

Abstract
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease characterized by progressive motor neuron loss, paralysis and death within 2–5 years of diagnosis. Currently, no effective pharmacological agents exist for the treatment of this devastating disease. Neuroinflammation may accelerate the progression of ALS. Cannabinoids produce anti-inflammatory actions via cannabinoid receptor 1 (CB1) and cannabinoid receptor 2 (CB2), and delay the progression of neuroinflammatory diseases. Additionally, CB2 receptors, which normally exist primarily in the periphery, are dramatically up-regulated in inflamed neural tissues associated with CNS disorders. In G93A-SOD1 mutant mice, the most well-characterized animal model of ALS, endogenous cannabinoids are elevated in spinal cords of symptomatic mice. Furthermore, treatment with non-selective cannabinoid partial agonists prior to, or upon, symptom appearance minimally delays disease onset and prolongs survival through undefined mechanisms. We demonstrate that mRNA, receptor binding and function of CB2, but not CB1, receptors are dramatically and selectively up-regulated in spinal cords of G93A-SOD1 mice in a temporal pattern paralleling disease progression. More importantly, daily injections of the selective CB2 agonist AM-1241, initiated at symptom onset, increase the survival interval after disease onset by 56%. Therefore, CB2 agonists may slow motor neuron degeneration and preserve motor function, and represent a novel therapeutic modality for treatment of ALS.
Keywords: AM-1241; amyotrophic lateral sclerosis; CB1 cannabinoid receptor; CB2 cannabinoid receptor; G93A-SOD1 transgenic mice; WIN-55,212
Amyotrophic lateral sclerosis (ALS) is a neurological disorder which first presents during mid-life as small tremors or muscle weakness, rapidly develops to full paralysis without affecting cognition, and ultimately results in death by respiratory arrest within 2–5 years after symptom onset (Strong and Rosenfeld 2003). ALS exists in two forms, familial amyotrophic lateral sclerosis (FALS) and sporadic amyotrophic lateral sclerosis (SALS) (Bruijn et al. 2004). Familial ALS comprises only 5–10% of all ALS cases. At least six genes have been identified as causes of FALS, the most common being that which codes for the cytosolic copper–zinc superoxide dismutase (SOD1) protein. To date, multiple clinical trials of several candidate therapeutic compounds for ALS have been completed [reviewed in (Gordon 2005)]. Unfortunately, none of these pharmacological agents alters the inevitable outcome of ALS and only one drug, riluzole, has been approved by the US Food and Drug Administration (Traynor et al. 2003).
Recent evidence indicates that ALS is a disease characterized by chronic inflammation (McGeer and McGeer 2002; Weydt and Moller 2005). Microglia are the resident macrophages of the CNS (Streit 2002). In response to CNS injury, microglia quickly convert to an ‘active’ state during which they change to an amoeboid shape, up-regulate the cell-surface expression of a variety of surface antigens and secrete several pro-inflammatory molecules (Hanisch 2002). As such, it is commonly accepted that microglial activation in the CNS signifies a primary neuroinflammatory state with deleterious effects on surrounding neurons (Nelson et al. 2002). Postmortem studies of CNS tissues obtained from FALS and SALS patients indicate that activated microglia accumulate not only in areas of profound motor neuron degeneration, but also in areas of mild damage (Ince et al. 1996). Recent in vivo studies employing positron emission tomography (PET scan) also demonstrate the presence of activated microglia in living SALS patients (Turner et al. 2004).
Δ9-Tetrahydrocannabinol (Δ9-THC) is the main psychoactive constituent in the plant Cannabis sativa (marijuana) and produces its effects by activation of cannabinoid receptor 1 (CB1) (Matsuda et al. 1990) and cannabinoid receptor 2 (CB2) (Munro et al. 1993) cannabinoid receptors. CB1 receptors are expressed throughout the CNS (Herkenham et al. 1990), while CB2 receptors are expressed predominantly in immune cells and non-neuronal tissues (Galiegue et al. 1995). Therapeutic agents which modulate the cann-abinoid system are effective in treating a wide variety of disorders characterized by inflammation (Di Marzo et al. 2004; Klein 2005). More specifically, drugs which activate CB2 receptors successfully improve the symptoms of several inflammatory diseases, such as intestinal hypermotility because of endotoxic shock (Mathison et al. 2004) and atherosclerosis (Steffens et al. 2005). In an animal model of multiple sclerosis, a disorder characterized by inflamed neural tissues, administration of a non-selective cannabinoid or a CB2 selective agonist provides relief from acute and chronic symptoms [reviewed in (Walter and Stella 2004)]. Furthermore, in neuroinflammatory conditions such as Alzheimer’s disease, CB2 (but not CB1) receptors appear to be dramatically up-regulated specifically in activated microglia, and selective activation of these receptors blocks the elevation of characteristic neurotoxic markers (Benito et al. 2003; Ramirez et al. 2005).
Mice which overexpress human mutant G93A-SOD1 protein develop a progressive motor neuron disease which is similar to human ALS (Gurney 1997). In the spinal cords of G93A-SOD1 (G93A) mice, an increased presence of endocannabinoids correlates with presentation of symptoms, and levels continue to escalate until the end-stage of the disease (Witting et al. 2004; Bilsland et al. 2006). Pharmacological or genetic elevation of endocannabinoid levels also slightly delays disease progression in G93A mice, while having no effect on survival (Bilsland et al. 2006). Administration of the non-selective partial cannabinoid agonists Δ9-THC (Raman et al. 2004) or cannabinol (Weydt et al. 2005) are minimally successful in delaying motor impairment and prolonging survival in G93A mice after the onset of symptoms. Lastly, a recent study reported elevated levels of CB2 receptors in microglia isolated from post-mortem human spinal cords of ALS patients (Yiangou et al. 2006). Collectively, these studies suggest that cannabinoid receptors might serve as novel therapeutic targets for ALS drug development.
The basis for the therapeutic actions of cannabinoids in ALS is not known. Furthermore, although potentially involved in the pathogenesis of ALS, the expression and function of CB1 and CB2 receptors in the G93A mouse model have not been determined. Most importantly, selective CB2 agonists, which appear to be most efficacious for treatment of chronic neuroinflammatory conditions (Walter and Stella 2004; Ramirez et al. 2005), have yet to be examined in G93A mice. Therefore, the purpose of the present study was to test the hypothesis that in the early stages of disease progression in G93A mice, CB2 receptors are selectively up-regulated in spinal cords as a compensatory, protective measure. As such, daily treatment with CB2 agonists, even initiated as late as symptom onset, will significantly prolong survival of affected mice.
Materials and methods
Drugs evaluated
The non-selective CB1/CB2 agonists examined in this study were CP-55,940 (−)-cis-3-[2-Hydroxy-4-(1,1-dimethylheptyl)-phenyl]-trans-4-(3-hydroxypropyl)-cyclohexanol), WIN-55,212-2 [2,3-Dihydro-5-methyl-3-[(morpholinyl)methyl]pyrrolo[1,2,3-de]-1,4-benzoxazin-yl]-(1-naphthalenyl)methanone and HU-210 (−)-11-hydroxy-delta(8)-tetrahydrocannabinol-dimethylheptyl. The selective CB1 agonist employed was ACEA (all Z)-N-(2-cycloethyl)-5,8,11,14-eicosatetraenamide. The selective CB1 antagonists used were AM-251 (N-(Piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichloro-phenyl)-4-methyl-1H-pyrazole-3-carboxamide and O-2050 ((6aR, 10aR)-3-(1-Methanesulfonylamino-4-hexyn-6-yl)-6a,7,10,10a-tetrahydro-6,6,9-trimethyl-6H-dibenzo[b,d]pyran). The selective CB2 agonists examined were AM-1241 ((R,S)-(+)-(2-Iodo-5-nitrobenzoyl)-[1-(1- methyl-piperidin-2-ylmethyl)-1H-indole-3-yl] methanone) and GW-405833 (2,3-dichloro-phenyl)-[5-methoxy-2-methyl-3-(2-morpholin-4-yl-ethyl)-indol-1-yl]-methanone. The selective CB2 antagonists used were AM-630, 6-iodo-2-methyl-1-[2-(4-morpholinyl)ethyl]-1H-indol-3-yl] (4-methoxyphenyl)methanone and SR-144528, N-[(1S)-endo-1,3,3-Trimethylbicyclo[2.2.1]heptan-2-yl]-5-(4-chloro-3-methylphenyl)-1-(4-methoxybenzyl)-pyrazole-3-carbo xamide. All drugs were obtained from Tocris Bioscience (Ellisville, MO, USA) except HU-210 and SR-144528 that were generous gifts from the National Institute on Drug Abuse drug inventory supply and control system.
In-house G93A mouse colony
Hemizygous transgenic male mice with the G93A mutation of the human SOD1 gene (G1H/1) mutation [B6SJL-TgN (SOD1-G93A) 1 Gur] were obtained from Jackson Laboratories and were bred locally with female B6SJL mice (Jackson Laboratory, Bar Harbor, ME, USA), according to the protocol obtained from the vendor. To decrease the inherent variability in disease onset and survival with these mice, littermate transgenic males (rather than random males from several different litters) were selected to sire subsequent generations of mice. Within three generations, the variability was all but eliminated such that the transgenic mice develop characteristic hind limb weakness at 90 days of age (±1–2 days) and progress to end-stage disease (requiring killing) within 18–30 days after onset; this has remained relatively constant for the last eight generations of mice.
