

Abstract
Experimental autoimmune encephalomyelitis (EAE) is a T-cell–mediated, autoimmune disorder characterized by central nervous system inflammation and demyelination, features reminiscent of the human disease, multiple sclerosis (MS). Prior work in the EAE model has suggested that encephalitogenic T cells are of the T helper (Th)-1 phenotype. Our group has performed several studies in the EAE model that suggest that a strategy for treating autoimmune disorders is to convert the pathogenic cells from the Th1 to Th2 phenotype. Peroxisome proliferator-activated receptors (PPARs) are members of a nuclear hormone receptor superfamily that include receptors for steroids, retinoids, and thyroid hormone, all of which are known to affect the immune response. Recently, we examined the role of PPARγ in EAE and observed that administration of the PPARγ agonist 15-deoxy-Δ12,14 prostaglandin J2 exerted a significant therapeutic effect predominantly by inhibiting the activation and expansion of encephalitogenic T cells. One potential advantage in studying PPARα agonists is that they have been very well tolerated when used in humans to treat conditions such as elevated triglycerides. Building on prior work in immune deviation and with PPAR agonists, we have demonstrated that PPARα agonists can alter the cytokine phenotype of myelin-reactive T cells, alter their encephalitogenicity, and be useful in the treatment of EAE. The fact that PPARα agonists have been used as therapeutic agents in humans to treat metabolic conditions for over 25 years with little toxicity makes them attractive candidates for use as adjunctive therapies in MS.
Keywords: PPAR, autoimmunity, multiple sclerosis, cytokines, nuclear receptors
Approximately 350,000 people in the United States have physician-diagnosed multiple sclerosis (MS)4 (1,2). Following trauma it is the next leading cause of neurologic disability in the United States in young adults, and most patients suffer from the effects of MS for most of their adult life. The cause of MS remains unknown, but because the disease involves perivascular mononuclear cell infiltrates and demyelination [features also characteristic of experimental autoimmune encephalomyelitis (EAE)], an autoimmune process is hypothesized to be involved in disease pathogenesis (3,4). There are now five drugs approved for use in the treatment of MS by the FDA, however, none of these agents are a cure for the disease, and the need for better treatment strategies for MS remains (5–8). In addition, with the withdrawal of drugs such as nataluzimab and its potential for infectious toxicity, it is clear that long-term safety is an important consideration for new MS therapies.
Relevance of EAE to multiple sclerosis
Several animal models have been used to study MS (3,4). Our laboratory has exclusively used the murine model of EAE for a number of reasons. Murine EAE results in a relapsing-remitting disease, similar to the early phase of disease for most MS patients. In chronic murine EAE, the pathology observed in the white matter shows demyelination that is reminiscent of the pathology seen in the central nervous system of humans with MS. With the advent of transgenic and homologous recombination technology, it is increasingly clear that many powerful molecular tools are becoming available for studying the immune response in pathologic processes such as EAE.
T cell phenotypes and autoimmune demyelination
Organ-specific autoimmune diseases such as EAE are mediated by IFN-γ–producing T helper (Th)-1 type cells (9). An abundance of data suggests that inflammatory immune responses or delayed type hypersensitivity responses are primarily mediated by these Th1 cells, which produce cytokines such as lymphotoxin and IFN-γ, but little IL-4 (10). In contrast, CD4+ Th2 populations produce large amounts of IL-4 and IL-5, mediate immune responses characterized by high levels of noncomplement binding IgG, IgE, and eosinophil-mediated cytotoxicity, but produce no organ-specific tissue destruction in normal mice. The production by Th2 cells of macrophage-deactivating cytokines, such as IL-4, IL-10, and IL-13, provides a strong foundation for their anti-inflammatory effects in vivo (11). On the other hand, it is very clear that cytokines such as IL-12 and type-I IFN can activate a transcription factor called signal transducing activator of transcription (STAT)-4 in human T cells and instruct the differentiation of Th1 cells (12). STAT4 is recruited to the human IFN-α or -β receptor through an interaction with the C-terminus of human STAT2, and this portion of STAT2 is not conserved between mice and humans (13). Thus, the most commonly used therapy for MS may be able to potentiate the development of autoreactive T cells.
