHz) δ 7.44-7.13 (m, 16H), 6. 82 (s, 1H), 5.50 (brs, 1H), 5.14 (s, 1H), 5.05 (s, 4H), 4.35 (s, 1H), 3.31-3.08 (m, 4H); 13C NMR (CDCl3, 75 MHz) δ 166.6, 160.0, 136.8, 133.4, 132.8, 132.3, 129.6, 129.3, 129.0, 128.5, 128.0, 126.7, 126.7, 108.9, 107.4, 73.7, 70.6, 68.0, 35.0, 32.1.
Preparation of dibenzoates 15 and 20
To a solution of 12 (33 mg, 0.2 mmol) in CH2Cl2 (3.0 mL) were added the corresponding benzoic acid (0.42 mmol), 4-Dimethylaminopyridine (DMAP, 6 mg, 0.05 mmol) and dicyclohexylcarbodiimide (DCC, 87 mg, 0.42 mmol). The mixture was stirred at room temperature overnight. Then the mixture was concentrated, EA (1 mL) was added and cooled in fridge, the urea byproduct was filtered and the filtrate was evaporated. The resulting residue was purified by column chromatography (EA/n-Hex= 1: 6) to afford the desired compound as white amorphous solid.
Compound 15
white solid (59 % yield). 1H NMR (CDCl3, 300 MHz) δ 7.43-7.22 (m, 38H), 5.72 (brs, 1H), 5.01-4.96 (m, 12H), 3.38 (dd, J= 17.4 Hz, J= 4.8 Hz, 1H), 3.25 (dd, J= 17.4 Hz, J= 6.9 Hz, 1H); 13C NMR (CDCl3, 75 MHz) δ 165.7, 152.7, 142.7, 137.7, 136.7, 132.6, 129.4, 128.8, 128.7, 128.5, 128.3, 128.2, 127.8, 126.9, 125.2, 109.2, 75.3, 71.2, 70.5, 32.5.
Compound 20
white solid (71 % yield). 1H NMR (CDCl3, 300 MHz) δ 7.42-7.18 (m, 28H), 6.74 (s, 1H), 5.72 (brs, 1H), 4.91 (m, 9H), 3.34 (m, 4H); 13C NMR (CDCl3, 75 MHz) δ 165.9, 160.0, 136.6, 132.5, 132.2, 129.4, 128.8, 128.4, 127.9, 126.8, 108.6, 107.8, 70.6, 70.4, 32.3.
General procedures for palladium catalyzed hydrogenolysis: preparation of 5, 7, 16 and 21
To a solution of benzylated substrate (0.1 mmol) in THF/MeOH (3 mL, 1:2) was added palladium hydroxide (20 mg, 20% on carbon). The resulting mixture was stirred at room temperature until TLC showed that the reaction was completed (within 6h). Then the reaction mixture was filtered to remove the catalyst. The filtrate was evaporated to afford product as a white solid.
Compound 16
white solid (95 % yield). 1H NMR (CD3OD, 300 MHz) δ 7.13-7.01 (m, 6H), 5.39 (m, 1H), 4.25 (m, 1H), 3.14-3.06 (m, 4H); 13C NMR (CD3OD, 75 MHz) δ 167.1, 145.2, 138.6, 133.5, 132.8, 129.0, 128.8, 126.1, 126.0, 120.6, 109.0, 72.5, 67.4, 34.4, 32.1; HRMS m/z calculated for C17H16O6Na 339.0840, found 339.0839.
Compound 5
white solid (95 % yield). 1H NMR (CD3OD, 300 MHz) δ 7.21-7.16 (m, 4H), 7.09 (s, 4H), 5.61 (m, 2H), 3.37-3.25 (m, 4H); 13C NMR (CD3OD, 75 MHz) δ 165.3, 145.1, 138.0, 132.8, 129.0, 126.3, 120.9, 109.0, 69.7, 31.9; HRMS m/z calculated for C24H20O10Na 491.0947, found 491.0949.
Compound 21
white solid (95 % yield). 1H NMR (CD3OD, 300 MHz) δ 7.14-7.07 (m, 4H), 6. 92 (s, 2H), 6.44 (s, 1H), 5.43 (m, 1H), 4.27 (m, 1H), 3.17-3.10 (m, 4H); 13C NMR (CD3OD, 75 MHz) δ 170.6, 166.0, 134.2, 133.2, 132.9, 129.3, 129.1, 126.3, 126.2, 108.2, 107.4, 73.2, 67.2, 35.0, 32.1; HRMS m/z calculated for C17H16O5Na 323.0891, found 323.0890.