Determination of symptom onset, randomization and drug treatment of G93A mice
Symptom onset was assessed by blinded observation of changes in hind limb gait; these changes are related to the mouse’s inability to support its weight on its hind limbs. At onset, mice initially place their weight on the toes and then quickly fall to full foot placement (as opposed to healthy mice which walk and run on the hind toes); this ‘toe-to-heel’ walking pattern produces an asymmetric gait between hind and fore limbs and a characteristic ‘wobbling’ gait Mouse groups were randomized based on symptom onset and alternately placed in control and treated groups, e.g., the first mouse to develop hind limb weakness was placed in the control group, the second was injected with test compound and placed in the treatment group, and so on. The net effect of this type of randomization was to create groups with mean onset ages which are virtually identical, thereby allowing the use of smaller numbers of mice (typically 10–13 per group) and still maintain sufficient statistical power. By definition, the onset administration paradigm employed was focused on what we term the ‘survival interval’ – namely the time from onset to end-stage killing. Because both drug- and vehicle-treated groups were derived from the same groups of age-matched mice, survival results were properly normalized by comparing survival intervals of drug-treated to survival intervals of vehicle-treated groups to determine survival interval ratios (SIRs).
All drugs and vehicle were administered once daily by the i.p route beginning on the first day of symptom onset. AM-1241 and WIN-55,212 have very poor water solubility and require a vehicle which is both capable of dissolving the drug and is biocompatible (with chronic dosing). Other groups have used complex vehicles composed of polyethoxylated vegetable oils and/or ethanol/glycerol/water mixtures. We tested a number of traditional vehicles such as ethanol/water, glycerol, polyethylene glycol, and high purity olive oil. Stable dissolution of AM-1241 and WIN-55,212 was achieved only with olive oil, and thus it was selected as the vehicle for these studies. Two different concentrations of AM-1241 (1 and 0.1 mg/mL) and one WIN-55,212 concentration (1 mg/mL) were prepared in order to minimize the volume of olive oil that was injected i.p.
Determination of survival end points and euthanasia
Mice were killed when any of the following criteria were met: (i) inability to right themselves within 30 s when placed on their sides; (ii) inability to eat or drink, or move toward food and water placed in low-rimmed dishes on cage floor; (iii) loss of more than 10% of total body weight in 24 h; (iv) gross loss of grooming behavior; or (v) labored breathing. Criteria for death were confirmed by a second investigator who is blinded to the group identity of each mouse. The age of symptom onset was subtracted from the age at death for each mouse, and a mean survival interval was calculated for each group. By calculating the ratio of the survival interval of treated groups to the survival interval of untreated littermate controls, a X-fold increase in survival was readily determined.
Membrane preparation
Brain regions were dissected from fresh mouse brains placed on an ice-cooled surface. Spinal cords, individual brain regions or spleen were suspended in a homogenization buffer containing 50 mmol/L Hepes, pH 7.4, 3 mmol/L MgCl2, and 1 mmol/L EGTA. Using a 7 mL Dounce glass homogenizer (Wheaton, Millville, NJ, USA), samples were subjected to 10 complete strokes and centrifuged at 40,000 g for 10 min at 4°C. After repeating the homogenization procedure twice more, the samples were resuspended in Hepes buffer (50 mmol/L, pH 7.4) and subjected to 10 strokes utilizing a 7 mL glass homogenizer. Membranes were stored in aliquots of approximately 1 mg/mL at −80°C.
Quantitative real-time PCR
Total RNA was isolated from G93A and WT-OE tissues using an RNeasy minikit and QiaShredder columns (Qiagen, Valencia, CA, USA). Genomic DNA contamination was eliminated using DNAse-free (Ambion, Austin, TX, USA). Total RNA (1 μg) was reverse transcribed according to commercial instructions (iScript cDNA synthesis kit; Bio-Rad Hercules, CA, USA) to generate cDNA at 25°C for 5 min, followed by 42°C for 30 min and 85°C for 5 min. The cDNA sequences for the appropriate targets were amplified using the polymerase chain reaction and corresponding primers (Operon, Huntsville, AL, USA) (Table 1). The PCR mixture contained 1× iQ SYBR Green Supermix (Bio-Rad), 200 nmol/L each of forward and reverse primers, and 10 ng of template. After initial denaturation at 95°C for 3 min, the following temperature-cycling profile for the amplification was used (40 cycles): 95°C for 10 s denaturing and 62°C for 1 min for annealing and extension. Melting curve analysis was accomplished in 80 cycles. The steps included 95°C for 1 min for denaturation, 55°C for 1 min to permit final extension, and 0.5°C temperature increments for 10 s each cycle from 55 to 95°C. Amplified cDNA products were analyzed using iCycler software (Bio-Rad).
Table 1.
Primer sequences
| Target | Forward | Reverse | Product (bp) |
|---|---|---|---|
| CB1 | 5′-CTGAACTCCACCGTGAACC-3′ | 5′-TTATTGGCGTGCTTGTGC-3′ | 152 |
| CB2 | 5′-TGCACTGGCTCTCATGG-3′ | 5′-GAGCGAATCTCTCCACTCC-3′ | 144 |
| GAPDH | 5′-GGGAAGCTCACTGGCATGG-3′ | 5′-CTCTTGATGTCATCATCATACTTGGCAG-3′ | 123 |
Western blots
To identify CB1 and CB2 receptors, each sample containing 100 μg of spinal cord membrane protein was separated by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis on 10% (w/v) polyacrylamide mini-gels. Prior to separation, samples were re-suspended in 40 μL of electrophoresis loading buffer (0.065 mol/L Tris–HCl, pH 6.8, 2% SDS, 10% glycerol, 5% 2-mercaptoethanol), and heated at 90°C for 2 min. The enhanced chemiluminescence (ECL) method of immunoblotting was employed (GE Healthcare/Amersham Biosciences, Piscataway, NJ, USA). Gels were transferred to Hybond-ECL nitrocellulose membranes and incubated overnight at 4°C with 10% milk in blotting buffer (TBS-0.1%) (Tris–HCl, pH 7.6, 25 mmol/L; NaCl, 0.09%; Tween-20, 0.1%). Blots were then washed three times (5 min each) with TBS-0.1% and incubated with primary antibodies (1 : 200) overnight at 4°C while shaking. For selected blots, the appropriate blocking peptide (1 : 100) was incubated with the respective primary antibody for 1 h at room temperature (25°C) prior to incubation with blots. The primary antibody solutions were removed and blots washed as described previously. Secondary antibody (donkey anti-rabbit horseradish peroxidase, 1 : 5000) was added and incubated for 4 h, with shaking. The secondary antibody was removed and blots washed as described. Blots were incubated for 1 min with equal volumes of ECL detection reagents 1 and 2. Chemiluminescence was captured for 2 h and saved as a TIFF file by a Flurochem 8900 MultiImage Light Cabinet (Alpha Innotech Corp., San Leandro, CA, USA). The captured images were digitized and the relative cannabinoid receptor levels compared after densitometry analysis (Scion NIH Image 1.63, NIH Freeware, NIH, Bethesda, MD, USA). The relative protein levels were calculated by normalizing to actin immunoreactivity and subtracting the background intensity.
The primary antibodies and blocking peptides for both the CB1 and CB2 receptors were purchased from Cayman Chemical (Ann Arbor, MI, USA). The CB1 receptor polyclonal antibody (Catalog No. 10006590) was raised against the C-terminal amino acids 461–472 of the human CB1 receptor. This antigen is identical to the corresponding sequence in murine, rat, canine and bovine species. The CB2 receptor polyclonal antibody (Catalog No. 101550) was raised against amino acids 20–33 in a sequence between the N-terminus and the first transmembrane domain of the protein of the human CB2 receptor. Human and murine CB2 receptors exhibit 82% homology at the amino acid level over the complete protein. CB1 (Catalog No. 10006591) and CB2 (Catalog No. 301550) blocking peptides were derived from the CB1 and CB2 receptor sequences used as antigens for production of the respective polyclonal antiserum.
Cannabinoid receptor binding
Each binding assay contained 30 μg of spinal cord membrane protein in a final volume of 1 mL in binding buffer (50 mmol/L Tris, 0.1% bovine serum albumin, 5 mmol/L of MgCl2, pH 7.4), as described previously (Prather et al. 2000b). [3H]CP-55,940 (180 Ci/mmol, Perkin-Elmer Life and Analytical Sciences, Wellesley, MA, USA) binds with equivalent affinity to CB1 and CB2 receptors with an approximate Ki of 0.5 nmol/L (Shoemaker et al. 2005). Specific CB1 receptor binding was defined as the binding of a receptor saturating concentration of [3H]CP-55,940 (5 nmol/L) displaced by a receptor saturating concentration of the CB1 selective ligand AM-251 (200 nmol/L). AM-251 displays high affinity for CB1 receptors with a Ki value of about 7 nmol/L, whereas its affinity at CB2 receptors is over 300-fold weaker (Gatley et al. 1997). Specific CB2 binding was defined as the binding of 5 nmol/L [3H]CP-55,940 displaced by a receptor-saturating concentration of the CB2 selective ligand AM-630 (200 nmol/L). AM-630 binds CB2 receptors with high affinity [Ki value of about 20 nmol/L (Shoemaker et al. 2005)], whereas its affinity for CB1 receptors is more than 165-fold less (Hosohata et al. 1997). All binding experiments were performed in triplicate. Reactions were terminated by rapid vacuum filtration through Whatman GF/B glass fiber filters followed by two washes with ice-cold binding buffer. About 4 mL of Scintiverse® (Fisher Scientific, Pittsburgh, PA, USA) was added to the filters and radioactivity quantified by scintillation counting.