Evidence in the EAE model suggests that the production of Th2 cytokines such as IL-4 or IL-10 may play an important role in the remissions observed in this disease (3,14,15). The ability of Th2 cells to regulate an already initiated immune response in EAE is less clear (16). Interestingly, it appears that proteolipid protein-specific T-cell clones isolated from MS patients have different cytokine phenotypes depending on when they are isolated (17). T-cell clones derived during acute attacks were more likely to be of a Th1 phenotype, whereas during remission, these cytokine responses were much more variable but included T-cell clones of a Th2 phenotype.
In the Th1 differentiation pathway, both IL-12 and IFN-γ are important in the differentiation of naïve T cells (see Fig. 1). IFN-γ is necessary for translocation of the transcription factor STAT1 to the nucleus. T box expressed in T cells (T-bet), which has been proposed to be the master regulator of Th1 differentiation, is a strong transactivator of the IFN-γ gene (18,19). IL-12, which is produced by antigen-presenting cells, is necessary for STAT4 activation and full activation of the IFN-γ gene (20). In addition to IL-12, it is now clear there is a family of heterodimeric cytokines that may play a role in the regulation of T-cell responses (21). IL-12p40 associates not only with IL-12p35, but also with a p19 chain to form IL-23. An essential role is suggested for IL-23 because mice deficient in IL-12p40 could not develop EAE, whereas mice deficient in IL-12p35 developed more severe disease (22). IL-23p19–deficient mice are resistant to EAE development, but can develop a normal Th1 response (23), thus the role of cytokines in producing encephalitogenic T cells may be more complicated than previously appreciated.
FIGURE 1.
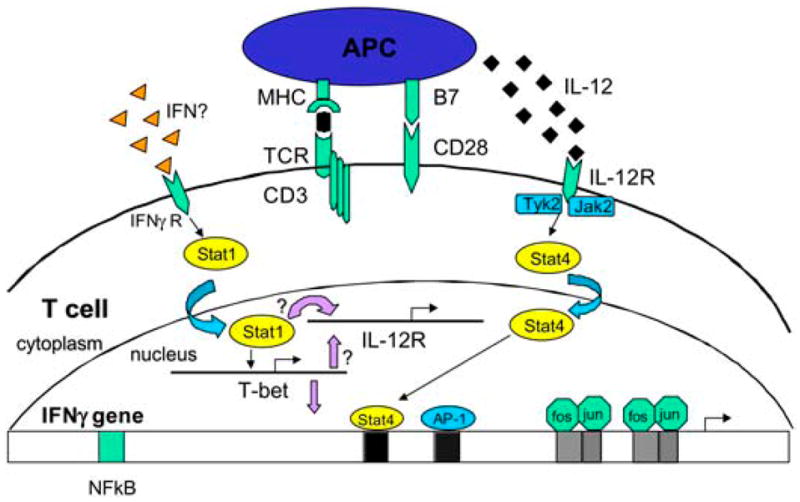
Transcriptional regulation of the differentiation of Th1 cells. IL-12 activates the STAT4 pathway while IFN-γ activates the STAT1 or T-bet pathway. It is unclear where PPARs regulate these pathways at a molecular level.
Our own work suggests that T-bet and STAT1 regulate each other in a feedback loop (24). Our studies suggest peroxisome proliferator-activated receptor-α (PPAR)α agonists affect the differentiation of myelin basic protein (MBP) Ac1-11–specific T cells into a Th2 phenotype, although the molecular mechanisms for this effect are still unclear (14). In the differentiation of Th2 cells, IL-4 is the critical cytokine that leads to activation and translocation of STAT6 to the nucleus and subsequent expression of GATA binding protein-3 (GATA-3) (see Fig. 2). GATA-3 is felt to be the master regulator of Th2 cell differentiation (25). Future studies will need to examine whether the increased IL-4 production we have observed in MBP Ac1-11–specific T cells due to PPARα agonists is the result of activating transcription factors of the Th2 differentiation pathway.