Compound 7
white solid (95 % yield). 1H NMR (CD3OD, 300 MHz) δ 7.16 (s, 4H), 6.88 (s, 4H), 6.46 (s, 2H), 5.66 (m, 2H), 3.31 (m, 4H); 13C NMR (CD3OD, 75 MHz) δ 166.3, 158.6, 132.5, 131.9, 128.9, 126.4, 107.7, 107.3, 70.4, 31.8; HRMS m/z calculated for C24H20O8 Na 459.1047, found 459.1050.
Preparation of the acetates 6, 8, 17 and 22
To a solution of the corresponding substrate (0.1 mmol) and Ac2O (0.5mL) in pyridine (0.5 mL) at room temperature. The resulting mixture was stirred dovernight. Then EA (50 mL) was added and 1N HCl (1 mL) and washed with CuSO4 solution (3×10 mL), water (2×10 mL) and brine (10 mL), dried over sodium sulfate and evaporated. The residue was purified by column chromatography over silica gel (EA/n-Hex= 3: 2) to afford the title product as white solid.
Compound 17
white solid (92 % yield). 1H NMR (CDCl3, 300 MHz) δ 7.74 (s, 2H), 7.21-7.11 (m, 4H), 5.64 (m, 1H), 5.42 (m, 1H), 3.26-3.16 (m, 4H), 2.30 (s, 9H), 2.07 (s, 3H); 13CNMR (CDCl3, 75 MHz) δ 170.9, 167.8, 166.6, 164.1, 143.7, 139.0, 132.6, 132.3, 129.3, 128.6, 126.8, 126.7, 122.5, 70.9, 69.7, 32.5, 31.7, 21.4, 20.8, 20.4; HRMS m/z calculated for C25H24O10Na 507.1266, found 507.1262.
Compound 6
white solid (94 % yield). 1H NMR (CDCl3, 300 MHz) δ 7.73-7.70 (m, 4H), 7.22-7.14 (m, 4H), 5.68 (m, 2H), 3.31 (m, 4H), 2.28 (s, 18H); 13C NMR (CDCl3, 75 MHz) δ 167.9, 166.6, 164.1, 143.7, 139.1, 132.2, 129.4, 128.4, 126.9, 122.6, 71.1, 32.1, 20.8, 20.4; HRMS m/z calculated for C36H32O16Na 743.1576, found 743.1583.
Compound 22
white solid (90 % yield). 1H NMR (CDCl3, 300 MHz) δ 7.60 (s, 2H), 7.21-7.11 (m, 5H), 5.64 (m, 1H), 5.44 (m, 1H), 3.26-3.17 (m, 4H), 2.31 (s, 6H), 2.08 (s, 3H),; 13CNMR (CDCl3, 75 MHz) δ 170.9, 169.0, 164.6, 151.1, 132.6, 132.5, 132.3, 129.3, 126.8, 126.7, 120.7, 120.6, 70.8, 69.7, 32.4, 31.8, 21.4, 21.3; HRMS m/z calculated for C23H22O8Na 449.1201, found 449.1207.
Compound 8
white solid (91 % yield). 1H NMR (CDCl3, 300 MHz) δ 7.58 (s, 4H), 7.23-7.14 (m, 6H), 5.70 (m, 2H), 3.32 (m, 4H), 2.28 (s, 12H); 13C NMR (CDCl3, 75 MHz) δ 169.0, 164.6, 151.1, 132.3, 132.2, 129.4, 126.9, 120.8, 120.6, 71.0, 32.1, 21.2; HRMS m/z calculated for C32H28O12Na 627.1468, found 627.1473.
Biological assays
Materials
Purified rabbit 20S proteasome and fluorogenic substrate Suc-LLVY-AMC for the proteasomal chymotrypsinlike (CT-like) activity were obtained from Calbiochem Inc. (San Diego, CA). Fetal bovine serum (FBS) was from Tissue Culture Biologicals (Tulare, CA). Penicillin and streptomycin were purchased from Invitrogen Co. (Carlsbad, CA). RPMI 1640 medium was purchased from Invitrogen Co. (Carlsbad, CA). MTT (3–4, 5-dimethyltiazol-2-yl-2.5-diphenyl-tetrazolium bromide) was purchased from Sigma-Aldrich.
Cell culture
Human breast cancer MDA-MB-231 cells were purchased from American Type Culture Collection (Manassas, VA) and grown in RPMI 1640 medium supplemented with 10% FBS, 100 units/ml of penicillin, and 100 μg/ml of streptomycin. Cells were maintained at 37°C and 5% CO2.