[35S]GTPγS binding
[35S]GTPγS binding assays were performed as described previously (Prather et al. 2000a) in a buffer containing 20 mmol/L Hepes, 100 mmol/L NaCl, and 10 mmol/L MgCl2 at pH 7.4. Each binding reaction contained 10 μg of spinal cord membrane protein, the presence or absence of cannabinoid ligands (see the following), plus 0.1 nmol/L [35S]GTPγS (1250 Ci/mmol, Perkin-Elmer Life and Analytical Sciences) and 10 μmol/L of GDP to suppress basal G-protein activation. Reactions were incubated for 2 h at 30°C. Non-specific binding was defined by binding observed in the presence of 10 μmol/L of non-radioactive GTPγS. The reaction was terminated by rapid vacuum filtration through glass fiber filters followed by two washes with ice-cold assay buffer. About 4 mL of Scintiverse (Fisher Scientific) was added to the filters and radioactivity quantified by scintillation counting.
Cannabinoid-mediated G-protein activation in spinal cord membranes was measured by selective antagonism of the [35S]GTPγS binding produced by a receptor-saturating concentration (100 nmol/L) of the full, non-selective CB1/CB2 agonist HU-210. HU-210 binds with equivalent affinity to CB1 and CB2 receptors with an approximate Ki of 0.5 nmol/L (Felder et al. 1995; Breivogel et al. 2001). In these studies, we first determined the minimal concentration of the neutral CB1 antagonist O-2050 (Gardner and Mallet 2006) required to completely block CB1-mediated G-protein activation by HU-210. This was accomplished by antagonism experiments employing membranes prepared from mouse cortex as a relatively pure source of CB1 receptors. In these studies, it was determined that 3 μmol/L of O-2050 was the minimal concentration required to completely block HU-210-mediated (100 nmol/L) G-protein activation by CB1 receptors in cortical membranes (data not shown). Next, the minimal concentration of the selective CB2 antagonist SR-144528 (Rinaldi-Carmona et al. 1998) required to completely block CB2-mediated G-protein activation by HU-210 was determined. This was accomplished by antagonism experiments employing membranes prepared from CHO–CB2 cells (Shoemaker et al. 2005) as a pure source of CB2 receptors. In these studies, it was shown that 3 μmol/L of SR-144528 was the minimal concentration required to completely block HU-210-mediated (100 nmol/L) G-protein activation by CB2 receptors in CHO–CB2 membranes (data not shown). Therefore, employing spinal cord membranes harvested from WT-OE and G93A mice, CB1-selective stimulation was defined as the amount of O-2050 (3 μmol/L) sensitive G-protein stimulation produced by HU-210 (100 nmol/L). CB2-selective activation was defined as the amount of SR-144528 (3 μmol/L) sensitive G-protein stimulation produced by HU-210 (100 nmol/L).
The selective antagonism method described here was developed in response to many failed attempts to demonstrate consistent, measurable G-protein activation with the selective CB1 agonist ACEA (Hillard et al. 1999) or the CB2 agonists GW-405833 (Valenzano et al. 2005) and AM-1241 (Yao et al. 2006) in mouse spinal cord membranes (data not shown). While these observations were surprising for the full CB1 agonist ACEA (Hillard et al. 1999), both GW-405833 and AM-1241 have been reported to act as partial agonists in several in vitro assays (Valenzano et al. 2005; Yao et al. 2006). In any case, it is likely that the poor G-protein stimulation produced by partial agonists in the present study is due to less than optimal experimental conditions and/or a relatively low density of cannabinoid receptors expressed in spinal cord membranes, leading to reduced receptor-mediated responses.
Statistical analysis
All curve-fitting and statistical analysis were conducted by employing the computer program GraphPad Prism® version 4.0b (Graph-Pad Software Inc., San Diego, CA, USA). All data are expressed as mean ± SEM. To compare three or more groups of data that follow a Gaussian distribution, statistical significance of the data was determined by a one-way anova, followed by a post hoc comparison using a Dunnett’s test. To compare two groups of data that follow a Gaussian distribution, the non-paired Student’s t-test was utilized. To compare three or more groups of data that do not follow a Gaussian distribution, statistical significance of the data was determined by the non-parametric Kruskal–Wallis test, followed by post hoc comparisons using a Dunn’s test. Kaplan–Meier survival analysis and the log rank (Mantel–Cox) test were used for survival comparisons.
Results
Initial experiments examined the spatial and temporal expression of CB2 receptors in the CNS of G93A mice (Figs 1 and 2). First, quantitative real-time polymerase chain reaction compared CB1 and CB2 receptor mRNA expressions in the spinal cords of G93A mice relative to age-matched mice overexpressing the human wild-type-SOD1 gene (WT-OE) (Fig. 1a). The amplification efficiency of the primers designed for the targets (CB1, CB2) and reference glyceraldehyde-3-phosphate dehydrogenase (GAP-DH) cDNAs was equivalent (data not shown) and the PCR products were of the predicted size (Fig. 1a, inset). Therefore, the comparative Ct method was employed for mRNA comparison (Giulietti et al. 2001). The expression level of CB1 mRNA (right panel) is slightly elevated in the spinal cords of 100 (3.7 ± 0.4-fold, p < 0.05, n = 6), but not 60-(2.7 ± 0.8-fold, n = 3) or 120- (1.1 ± 0.2-fold, n = 3) day-old G93A mice, compared with age-matched WT-OE control animals. In addition, a small but significant (p < 0.05) decrease of CB1 mRNA occurs in end-stage G93A mice (120 days of age), relative to 100-day-old G93A mice. In contrast, CB2 mRNA (left panel) is significantly elevated in the spinal cords of 60- (3.6 ± 0.3-fold, p < 0.01, n = 3), 100-(12.6 ± 2.0-fold, p < 0.01, n = 6) and 120- (27.9 ± 6.5-fold, p < 0.01, n = 3) day-old G93A mice relative to age-matched WT-OE controls. Furthermore, the elevation in CB2 mRNA is age-dependent, increasing slightly in 60-day-old mice prior to symptom onset and rising to the highest levels in 120-day-old mice (p < 0.01).
Fig. 1.
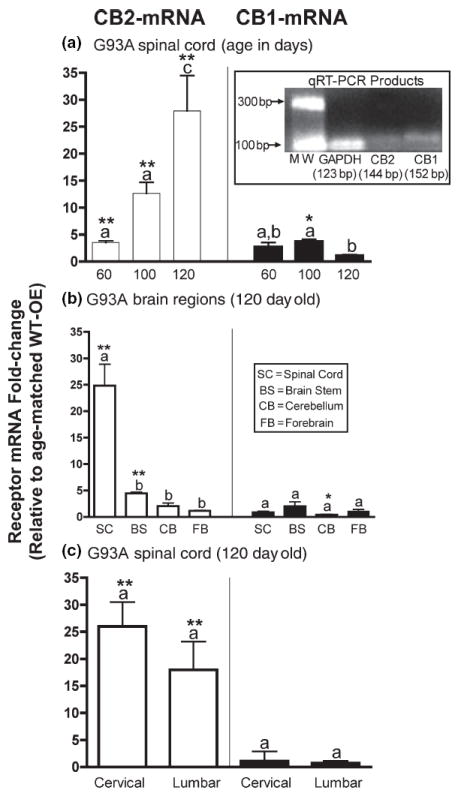
CB2, but not CB1, receptor mRNA is dramatically and selectively up-regulated in the spinal cords of G93A mice in a temporal pattern closely paralleling disease progression. (a) Comparison of CB2 (left panel) and CB1 (right panel) mRNA expression in spinal cords of G93A mice at various ages, relative to age-matched WT-OE control mice. (inset) Separation of PCR products by 1% agarose gel electrophoresis. (b) Comparison of CB2 (left panel) and CB1 (right panel) mRNA expression in various brain regions of 120-day-old G93A mice, relative to age-matched WT-OE control mice. (c) Comparison of CB2 (left panel) and CB1 (right panel) mRNA expression in cervical and lumbar regions of spinal cords of 120-day-old G93A mice, relative to age-matched WT-OE control mice. *, **Significantly different from mRNA expression in WT-OE control mice, p < 0.05, 0.01 (non-paired Student’s t-test). a–cFold-changes that are designated with different letters are significantly different, p < 0.05 (anova followed by a Dunnett’s post hoc comparison).
Fig. 2.