FIGURE 2.
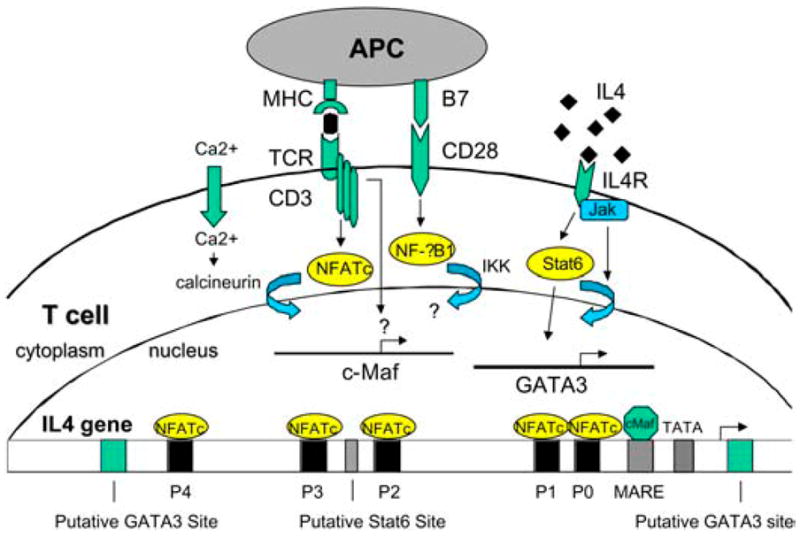
Transcriptional regulation of the differentiation of Th2 cells. IL-4 turns on the STAT6 pathway and GATA-3. It is unclear where PPARs such as PPARα turns on Th2 differentiation.
PPARγ: Role in inflammatory diseases
PPARs are members of the nuclear hormone receptor superfamily that also include steroid, retinoid, and thyroid hormone receptors. PPARs have been studied most extensively in the regulation of genes involved in glucose and lipid metabolism (26). Three PPAR subtypes exist (α, δ, and γ) and they exhibit different tissue distribution as well as different ligand specificities. In addition to adipocytes, it was recently shown that cells of the monocyte and/or macrophage lineage express PPARγ and PPARα, suggesting a possible role for these receptors in the function of these cells. Subsequently, PPARγ ligands, which include the naturally occurring prostaglandin metabolite 15-deoxy-Δ12,14 prostaglandin J2 as well as thiazolidinediones, have been shown to have anti-inflammatory activity (27). 15-Deoxy-Δ12,14 prostaglandin J2 was shown to inhibit inducible nitric oxide synthase, matrix metalloproteinase-9, IL-1β, IL-6, and tumor necrosis factor (TNF)-α production by monocytes and/or macrophages (28,29). Both PPARγ and PPARα have been shown to be expressed on T cells and their ligands inhibit IL-2 secretion and T-cell proliferation (30,31). PPARγ ligands likely block IL-2 production by a mechanism that inhibits the nuclear factor of activated T cells that bind to the IL-2 promoter (32). PPARγ ligands can also affect T-cell function indirectly by inhibiting endothelial-cell production of chemokines (33). The evidence that PPAR agonists can regulate inflammation is supported by several animal studies. These include suppression of adjuvant arthritis in rats (34), inhibition of atherosclerosis in mice (35), inhibition of the inflammatory response and stroke size following cerebrovascular occlusion in rats, and amelioration of inflammatory bowel disease in mice (36). As mentioned previously, our studies and those of others show that PPARγ agonists can inhibit the clinical signs of EAE (27,37–39).