Inhibition of purified 20S proteasome activity by EGCG and its analogs
A purified rabbit 20S proteasome (35 ng) was incubated with 20 μM of substrate Suc-LLVY-AMC in 100 μl assay buffer (20 mM Tris–HCl, pH 7.5), in the presence of EGCG or EGCG analogs at different concentrations or the solvent for 2 h at 37°C, followed by measurement of hydrolysis of the fluorogenic substrates using a Wallac Victor3™ multi-label counter with 355-nm excitation and 460-nm emission wavelengths.
Inhibition of 26S proteasome activity by EGCG and its analogs in vitro
Human breast cancer MDA-MB-231 cell extract (5.7 μg) was incubated in 20 μM of substrate Suc-LLVY-AMC in 100 μl assay buffer (20 mM Tris–HCl, pH 7.5) in the presence of EGCG or EGCG analogs at different concentrations or the solvent for 2 h at 37°C, followed by measurement of hydrolysis of the fluorogenic substrates as described above.
Inhibition of cellular 26S proteasome by EGCG and its analogs
Human breast cancer MDA-MB-231 cells were treated with compound 5 or 7 for 24 hours. Cell lysates were subjected to chymotrypsin activity assay and Western blotting analysis as described before.20
MTT assay
Cells were grown in a 96-well plate. Triplicate wells of cells were treated with indicated concentrations of EGCG or EGCG analogs for 24 h. After aspiration of medium, MTT (1 mg/ml) was then added to the cell cultures, followed by incubation for 3 h at 37°C. After cells were crystallized, MTT was removed and DMSO was added to dissolve the metabolized MTT product. The absorbance was then measured on a Wallac Victor3 1420 Multi-label counter at 540 nm.
Acknowledgement
Funding support from the Natural Sciences and Engineering Research Council of Canada (NSERC) and the National Cancer Institute (Grant Number: 1R01CA120009 and 3R01CA120009-04S1) is gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Guo S, Lu J, Subramanian A, Sonenshein GE. Cancer Res. 2006;66:5322. doi: 10.1158/0008-5472.CAN-05-4287. [DOI] [PubMed] [Google Scholar]
- 2.Sun CL, Yuan JM, Koh WP, Yu MC. Carcinogenesis. 2006;27:1310. doi: 10.1093/carcin/bgi276. [DOI] [PubMed] [Google Scholar]
- 3.Sartippour MR, Heber D, Ma J, Lu Q, Go VL, Nguyen M. Nutr. Cancer. 2001;40:149. doi: 10.1207/S15327914NC402_11. [DOI] [PubMed] [Google Scholar]
- 4.Landau JM, Wang ZY, Yang GY, Ding W, Yang CS. Carcinogenesis. 1998;19:501. doi: 10.1093/carcin/19.3.501. [DOI] [PubMed] [Google Scholar]
- 5.Horie N, Hirabayashi N, Takahashi Y, Miyauchi Y, Taguchi H, Takeishi K. Biol. Pharm. Bull. 2005;28:574. doi: 10.1248/bpb.28.574. [DOI] [PubMed] [Google Scholar]
- 6.Bettuzzi S, Brausi M, Rizzi F, Castagnetti G, Peracchia G, Corti A. Cancer Res. 2006;66:1234. doi: 10.1158/0008-5472.CAN-05-1145. [DOI] [PubMed] [Google Scholar]
- 7.Kemberling JK, Hampton JA, Keck RW, Gomez MA, Selman SH. J. Urol. 2003;170:773. doi: 10.1097/01.ju.0000081278.64511.96. [DOI] [PubMed] [Google Scholar]
- 8.Nam S, Smith DM, Dou QP. J. Biol. Chem. 2001;276:13322. doi: 10.1074/jbc.M004209200. [DOI] [PubMed] [Google Scholar]
- 9.An B, Goldharb RH, Siman R, Dou QP. Cell Death Differ. 1998;5:1062. doi: 10.1038/sj.cdd.4400436. [DOI] [PubMed] [Google Scholar]
- 10.Lopes UG, Erhardt P, Yao R, Cooper GM. J. Biol. Chem. 1997;272:12893. doi: 10.1074/jbc.272.20.12893. [DOI] [PubMed] [Google Scholar]
- 11.Ciechanover A, Orian A, Schwartz AL. BioEssays. 2000;22:443. doi: 10.1002/(SICI)1521-1878(200005)22:5<442::AID-BIES6>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 12.Smith DM, Daniel KG, Wang Z, Guida WC, Chan TH, Dou QP. Proteins: Structure, Function, and Bioinformatics. 2004;54:58. doi: 10.1002/prot.10504. [DOI] [PubMed] [Google Scholar]