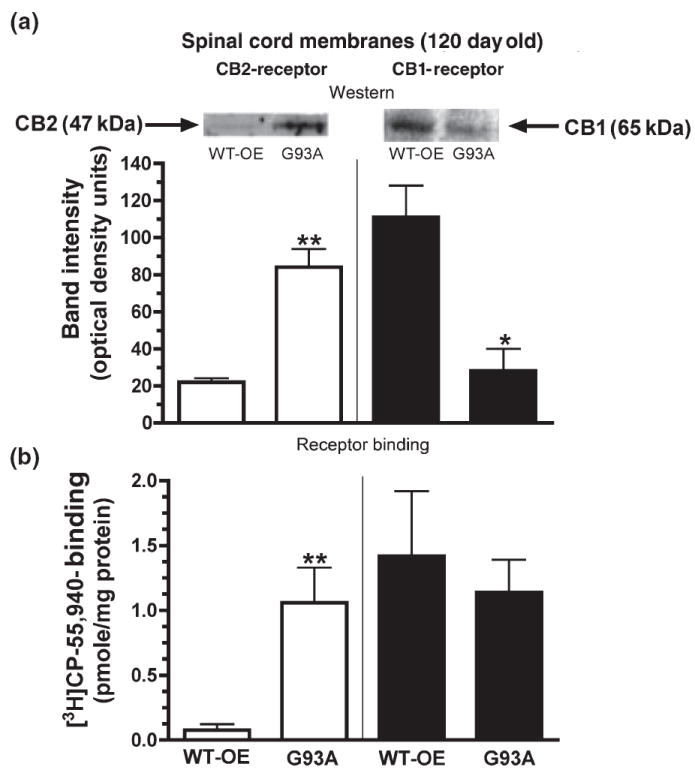
Up-regulation of CB2 receptor mRNA in spinal cords of symptomatic G93A mice is reflected by increased CB2 receptor immunoreactivity and binding. (a) Comparison of CB2 (left panel) and CB1 (right panel) protein levels by western analysis in spinal cord membranes of 120-day-old G93A and age-matched WT-OE control mice. The relative protein levels were calculated by normalizing to actin immunoreactivity and subtracting the background intensity (insets). Representative western blot of CB2 (left panel) and CB1 (right panel) receptors. (b) Comparison of specific receptor binding of CB2 (left panel) and CB1 (right panel) receptors in spinal cord membranes of 120-day-old G93A and age-matched WT-OE control mice. Specific CB1 receptor binding was defined as the binding of a receptor-saturating concentration of [3H]CP-55 940 (5 nmol/L) displaced by a receptor-saturating concentration of the CB1 selective ligand AM-251 (200 nmol/L). Specific CB2 binding was defined as the binding of 5 nmol/L [3H]CP-55 940 displaced by a receptor-saturating concentration of the CB2 selective ligand AM-630 (200 nmol/L). *, **Significantly different from the value obtained for WT-OE control mice, p < 0.05, 0.01 (non-paired Student’s t-test).
To determine whether CB2 mRNA up-regulation in the CNS of G93A mice is correlated in any way to disease pathology, cannabinoid receptor mRNA expression was examined in the spinal cord (SC), brainstem (BS), cerebellum (CB) and forebrain (FB) of end-stage (120-day-old) G93A mice, relative to age-matched WT-OE controls (Fig. 1b). While CB1 mRNA (right panel) is slightly decreased in the cerebellum of end-stage G93A mice relative to WT-OE controls (0.4 ± 0.1-fold, p < 0.05, n = 3), this reduction is not significantly different when compared with CB1 mRNA changes in all other brain regions of G93A mice (SC: 0.8 ± 0.3-fold, n = 6; BS: 2.0 ± 0.9-fold, n = 3; FB: 1.0 ± 0.4-fold, n = 4). In sharp contrast, CB2 mRNA is significantly increased only in the spinal cord (24.8 ± 4.1-fold, p < 0.01, n = 5) and brainstem (4.5 ± 0.3-fold, p < 0.01, n = 3), but not in cerebellum (2.0 ± 0.6-fold, n = 3) or forebrain (1.1 ± 0.4-fold, n=4). CB2 mRNA up-regulation is much greater in the spinal cord than in the brainstem (p < 0.01) of G93A mice, consistent with disease pathogenesis.
Cannabinoid receptor mRNA expression in lumbar and cervical regions of spinal cords of end-stage G93A mice was next examined (Fig. 1c). CB1 mRNA levels (right panel) are unchanged in either the cervical (1.1 ± 1.8-fold, n = 3) or lumbar (0.72 ± 0.4-fold, n = 3) spinal cord regions. Unlike the reported regional distribution of endocannabinoids (Witting et al. 2004), CB2 receptor mRNA up-regulation is similar in both the cervical (25.6 ± 4.5-fold, n = 3) and lumbar (18.0 ± 5.2-fold, n = 3) regions of G93A spinal cords when compared with age-matched WT-OE control mice.
The density and function of cannabinoid receptors was next examined in membranes prepared from spinal cords using western analysis (Fig. 2a), receptor binding (Fig. 2b) and [35S]GTPγS binding (Fig. 3) assays. In initial optimization studies, the CB1 receptor antibody identified an immunoreactive band in membranes prepared from mouse cortex (a relatively pure source of CB1 receptors), but not from CHO–CB2 membranes, with a molecular weight predicted for CB1 receptors of approximately 65 kDa (data not shown). In contrast, a 47-kDa immunoreactive band corresponding to the predicted molecular weight for CB2 receptors was recognized by the CB2 receptor antibody in membranes prepared from CHO–CB2 cells (a pure source of CB2 receptors), but not from mouse cortex (data not shown). In spinal cord membranes prepared from WT-OE and G93A mice (Fig. 2a), selective antibodies identified immunoreactive bands with the predicted molecular weight for CB2 (left panel inset) or CB1 (right panel inset) receptors. Furthermore, the band recognized by both antibodies was eliminated upon pre-incubation of antibodies with an excess of the appropriate blocking peptide (data not shown). Although little CB2 receptor immunoreactivity is present in spinal cords of 120-day-old WT-OE mice (22.0 ± 2.1 OD units, n = 3), approximately fourfold greater CB2 receptor density (p < 0.01) is observed in end-stage G93A animals (84.0 ± 9.9 OD units, n = 3). In contrast, CB1 receptor immunoreactivity is decreased (p < 0.05) almost fourfold in spinal cord membranes of 120-day-old G93A (28.0 ± 12.0 OD units, n = 3), relative to WT-OE (111.0 ± 17.0 OD units, n = 3) control mice.
Fig. 3.
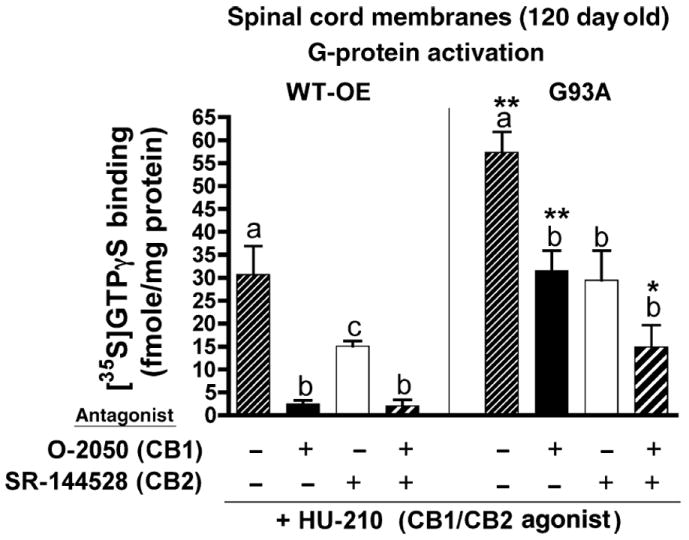
Up-regulation of CB2 receptor mRNA and protein levels in spinal cords of symptomatic G93A mice is reflected by increased function of CB2 receptors. Comparison of CB1 and CB2 receptor stimulation of [35S]GTPγS binding in spinal cord membranes prepared from WT-OE (left panel) or G93A (right panel) mice. Cannabinoid-mediated G-protein activation in spinal cord membranes was measured by selective antagonism of the [35S]GTPγS binding produced by a receptor-saturating concentration (100 nmol/L) of the full, non-selective CB1/CB2 agonist HU-210 (narrow hatched bars). CB1 stimulation was defined as the amount of HU-210 (100 nmol/L) stimulated G-protein stimulation blocked by the CB1-selective antagonist O-2050 (3 μmol/L) (filled bars). CB2 stimulation was defined as the amount of HU-210 (100 nmol/L) stimulated G-protein stimulation blocked by the CB2-selective antagonist SR-144528 (3 μmol/L) (open bars). CB1/CB2-stimulation was defined as the amount of HU-210 (100 nmol/L) stimulated G-protein stimulation blocked by concurrent incubation with O-2050 (3 μmol/L) and SR-144528 (3 μmol/L) (wide hatched bars). *, **Significantly different from levels of [35S]GTPγS binding (fmoles/mg protein) produced in response to identical conditions in WT-OE spinal cord membranes, p <0.05, 0.01 (non-paired Student’s t-test). a–cLevels of [35S]GTPγS binding (fmoles/mg protein) that are designated with different letters are significantly different, p < 0.05 (anova followed by a Dunnett’s post hoc comparison).
Cannabinoid receptor binding experiments (Fig. 2b) were conducted to confirm the results observed from western analysis. Similar to results reported for mRNA and western analysis, predominantly CB1 (1.42 ± 0.5 pmol/mg, n = 3) and much less CB2 (0.077 ± 0.046 pmol/mg, n = 3) receptors are present in spinal cord membranes of 120-day-old WT-OE control mice. In agreement with elevated CB2 mRNA and immunoreactivity, CB2 receptor density also is increased over 13-fold in the spinal cords of 120-day-old G93A mice (1.06 ± 0.27 pmol/mg, p < 0.01, n = 3), relative to that observed in age-matched WT-OE controls. Similar to decreased immunoreactivity, CB1 receptor density also is reduced slightly, although not significantly, by 20% (to 1.14 ± 0.25 pmol/mg, n = 3) in 120-day-old G93A relative to age-matched WT-OE control mice.