PPARα: Role in inflammatory diseases
PPARα ligands have also been shown to regulate inflammatory responses, although there is clearly less evidence along this line compared with PPARγ agonists. PPARα-deficient mice have abnormally prolonged responses to inflammatory stimuli such as arachidonic acid and leukotrienes (40). The expression of IL-6, the vascular cell adhesion molecule, and cyclooxygenase-2 in response to cytokine activation can be inhibited by PPARα ligands (41). PPARα ligands were also shown to decrease nuclear factor-κB (NF-κB) activation, IL-12, and IL-6 production in aged mice (42). PPARα ligands may inhibit the functional expression of NF-κB, in part by augmenting the expression of inhibitor of NF-κB (IκBα) (43). Recently, the PPARα ligand WY14,643 was shown to inhibit IgG responses in myelin oligodendrocyte glycoprotein 35–55/complete Freund’s adjuvant FA-immunized mice (44). When mice were fed this agonist, splenocytes demonstrated impaired production of IFN-γ, IL-6, and TNFα. Interestingly, the authors had hoped to examine the effect of fibrates on EAE; however, the combination of immunization with myelin oligodendrocyte glycoprotein 35–55/complete Freund’s adjuvant, pertussis toxin and WY14,643 treatment consistently induced mortality 5–10 d after immunization. Several PPARα agonists have been shown to potentiate TNFα secretion in response to endotoxins; however, this was observed in mice and rats, and not in guinea pigs, monkeys, or humans (45,46).
Regulation of cytokine expression by PPAR
Recently, several studies have examined the effect of PPARs to mediate anti-inflammatory activity. The majority of these studies have focused on PPARγ and have examined their effects on cells of the macrophage and/or monocyte lineage (47). As noted above, the majority of these studies have shown that PPARγ agonists inhibit the expression of inflammatory mediators such as inducible nitric oxide synthase, presumably by antagonizing activation of transcription factors such as NF-κB. PPARα is expressed in human monocytes that differentiate into macrophages, and PPARα agonists have been shown to induce apoptosis in macrophages (48).
Our work and the work of others demonstate that PPARγ ligands can inhibit T-cell proliferation or the production of IL-2 and IFN-γ by T cells stimulated with antigen (27,30,31). Studies that compare the ability of PPARα ligands with those of PPARγ ligands suggest that the effect on the inhibition of T-cell proliferation and IFN-γ secretion is more marked with PPARγ agonists (30). Some fibrates, such as gemfibrozil and ciprofibrate, are able to induce IL-4 secretion by splenocytes stimulated with concanavalin A (Con A). However, another PPARα agonist, GW7647, did not increase IL-4 secretion.
Recently, a study examining the regulation of T-bet by PPARα concluded that it is unliganded PPARα that suppresses T-bet expression and subsequent IFN-γ expression in T cells (49). In that study, CD4+ T cells deficient in PPARα showed that unliganded PPARα was important in regulating the phosphorylation of p38 mitogen-activated protein kinase and T-bet expression. PPARα agonists resulted in suppressed p38 mitogen-activated protein kinase phosphorylation and promoted T-bet expression. Studies by our group are examining the effects of PPARα activation on T cell differentiation and determining the effects on the T-bet or STAT1 pathway as well as on Th2 differentiation pathways.
PPAR activators in human T lymphocytes
Recent work has shown that both PPARα and PPARγ are expressed by human CD4+ T cells at both the mRNA and protein levels (31). Activation of human CD4+ T cells with anti-CD3 increased IFN-γ production, but this was dramatically inhibited in the presence of either PPARα or PPARγ agonists. The PPARα agonist WY14643 was also quite effective in inhibiting TNFα production and IL-2 secretion. It should be noted that these studies examined the effect of the PPARα agonists at clinically relevant concentrations and did not observe any evidence of effects on T-cell viability. However, these studies did not examine the effect of PPARα agonists on Th2 cytokines such as IL-4. Our studies have examined the effect of PPARα agonists gemfibrozil and fenofibrate on promoting Th2 cytokine production by human myelin-specific T cells (14). Human MBP-specific T-cell lines secreted increased IL-4 when generated in the presence of gemfibrozil (Fig. 3).