- 13.Li L, Chan TH. Org. Lett. 2001;3:739. doi: 10.1021/ol000394z. [DOI] [PubMed] [Google Scholar]
- 14.Smith DM, Wang Z, Kazi A, Li LH, Chan TH, Dou QP. Mol. Med. 2002;8:382. [PMC free article] [PubMed] [Google Scholar]
- 15.Wan SB, Chen D, Dou QP, Chan TH. Bioorg. Med. Chem. 2004;12:3521. doi: 10.1016/j.bmc.2004.04.033. [DOI] [PubMed] [Google Scholar]
- 16.Lu H, Meng X, Yang CS. Drug Metab Dispos. 2003;31:572. doi: 10.1124/dmd.31.5.572. [DOI] [PubMed] [Google Scholar]
- 17.Okushio K, Suzuki M, Matsumoto N, et al. Biosci Biotechnol Biochem. 1999;63:430. doi: 10.1271/bbb.63.430. [DOI] [PubMed] [Google Scholar]
- 18.Lam WH, Kazi A, Kuhn DJ, Chow LMC, Chan ASC, Dou QP, Chan TH. Bioorg. Med. Chem. 2004;12:5587. doi: 10.1016/j.bmc.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 19.Lambert JD, Sang S, Hong J, Kwon SJ, Lee MJ, Ho CT, Yang CS. Drug Metab. Dispos. 2006;34:2111. doi: 10.1124/dmd.106.011460. [DOI] [PubMed] [Google Scholar]
- 20.Landis-Piwowar KR, Huo CD, Chen D, Cui QC, Minic V, Shi GQ, Chan TH, Dou QP. Cancer Res. 2007;67:4303. doi: 10.1158/0008-5472.CAN-06-4699. [DOI] [PubMed] [Google Scholar]
- 21.Lee SK, Chan WK, Lee TW, Lam WH, Wang X, Chan TH, Wong YC. Nutri. Cancer. 2008;60:483. doi: 10.1080/01635580801947674. [DOI] [PubMed] [Google Scholar]
- 22.Mannisto PT, Kaakkola S. Pharmacol Rev. 1999;51:593. [PubMed] [Google Scholar]
- 23.Lachman HM, Papolos DF, Saito T, et al. Pharmacogenetics. 1996;6:243. doi: 10.1097/00008571-199606000-00007. [DOI] [PubMed] [Google Scholar]
- 24.Weinshilboum RM, Otterness DM, Szumlanski CL. Annu Rev Pharmacol Toxicol. 1999;39:19. doi: 10.1146/annurev.pharmtox.39.1.19. [DOI] [PubMed] [Google Scholar]
- 25.Wu AH, Tseng CC, Van Den Berg D, et al. Cancer Res. 2003;63:7526. [PubMed] [Google Scholar]
- 26.Zhu BT, Patel UK, Cia MX, Conney AH. Drug Metab. Dispos. 2000;28:1024. [PubMed] [Google Scholar]
- 27.Meng X, Sang S, Zhu N, Lu H, et al. Chem. Res. Toxicol. 2002;15:42. doi: 10.1021/tx010184a. [DOI] [PubMed] [Google Scholar]
- 28.Wan SB, Dou QP, Chan TH. Tetrahedron. 2006;62:5897. [Google Scholar]
- 29.Landis-Piwowar KR, Wan SB, Wiegand RA, Kuhn DJ, Chan TH, Dou QP. J. Cell Physiol. 2007;213:252. doi: 10.1002/jcp.21124. [DOI] [PubMed] [Google Scholar]
- 30.Landis-Piwowar KR, Kuhn DJ, Wan SB, Chen D, Chan TH, Dou QP. Internat. J. Mol. Med. 2005;15:735. [PubMed] [Google Scholar]
- 31.Hara Y. Green Tea: Health Benefits and Applications. Marcel Dekker Inc.; New York: 2001. [Google Scholar]
- 32.U. S. Food and Drug Administration . Letter Responding to Health Claim Petition dated January 27, 2004: Green Tea and Reduced Risk of Cancer Health Claim. Jun 30, 2005. Docket number 2004Q-0083. [Google Scholar]
- 33.Yang CS, Chen L, Lee MJ, Balentine D, et al. Cancer Epidemiol. Biomarkers Prev. 1998;7:351. [PubMed] [Google Scholar]
- 34.Chen Z, Zhu QY, Tsang D, Huang Y. J. Agric. Food Chem. 2001;49:477. doi: 10.1021/jf000877h. [DOI] [PubMed] [Google Scholar]
- 35.Chen D, Milacic V, Chen MS, Wan SB, et al. Histol. Histopathol. 2008;23:487. doi: 10.14670/hh-23.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dou QP, Landis-Piwowar KR, Chen D, Huo C, Wan SB, Chan TH. Inflammopharmacology. 2008;16:208. doi: 10.1007/s10787-008-8017-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Landis-Piwowar K, Chen D, Chan TH, Dou QP. submitted [Google Scholar]