To determine whether the up-regulated CB2 receptors in G93A spinal cord membranes are functional, G-protein activation assays were conducted (Fig. 3). We initially attempted to compare CB1 and CB2 receptor activation of G-proteins between WT-OE and G93A spinal cord membranes by conducting [35S]GTPγS binding assays in the presence of selective agonists. However, after considerable effort, we were unable to demonstrate consistent, measurable G-protein activation with the selective CB1 agonist ACEA (Hillard et al. 1999) or the CB2 agonists GW-405833 (Valenzano et al. 2005) and AM-1241 (Yao et al. 2006) in mouse spinal cord membranes (data not shown). Therefore, G-protein activation produced by CB1 and CB2 receptors was instead quantified by selectively antagonizing the [35S]GTPγS binding produced by the CB1/CB2 full agonist HU-210 (Felder et al. 1995; Breivogel et al. 2001) with the CB1 antagonist 0–2050 (Gardner and Mallet 2006) or the CB2 antagonist SR-144528 (Rinaldi-Carmona et al. 1998).
In WT-OE spinal cord membranes (Fig. 3, left panel), stimulation of CB1/CB2 receptors by HU-210 produces 30.7 ± 6.2 fmol/mg protein (n = 4) of [35S]GTPγS binding to G-proteins. Co-incubation with the CB1 selective antagonist O-2050 almost completely blocks G-protein stimulation by HU-210 (to 2.5 ± 0.8 fmol/mg protein, p < 0.01, n = 4). Interestingly, the CB2 selective antagonist SR-144528 also significantly reduces HU-210 stimulation by approximately 50% (to 15.1 ± 1.1 fmol/mg protein, p < 0.05, n = 4). As might have been anticipated, co-incubation of HU-210 with both antagonists concurrently also reduces G-protein activation by over 90% (to 2.1 ± 1.3 fmol/mg protein, p < 0.01, n = 4). Collectively, these data indicate that the stimulation of G-proteins produced by HU-210 in WT-OE spinal cord membranes occurs primarily via activation of CB1 receptors. Although the partial reduction of G-protein stimulation by HU-210 in the presence of the CB2 selective antagonist SR-144528 suggests that CB2 receptors may also participate, it is possible that the observed results might be due to non-selective blockade of CB1 receptors by the 3 μmol/L concentration of SR-144528 employed in the assay.
In G93A spinal cord membranes (Fig. 3, right panel), stimulation of CB1/CB2 receptors by HU-210 produces a significantly greater increase in [35S]GTPγS binding to G-proteins (57.4 ± 4.4 fmol/mg protein, p < 0.01, n = 4) relative to that observed in WT-OE membranes. Furthermore, in G93A membranes, co-incubation of HU-210 with the CB1 selective antagonist O-2050 reduces G-protein stimulation by only 46% (to 31.5 ± 4.4 fmol/mg protein, p < 0.05, n = 4), compared with near-complete blockade in WT-OE membranes. Importantly, although the percent blockade of HU-210-induced G-protein activation by O-2050 in G93A membranes is half of that observed in WT-OE membranes (45% vs. 92%), the net reduction in fmoles of activated G-proteins by O-2050 is almost identical between membrane preparations. In other words, O-2050 reduced HU-210-induced G-protein activation by 28.3 fmol/mg protein in WT-OE membranes (30.7 − 2.5 = 28.3 fmol/mg protein) and 25.9 fmol/mg protein in G93A membranes (57.4 - 31.5 = 25.9 fmol/mg protein). This indicates that CB1 receptors activate similar levels of G-proteins in both WT-OE and G93A tissues. The CB2 selective antagonist SR-144528 also significantly (p < 0.05, n = 4) reduces HU-210 G-protein stimulation in G93A membranes by 49%, to 29.5 ± 6.4 fmol/mg protein. In contrast to that observed for CB1 receptors, the net reduction in fmoles of activated G-proteins by SR-144528 is markedly different between membrane preparations. For example, SR-144528 reduces G-protein activation by 15.6 fmol/mg protein in WT-OE membranes (30.7 − 15.1 = 15.6 fmol/mg protein) and 27.9 fmol/mg protein in G93A membranes (57.4 − 29.5 = 27.9 fmol/mg protein). This suggests that CB2 receptors activate approximately twice the amount of G-proteins in G93A, relative to WT-OE spinal cord membranes. Very interestingly, although coincubation of HU-210 with both antagonists concurrently reduces G-protein activation to a level lower than that obtained with either antagonist alone, a significant level of HU-210-activated G-proteins cannot be blocked under these conditions (14.9 ± 4.8 fmol/mg protein, n = 4). These data indicate that HU-210 may activate G-proteins via a non-CB1/CB2 receptor in spinal cord membranes prepared from G93A, but not WT-OE mice.
The effect of chronic administration of cannabinoids on the survival of G93A mice was next examined (Fig. 4). Two cannabinoid agonists were tested, WIN-55,212 and AM-1241. WIN-55,212 exhibits a slightly higher affinity for human CB2 (3.3 nmol/L), when compared with CB1 (62.3 nmol/L) receptors (Felder et al. 1995). In contrast, AM-1241 displays over an 80-fold higher affinity for CB2 (7 nmol/L), relative to CB1 (580 nmol/L) receptors (Yao et al. 2006). Mice were administered daily i.p. injections, beginning at onset of symptoms, with one of four treatments: vehicle (Fig. 4a–c, n = 9), the relatively non-selective CB1/CB2 agonist WIN-55,212 (5 mg/kg, Fig. 4a, n = 6), the selective CB2 agonist AM-1241 (0.3 mg/kg, Fig. 4b, n = 14) or AM-1241 (3 mg/kg, Fig. 4c, n = 14). The number of days between symptom onset and animal killing was measured (e.g., survival interval). In humans, this is analogous to the time between diagnosis of ALS and death, ranging from 2 to 5 years. Mice injected with vehicle (open squares in all panels) survive from 18 to 30 days following symptom onset, with an average survival interval of 23.7 ± 1.7 days (Fig. 4d). Treatment at onset with the non-selective CB1/CB2 agonist WIN-55,212 produces a significant rightward shift in the survival curve (p < 0.0249), reflected by an increase of 8.8 days in the survival interval (32.5 ± 3.6 days, p < 0.0496) (Fig. 4a). Onset administration with either 0.3 (Fig. 4b, p < 0.0017) or 3.0 mg/kg (Fig. 4c, p < 0.0005) of the selective CB2 agonist AM-1241 results in a highly significant extension of survival. Mice receiving daily injections of 0.3 and 3 mg/kg AM-1241 live an average of 9.7 (33.4 ± 2.2 days, p < 0.0081) and 13.2 (36.9 ± 2.8 days, p < 0.0022) days longer after symptom onset than vehicle-treated controls, respectively (Fig. 4d).
Fig. 4.
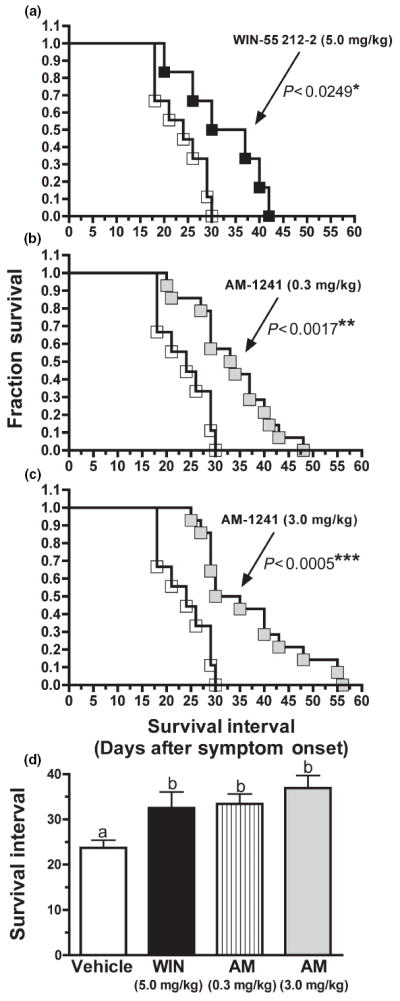
Treatment with the selective CB2 agonist AM-1241, initiated at symptom onset, produces a pronounced increase in survival of G93A-SOD1 mice. (a–c) Comparison of the effects of daily treatment, initiated at symptom onset, on survival of G93A mice with (a) 5 mg/kg WIN-55 212 (n = 6), (b) 0.3 mg/kg AM-1241 (n = 14) or (c) 3.0 mg/kg AM-1241 (n = 14). The response of vehicle-treated control G93A mice (n = 9) is represented by the open squares in each panel. (d) Comparison of the survival interval of G93A mice treated daily with vehicle or the listed drugs. *, **, ***Significantly different from the vehicle-treated survival curve, p < 0.0249, 0.0017, 0.0005 (Kaplan–Meier survival analysis and log rank test (Mantel–Cox). a–bSurvival intervals that are designated with different letters are significantly different, p < 0.05 (Kruskal–Wallis test followed by a Dunn’s post hoc comparison).
When compared with the efficacy of other drugs evaluated in the G93A mouse model (Table 2), the magnitude of effect produced by AM-1241 initiated at symptom onset rivals the best yet reported for any pharmacological agent, even those given pre-symptomatically. The most effective dose of AM-1241 produced a SIR of 1.56, with mice living 56% longer after symptom onset than controls. If extension of total life span is considered, AM-1241 produced a total life span ratio of 1.11 (e.g., mice live 11% longer than controls).
Table 2.