FIGURE 3.
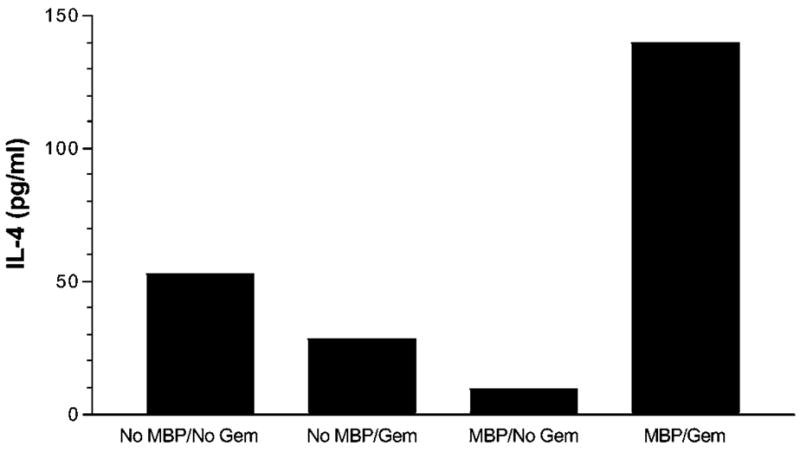
Gemfibrozil increases IL-4 secretion by a human T-cell line. The MBP-specific T-cell line was generated by weekly stimulation with antigen in the presence or absence of gemfibrozil (100 μM). IL-4 was measured at 48 h time point by ELISA. Means of quadruplicate cultures are shown. SD were <10% of the mean.
Relevance to multiple sclerosis
Although there are presently several medications approved by the FDA for the treatment of MS, none of them are a cure. Similar to the situation that has occurred in oncology, it is likely that improved treatment protocols will eventually arise from combinations of therapies targeting different aspects of the disease process. Future studies will determine whether PPARα agonists, which are presently being used to treat humans, can also be beneficial in autoimmune demyelination. As several groups continue to gather data on various PPAR agonists, it is expected that there will be an attempt to launch an investigator-initiated trial to test one or several of these compounds in patients with MS.
Footnotes
Published in a supplement to The Journal of Nutrition. Presented at the “Nutrients, Nuclear Receptors, Inflammation, and Immunity” symposium held April 3, 2005 at Experimental Biology 2005 in San Diego, California. The symposium was sponsored by the National Institutes of Health Office of Dietary Supplements, the U.S. Department of Agriculture, Wyeth Nutrition, and the American Society for Nutrition. The symposium was chaired by Charles Stephensen of the U.S. Department of Agriculture Western Human Nutrition Research Center at the University of California, Davis, and by Margherita Cantorna of the Nutrition Department at the Pennsylvania State University.
This work was supported by grants from the National Institutes of Health and National Multiple Sclerosis Society (M.K.R. and P.D.D.). This work was also supported by a Harry Weaver Neuroscience Scholarship from the National Multiple Sclerosis Society (A.E.L.R.) and a Doris Duke Predoctoral Fellowship (M.M.).
Abbreviations used: CFA, complete Freund’s adjuvant; EAE, experimental autoimmune encephalomyelitis; GATA-3, GATA binding protein-3; MBP, myelin basic protein; MS, multiple sclerosis; NF-κB, nuclear factor-κB; PPAR, peroxisome proliferator-activated receptor; STAT, signal transducing activator of transcription; Th, T helper; TNF, tumor necrosis factor.