Comparison of the effect of drugs on survival interval and total life-span in G93A mice
| Drug | Route | Began | Survival interval ratio | Total life-span ratio | Reference |
|---|---|---|---|---|---|
| Drugs given at onset of symptoms | |||||
| MnPorphyrin | i.p. | Onset | 2.96 | 1.26 | Crow et al. (2005) |
| Cyclosporine-A | i.t. | Onset | 2.05 | 1.11 | Kirkinezos et al. (2004) |
| AM-1241 (3.0 mg/kg) | i.p. | Onset | 1.56 | 1.11 | Fig. 4c |
| FeTCPP | i.p. | Onset | 1.55 | 1.05 | Wu et al. (2003) |
| Arimoclomol | i.p. | Onset | 1.42 | 1.18 | Kieran et al. (2004) |
| AM-1241 (0.3 mg/kg) | i.p. | Onset | 1.41 | 1.07 | Fig. 4b |
| WIN-55 212 | i.p. | Onset | 1.37 | 1.04 | Fig. 4a |
| ZK18763870 (140 mg/kg) | Diet | Onset | 1.34 | 1.11 | Tortarolo et al. (2006) |
| NDGA | Oral | Onset | 1.32 | 1.10 | West et al. (2004) |
| Ceftriaxone | i.p. | Onset | 1.26 | 1.08 | Rothstein et al. (2005) |
| ZK18763870 (70 mg/kg) | Diet | Onset | 1.22 | 1.07 | Tortarolo et al. (2006) |
| Δ9-THC (20 mg/kg) | i.p. | Onset | 1.13 | 1.05 | Raman et al. (2004) |
| Drugs given prior to onset of symptoms | |||||
| EUK-134 | i.p. | 60 | 1.57 | 1.10 | Jung et al. (2001) |
| Arimoclomol | i.p. | 35 | 1.51 | 1.22 | Kieran et al. (2004) |
| Ceftriaxone | i.p. | 42 | 1.34 | 1.11 | Rothstein et al. (2005) |
| Minocycline (25 mg/kg) | i.p. | 70 | 1.32 | 1.10 | Van Den Bosch et al. (2002) |
| EUK-8 | i.p. | 60 | 1.22 | 1.08 | Jung et al. (2001) |
| Pioglitazone | Diet | 30 | 1.18 | 1.13 | Kiaei et al. (2005) |
| Memantine | s.c. | 70 | 1.17 | 1.07 | Wang and Zhang (2005) |
| Δ9-THC (10 mg/kg) | i.p. | 60 | 1.12 | 1.04 | Raman et al. (2004) |
| Minocycline (50 mg/kg) | i.p. | 70 | 1.12 | 1.16 | Van Den Bosch et al. (2002) |
| Minocycline + creatine | i.p./diet | 28 | 1.09 | 1.24 | Zhang et al. (2003) |
| Pioglitazone | Diet | 57 | 1.0 | 1.08 | Kiaei et al. (2005) |
| Creatine | Diet | 28 | 0.96 | 1.12 | Zhang et al. (2003) |
| Minocycline | i.p. | 28 | 0.91 | 1.13 | Zhang et al. (2003) |
| Cannabinol | s.c. | 40 | N/D | 1.0 | Weydt et al. (2005) |
| Lenalidomide (100 mg/kg) | Gavage | 30 | N/D | 1.18 | Kiaei et al. (2006) |
| Thalidomide (50 mg/kg) | Gavage | 30 | N/D | 1.11 | Kiaei et al. (2006) |
| Thalidomide (100 mg/kg) | Gavage | 30 | N/D | 1.16 | Kiaei et al. (2006) |
Discussion
In G93A mutant mice, the most well-characterized animal model of ALS (Gurney 1997), endocannabinoids are elevated in spinal cords of affected animals (Witting et al. 2004; Bilsland et al. 2006). Additionally, treatment with non-selective cannabinoid partial agonists prior to, or upon, symptom appearance minimally delays disease onset and prolongs survival (Raman et al. 2004; Weydt et al. 2005). However, the basis of the therapeutic effect of cannabinoids and the role of CB1 and CB2 receptors in relation to disease progression in G93A mice have not been determined. Furthermore, the potential therapeutic effect of selective CB2 agonists, which appear to be most efficacious for treatment of chronic neuroinflammatory conditions (Walter and Stella 2004; Ramirez et al. 2005), have yet to be examined in this animal model of ALS. We demonstrate that mRNA, receptor binding and function of CB2, but not CB1, receptors are dramatically and selectively up-regulated in the spinal cords of G93A mice in a temporal pattern closely paralleling disease progression. More importantly, we show for the first time that daily i.p. injections of mice with the selective CB2 agonist AM-1241, initiated at symptom appearance, increase the survival interval after symptom onset by 56%. Taken collectively, findings from this study indicate that CB2 agonists may ultimately be developed as novel therapeutic drugs that can be administered alone or in combination with other agents at symptom onset for the treatment of ALS in human patients.
Recent evidence indicates that ALS is a disease characterized by chronic inflammation (McGeer and McGeer 2002; Weydt and Moller 2005). Furthermore, CB2 receptors are up-regulated in the target tissues of several neuroinflammatory diseases (Benito et al. 2003; Ramirez et al. 2005). The primary site of pathology in ALS patients is the spinal cord, with involvement of lower brain-stem regions late in the disease process (Cleveland and Rothstein 2001). In G93A mice, CB2 receptor mRNA is selectively up-regulated in the spinal cord in a temporal pattern closely paralleling disease progression. Furthermore, increased mRNA levels are correlated with elevated CB2 receptor protein levels in the spinal cords of end-stage G93A mice. These findings suggest that, similar to other neuroinflammatory diseases, components of the cannabinoid system are selectively altered in the target tissue associated with ALS pathogenesis. In addition, low (but demonstrable) levels of both CB2 receptor mRNA and protein observed in WT-OE spinal cords reported here are in agreement with recent studies demonstrating the presence of functional CB2 receptors in the CNS of rodents (Van Sickle et al. 2005; Gong et al. 2006).
Drugs which activate CB2 receptors, successfully improve the symptoms of several inflammatory diseases including intestinal hypermotility because of endotoxic shock (Mathison et al. 2004), atherosclerosis (Steffens et al. 2005), multiple sclerosis (Walter and Stella 2004) and Alzheimer’s disease (Benito et al. 2003; Ramirez et al. 2005). Recent in vitro studies demonstrate that CB2 receptors are up-regulated in microglia in response to inflammatory stimuli (Maresz et al. 2005) and that CB2 agonists suppress microglial activation (Ehrhart et al. 2005). In the present study, we demonstrate that not only are CB2 receptors dramatically up-regulated in the spinal cords of symptomatic G93A mice, they are also able to functionally stimulate G-proteins when activated by cannabinoid agonists. As such, the beneficial effects of cannabinoids reported here could potentially be mediated via CB2 receptor-mediated suppression of microglial/macrophage activation in the spinal cords of symptomatic G93A mice. Specifically, we propose that in the early stages of motor neuron degeneration, endocannabinoids and CB2 receptors are selectively up-regulated in spinal microglia as a compensatory, protective measure to reduce inflammation.
In contrast to the above hypothesis, it is important to note that at least one study has indicated that the CB2 selective agonist AM-1241 might act as a ‘protean agonist’, displaying antagonist, inverse agonist or partial agonist activity depending on the assay and/or tissue examined (Yao et al. 2006). Furthermore, in the present study, AM-1241 produced little to no stimulation of G-proteins in symptomatic G93A spinal cord membranes. Although the lack of agonist activity reported here might be the result of less-than-optimal experimental conditions, it is also possible that the therapeutic effect of AM-1241 in this animal model might instead result from antagonism of CB2 receptor stimulation produced by the endogenous cannabinoid agonists 2-arachido-noyl glycerol and/or anandamide, known to be elevated in the spinal cords of symptomatic G93A mice (Witting et al. 2004; Bilsland et al. 2006). Future experiments employing treatment of G93A mice with selective CB2 antagonists and/or inverse agonists should readily resolve this issue.
Increasing evidence suggests that some cannabinoids mediate their effects via action at a non-CB1/CB2 receptor [reviewed in (Baker et al. 2006)]. Very interestingly, in the present study, we demonstrate that approximately 25% of the G-proteins activated by the full cannabinoid agonist HU-210 in spinal cord membranes prepared from symptomatic G93A mice cannot be blocked by concurrent, co-incubation with receptor-saturating concentrations of CB1 and CB2 antagonists. In contrast, complete blockade of HU-210-induced G-protein stimulation is observed in WT-OE membranes co-incubated with both antagonists. This suggests that in addition to CB2 receptor up-regulation occurring during end-stage disease in G93A mice, a novel non-CB1/CB2 receptor might be induced as well.
Results for the present study also reveal a trend indicating that the density and function of CB1 receptors are possibly down-regulated in the spinal cords of end-stage G93A mice. If CB1 receptor signaling is indeed reduced, it is likely that the observed therapeutic effect of WIN-55,212 in G93A mice is mediated via CB2, and not CB1, receptors. While it is unknown whether reduced CB1 receptor signaling contributes to ALS pathogenesis, a similar reduction in CB1 receptor density (with concurrent CB2 up-regulation) has been reported in the brains of Alzheimer’s patients (Ramirez et al. 2005). A recent study also demonstrated that while knock-out of CB1 receptors in G93A mice had no effect on disease onset, it significantly extended life-span (Bilsland et al. 2006). These studies indicate that CB1 receptor activation might actually exacerbate disease progression in G93A mice. As such, future experiments are planned to examine the therapeutic potential of CB1 antagonists/inverse agonists, administered alone or in combination with CB2 agonists, on disease progression in this ALS animal model.
To date, multiple clinical trials of several candidate therapeutic compounds have been completed [reviewed in (Gordon 2005)]. Unfortunately, none of these pharmacological agents alters the inevitable outcome of ALS and only one drug, riluzole, has been approved by the US Food and Drug Administration (Traynor et al. 2003). In addition to only modest efficacy, 15–18% of patients taking riluzole experience significant adverse effects (Bensimon and Doble 2004). In contrast to the many drawbacks of current drug therapy for ALS, data presented here provide evidence that CB2 agonists may instead act as efficacious pharmacological agents with several distinct advantages for the management of this devastating disease. The most important benefit of potential CB2 agonist therapy for ALS, suggested by this study, is that significant therapeutic effects are observed even when agonists are initiated at symptom onset. In human ALS patients, drug treatment cannot begin until onset of symptoms has been established (e.g., by muscle weakness and differential diagnosis) (Strong and Rosenfeld 2003). In addition, our results suggest that AM-1241 might provide improved efficacy, relative to other recently tested pharmacological agents (Table 2). Lastly, because of selective CB2 receptor up-regulation in the affected neural tissues (e.g., spinal cord), it could be predicted that CB2 agonist therapy for ALS will provide enhanced therapeutic efficacy with a potential reduction in adverse effects.
Acknowledgments
This work was supported in part by National Institute of on Drug Abuse grant RO1-DA13660 (P.L.P.), National Institute of Neurological Disorders and Stroke grant RO1-NS040819 (J.P.C) and University of Arkansas for Medical Sciences Tobacco Award (P.L.P.).
Abbreviations used
- Δ9-THC
Δ9-tetrahydrocannabinol
- ALS
amyo-trophic lateral sclerosis
- CB1
cannabinoid receptor 1
- CB2
cannabinoid receptor 2
- FALS
familial amyotrophic lateral sclerosis
- G93A
G93A-SOD1 overexpressing transgenic mice
- SALS
sporadic amyotrophic lateral sclerosis
- SIR
survival interval ratio
- SOD
superoxide dismutase
- WT-OE
wild-type SOD1 overexpressing transgenic mice
References
- Baker D, Pryce G, Davies WL, Hiley CR. In silico patent searching reveals a new cannabinoid receptor. Trends Pharmacol Sci. 2006;27:1–4. doi: 10.1016/j.tips.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Benito C, Nunez E, Tolon RM, Carrier EJ, Rabano A, Hillard CJ, Romero J. Cannabinoid CB2 receptors and fatty acid amide hydrolase are selectively overexpressed in neuritic plaque-associated glia in Alzheimer’s disease brains. J Neurosci. 2003;23:11136–11141. doi: 10.1523/JNEUROSCI.23-35-11136.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensimon G, Doble A. The tolerability of riluzole in the treatment of patients with amyotrophic lateral sclerosis. Expert Opin Drug Saf. 2004;3:525–534. doi: 10.1517/14740338.3.6.525. [DOI] [PubMed] [Google Scholar]
- Bilsland LG, Dick JR, Pryce G, Petrosino S, Di Marzo V, Baker D, Greensmith L. Increasing cannabinoid levels by pharmacological and genetic manipulation delay disease progression in SOD1 mice. FASEB J. 2006;20:1003–1005. doi: 10.1096/fj.05-4743fje. [DOI] [PubMed] [Google Scholar]
- Breivogel CS, Griffin G, Di Marzo V, Martin BR. Evidence for a new G protein-coupled cannabinoid receptor in mouse brain. Mol Pharmacol. 2001;60:155–163. [PubMed] [Google Scholar]
- Bruijn LI, Miller TM, Cleveland DW. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci. 2004;27:723–749. doi: 10.1146/annurev.neuro.27.070203.144244. [DOI] [PubMed] [Google Scholar]
- Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci. 2001;2:806–819. doi: 10.1038/35097565. [DOI] [PubMed] [Google Scholar]
- Crow JP, Calingasan NY, Chen J, Hill JL, Beal MF. Manganese porphyrin given at symptom onset markedly extends survival of ALS mice. Ann Neurol. 2005;58:258–265. doi: 10.1002/ana.20552. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Bifulco M, De Petrocellis L. The endocannabinoid system and its therapeutic exploitation. Nat Rev Drug Discov. 2004;3:771–784. doi: 10.1038/nrd1495. [DOI] [PubMed] [Google Scholar]
- Ehrhart J, Obregon D, Mori T, Hou H, Sun N, Bai Y, Klein T, Fernandez F, Tan J, Shytle D. Stimulation of cannabinoid receptor 2 (CB2) suppresses microglial activation. J Neuroinflammation. 2005;2:29. doi: 10.1186/1742-2094-2-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felder CC, Joyce KE, Briley EM, Mansouri J, Mackie K, Blond O, Lai Y, Ma AL, Mitchell RL. Comparison of the pharmacology and signal transduction of the human cannabinoid CB1 and CB2 receptors. Mol Pharmacol. 1995;48:443–450. [PubMed] [Google Scholar]
- Galiegue S, Mary S, Marchand J, Dussossoy D, Carriere D, Carayon P, Bouaboula M, Shire D, Le Fur G, Casellas P. Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. Eur J Biochem. 1995;232:54–61. doi: 10.1111/j.1432-1033.1995.tb20780.x. [DOI] [PubMed] [Google Scholar]
- Gardner A, Mallet PE. Suppression of feeding, drinking, and locomotion by a putative cannabinoid receptor ‘silent antagonist’. Eur J Pharmacol. 2006;530:103–106. doi: 10.1016/j.ejphar.2005.11.032. [DOI] [PubMed] [Google Scholar]
- Gatley SJ, Lan R, Pyatt B, Gifford AN, Volkow ND, Makriyannis A. Binding of the non-classical cannabinoid CP 55,940, and the diarylpyrazole AM251 to rodent brain cannabinoid receptors. Life Sci. 1997;61:PL191–PL197. doi: 10.1016/s0024-3205(97)00690-5. [DOI] [PubMed] [Google Scholar]
- Giulietti A, Overbergh L, Valckx D, Decallonne B, Bouillon R, Mathieu C. An overview of real-time quantitative PCR: applications to quantify cytokine gene expression. Methods. 2001;25:386–401. doi: 10.1006/meth.2001.1261. [DOI] [PubMed] [Google Scholar]
- Gong JP, Onaivi ES, Ishiguro H, Liu QR, Tagliaferro PA, Brusco A, Uhl GR. Cannabinoid CB2 receptors: immunohistochemical localization in rat brain. Brain Res. 2006;1071:10–23. doi: 10.1016/j.brainres.2005.11.035. [DOI] [PubMed] [Google Scholar]
- Gordon PH. Advances in clinical trials for amyotrophic lateral sclerosis. Curr Neurol Neurosci Rep. 2005;5:48–54. doi: 10.1007/s11910-005-0023-2. [DOI] [PubMed] [Google Scholar]
- Gurney ME. Transgenic animal models of familial amyotrophic lateral sclerosis. J Neurol. 1997;244(Suppl 2):S15–20. doi: 10.1007/BF03160575. [DOI] [PubMed] [Google Scholar]
- Hanisch UK. Microglia as a source and target of cytokines. Glia. 2002;40:140–155. doi: 10.1002/glia.10161. [DOI] [PubMed] [Google Scholar]
- Herkenham M, Lynn AB, Little MD, Johnson MR, Melvin LS, de Costa BR, Rice KC. Cannabinoid receptor localization in brain. Proc Natl Acad Sci USA. 1990;87:1932–1936. doi: 10.1073/pnas.87.5.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillard CJ, Manna S, Greenberg MJ, Di Camelli R, Ross RA, Stevenson LA, Murphy V, Pertwee RG, Campbell WB. Synthesis and characterization of potent and selective agonists of the neuronal cannabinoid receptor (CB1) J Pharmacol Exp Ther. 1999;289:1427–1433. [PubMed] [Google Scholar]
- Hosohata K, Quock RM, Hosohata Y, Burkey TH, Makriyannis A, Consroe P, Roeske WR, Yamamura HI. AM630 is a competitive cannabinoid receptor antagonist in the guinea pig brain. Life Sci. 1997;61:PL115–PL118. doi: 10.1016/s0024-3205(97)00596-1. [DOI] [PubMed] [Google Scholar]
- Ince PG, Shaw PJ, Slade JY, Jones C, Hudgson P. Familial amyotrophic lateral sclerosis with a mutation in exon 4 of the Cu/Zn superoxide dismutase gene: pathological and immunocytochemical changes. Acta Neuropathol (Berl) 1996;92:395–403. doi: 10.1007/s004010050535. [DOI] [PubMed] [Google Scholar]
- Jung C, Rong Y, Doctrow S, Baudry M, Malfroy B, Xu Z. Synthetic superoxide dismutase/catalase mimetics reduce oxidative stress and prolong survival in a mouse amyotrophic lateral sclerosis model. Neurosci Lett. 2001;304:157–160. doi: 10.1016/s0304-3940(01)01784-0. [DOI] [PubMed] [Google Scholar]
- Kiaei M, Kipiani K, Chen J, Calingasan NY, Beal MF. Peroxisome proliferator-activated receptor-gamma agonist extends survival in transgenic mouse model of amyotrophic lateral sclerosis. Exp Neurol. 2005;191:331–336. doi: 10.1016/j.expneurol.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Kiaei M, Petri S, Kipiani K, et al. Thalidomide and lenalidomide extend survival in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci. 2006;26:2467–2473. doi: 10.1523/JNEUROSCI.5253-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieran D, Kalmar B, Dick JR, Riddoch-Contreras J, Burnstock G, Greensmith L. Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice. Nat Med. 2004;10:402–405. doi: 10.1038/nm1021. [DOI] [PubMed] [Google Scholar]
- Kirkinezos IG, Hernandez D, Bradley WG, Moraes CT. An ALS mouse model with a permeable blood-brain barrier benefits from systemic cyclosporine A treatment. J Neurochem. 2004;88:821–826. doi: 10.1046/j.1471-4159.2003.02181.x. [DOI] [PubMed] [Google Scholar]
- Klein TW. Cannabinoid-based drugs as anti-inflammatory therapeutics. Nat Rev Immunol. 2005;5:400–411. doi: 10.1038/nri1602. [DOI] [PubMed] [Google Scholar]
- Maresz K, Carrier EJ, Ponomarev ED, Hillard CJ, Dittel BN. Modulation of the cannabinoid CB2 receptor in microglial cells in response to inflammatory stimuli. J Neurochem. 2005;95:437–445. doi: 10.1111/j.1471-4159.2005.03380.x. [DOI] [PubMed] [Google Scholar]
- Mathison R, Ho W, Pittman QJ, Davison JS, Sharkey KA. Effects of cannabinoid receptor-2 activation on accelerated gastrointestinal transit in lipopolysaccharide-treated rats. Br J Pharmacol. 2004;142:1247–1254. doi: 10.1038/sj.bjp.0705889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. Inflammatory processes in amyotrophic lateral sclerosis. Muscle Nerve. 2002;26:459–470. doi: 10.1002/mus.10191. [DOI] [PubMed] [Google Scholar]
- Munro S, Thomas K, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- Nelson PT, Soma LA, Lavi E. Microglia in diseases of the central nervous system. Ann Med. 2002;34:491–500. doi: 10.1080/078538902321117698. [DOI] [PubMed] [Google Scholar]
- Prather PL, Martin NA, Breivogel CS, Childers SR. Activation of cannabinoid receptors in rat brain by WIN 55212-2 produces coupling to multiple G protein alpha-subunits with different potencies. Mol Pharmacol. 2000a;57:1000–1010. [PubMed] [Google Scholar]
- Prather PL, Song L, Piros ET, Law PY, Hales TG. δ-Opioid receptors are more efficiently coupled to adenylyl cyclase than to L-type Ca2+ channels in transfected rat pituitary cells. J Pharmacol Exp Ther. 2000b;295:552–562. [PubMed] [Google Scholar]
- Raman C, McAllister SD, Rizvi G, Patel SG, Moore DH, Abood ME. Amyotrophic lateral sclerosis: delayed disease progression in mice by treatment with a cannabinoid. Amyotroph Lateral Scler Other Motor Neuron Disord. 2004;5:33–39. doi: 10.1080/14660820310016813. [DOI] [PubMed] [Google Scholar]
- Ramirez BG, Blazquez C, Gomez del Pulgar T, Guzman M, de Ceballos ML. Prevention of Alzheimer’s disease pathology by cannabinoids: neuroprotection mediated by blockade of microglial activation. J Neurosci. 2005;25:1904–1913. doi: 10.1523/JNEUROSCI.4540-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaldi-Carmona M, Barth F, Millan J, et al. SR 144528, the first potent and selective antagonist of the CB2 cannabinoid receptor. J Pharmacol Exp Ther. 1998;284:644–650. [PubMed] [Google Scholar]
- Rothstein JD, Patel S, Regan MR, et al. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433:73–77. doi: 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- Shoemaker JL, Joseph BK, Ruckle MB, Mayeux PR, Prather PL. The endocannabinoid noladin ether acts as a full agonist at human CB2 cannabinoid receptors. J Pharmacol Exp Ther. 2005;314:868–875. doi: 10.1124/jpet.105.085282. [DOI] [PubMed] [Google Scholar]
- Steffens S, Veillard NR, Arnaud C, Pelli G, Burger F, Staub C, Karsak M, Zimmer A, Frossard JL, Mach F. Low dose oral cannabinoid therapy reduces progression of atherosclerosis in mice. Nature. 2005;434:782–786. doi: 10.1038/nature03389. [DOI] [PubMed] [Google Scholar]
- Streit WJ. Microglia as neuroprotective, immunocompetent cells of the CNS. Glia. 2002;40:133–139. doi: 10.1002/glia.10154. [DOI] [PubMed] [Google Scholar]
- Strong M, Rosenfeld J. Amyotrophic lateral sclerosis: a review of current concepts. Amyotroph Lateral Scler Other Motor Neuron Disord. 2003;4:136–143. doi: 10.1080/14660820310011250. [DOI] [PubMed] [Google Scholar]
- Tortarolo M, Grignaschi G, Calvaresi N, et al. Glutamate AMPA receptors change in motor neurons of SOD1(G93A) transgenic mice and their inhibition by a noncompetitive antagonist ameliorates the progression of amytrophic lateral sclerosis-like disease. J Neurosci Res. 2006;83:134–146. doi: 10.1002/jnr.20715. [DOI] [PubMed] [Google Scholar]
- Traynor BJ, Alexander M, Corr B, Frost E, Hardiman O. An outcome study of riluzole in amyotrophic lateral sclerosis – a population-based study in Ireland, 1996–2000. J Neurol. 2003;250:473–479. doi: 10.1007/s00415-003-1026-z. [DOI] [PubMed] [Google Scholar]
- Turner MR, Cagnin A, Turkheimer FE, Miller CC, Shaw CE, Brooks DJ, Leigh PN, Banati RB. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C](R)-PK11195 positron emission tomography study. Neurobiol Dis. 2004;15:601–609. doi: 10.1016/j.nbd.2003.12.012. [DOI] [PubMed] [Google Scholar]
- Valenzano KJ, Tafesse L, Lee G, et al. Pharmacological and pharmacokinetic characterization of the cannabinoid receptor 2 agonist, GW405833, utilizing rodent models of acute and chronic pain, anxiety, ataxia and catalepsy. Neuropharmacology. 2005;48:658–672. doi: 10.1016/j.neuropharm.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Van Den Bosch L, Tilkin P, Lemmens G, Robberecht W. Minocycline delays disease onset and mortality in a transgenic model of ALS. Neuroreport. 2002;13:1067–1070. doi: 10.1097/00001756-200206120-00018. [DOI] [PubMed] [Google Scholar]
- Van Sickle MD, Duncan M, Kingsley PJ, et al. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science. 2005;310:329–332. doi: 10.1126/science.1115740. [DOI] [PubMed] [Google Scholar]
- Walter L, Stella N. Cannabinoids and neuroinflammation. Br J Pharmacol. 2004;141:775–785. doi: 10.1038/sj.bjp.0705667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Zhang D. Memantine prolongs survival in an amyotrophic lateral sclerosis mouse model. Eur J Neurosci. 2005;22:2376–2380. doi: 10.1111/j.1460-9568.2005.04431.x. [DOI] [PubMed] [Google Scholar]
- West M, Mhatre M, Ceballos A, et al. The arachidonic acid 5-lipoxygenase inhibitor nordihydroguaiaretic acid inhibits tumor necrosis factor alpha activation of microglia and extends survival of G93A-SOD1 transgenic mice. J Neurochem. 2004;91:133–143. doi: 10.1111/j.1471-4159.2004.02700.x. [DOI] [PubMed] [Google Scholar]
- Weydt P, Moller T. Neuroinflammation in the pathogenesis of amyotrophic lateral sclerosis. Neuroreport. 2005;16:527–531. doi: 10.1097/00001756-200504250-00001. [DOI] [PubMed] [Google Scholar]
- Weydt P, Hong S, Witting A, Moller T, Stella N, Kliot M. Cannabinol delays symptom onset in SOD1 (G93A) transgenic mice without affecting survival. Amyotroph Lateral Scler Other Motor Neuron Disord. 2005;6:182–184. doi: 10.1080/14660820510030149. [DOI] [PubMed] [Google Scholar]
- Witting A, Weydt P, Hong S, Kliot M, Moller T, Stella N. Endocannabinoids accumulate in spinal cord of SOD1 G93A transgenic mice. J Neurochem. 2004;89:1555–1557. doi: 10.1111/j.1471-4159.2004.02544.x. [DOI] [PubMed] [Google Scholar]
- Wu AS, Kiaei M, Aguirre N, Crow JP, Calingasan NY, Browne SE, Beal MF. Iron porphyrin treatment extends survival in a transgenic animal model of amyotrophic lateral sclerosis. J Neurochem. 2003;85:142–150. doi: 10.1046/j.1471-4159.2003.01639.x. [DOI] [PubMed] [Google Scholar]
- Yao BB, Mukherjee S, Fan Y, Garrison TR, Daza AV, Grayson GK, Hooker BA, Dart MJ, Sullivan JP, Meyer MD. In vitro pharmacological characterization of AM1241: a protean agonist at the cannabinoid CB(2) receptor? Br J Pharmacol. 2006;149:145–154. doi: 10.1038/sj.bjp.0706838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yiangou Y, Facer P, Durrenberger P, Chessell IP, Naylor A, Bountra C, Banati RR, Anand P. COX-2, CB2 and P2X7-immunoreactivities are increased in activated microglial cells/macrophages of multiple sclerosis and amyotrophic lateral sclerosis spinal cord. BMC Neurol. 2006;6:12. doi: 10.1186/1471-2377-6-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Narayanan M, Friedlander RM. Additive neuroprotective effects of minocycline with creatine in a mouse model of ALS. Ann Neurol. 2003;53:267–270. doi: 10.1002/ana.10476. [DOI] [PubMed] [Google Scholar]