LITERATURE CITED
- 1.McFarlin DE, McFarland HF. Multiple sclerosis (Part I) N Engl J Med. 1982;307:1183–8. doi: 10.1056/NEJM198211043071905. [DOI] [PubMed] [Google Scholar]
- 2.Anderson DW, Ellenberg JH, Leventhal CM, Reingold SC, Rodriguez M, Silberberg DH. Revised estimate of the prevalence of multiple sclerosis in the United States. Ann Neurol. 1992;31:333–6. doi: 10.1002/ana.410310317. [DOI] [PubMed] [Google Scholar]
- 3.Martin R, McFarland HF, McFarlin DE. Immunological aspects of demyelinating diseases. Annu Rev Immunol. 1992;10:153–87. doi: 10.1146/annurev.iy.10.040192.001101. [DOI] [PubMed] [Google Scholar]
- 4.Arnason BG. Relevance of experimental allergic encephalomyelitis to multiple sclerosis. Neurol Clin. 1983;1:765–82. [PubMed] [Google Scholar]
- 5.IFNB Multiple Sclerosis Study Group. Interferon beta-1b is effective in relapsing-remitting multiple sclerosis I: clinical results of a multicenter, randomized, double-blind, placebo-controlled trial. 1993;43:655–666 [classical article] Neurology. 2001;57:S3–9. doi: 10.1212/wnl.43.4.655. [DOI] [PubMed] [Google Scholar]
- 6.Jacobs LD, Cookfair DL, Rudick RA Multiple Sclerosis Collaborative Research Group. Intramuscular interferon beta-1a for disease progression in relapsing multiple sclerosis. Ann Neurol. 1996;39:285–94. doi: 10.1002/ana.410390304. [DOI] [PubMed] [Google Scholar]
- 7.Johnson KP, Brooks BR, Cohen JA, Ford CC, Goldstein J, Lisak RP, Myers LW, Panitch HS, Rose JW, Schiffer RB. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: Results of a phase III multicenter, double-blind, placebo-controlled trial. The Copolymer 1 Multiple Sclerosis Study Group. Neurology. 1995;45:1268–76. doi: 10.1212/wnl.45.7.1268. [DOI] [PubMed] [Google Scholar]
- 8.Millefiorini E, Gasperini C, Pozzilli C, D’Andrea F, Bastianello S, Trojano M, Morino S, Morra VB, Bozzao A, et al. Randomized placebo-controlled trial of mitoxantrone in relapsing-remitting multiple sclerosis: 24-month clinical and MRI outcome. J Neurol. 1997;244:153–9. doi: 10.1007/s004150050066. [DOI] [PubMed] [Google Scholar]
- 9.Ando DG, Clayton J, Kono D, Urban JL, Sercarz EE. Encephalitogenic T cells in the B10.PL model of experimental allergic encephalomyelitis (EAE) are of the Th-1 lymphokine subtype. Cell Immunol. 1989;124:132–43. doi: 10.1016/0008-8749(89)90117-2. [DOI] [PubMed] [Google Scholar]
- 10.Mosmann TR, Coffman RL. TH1 and TH2 cells: Different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. 1989;7:145–73. doi: 10.1146/annurev.iy.07.040189.001045. [DOI] [PubMed] [Google Scholar]
- 11.Röcken M, Racke M, Shevach EM. IL-4-induced immune deviation as antigen-specific therapy for inflammatory autoimmune disease. Immunol Today. 1996;17:225–31. doi: 10.1016/0167-5699(96)80556-1. [DOI] [PubMed] [Google Scholar]
- 12.Farrar JD, Murphy KM. Type I interferons and T helper development. Immunol Today. 2000;21:484–9. doi: 10.1016/s0167-5699(00)01710-2. [DOI] [PubMed] [Google Scholar]
- 13.Farrar JD, Smith JD, Murphy TL, Leung S, Stark GR, Murphy KM. Selective loss of type I interferon-induced Stat4 activation caused by a minisatellite insertion in mouse Stat2. Nat Immunol. 2000;1:65–9. doi: 10.1038/76932. [DOI] [PubMed] [Google Scholar]
- 14.Lovett-Racke AE, Hussain RZ, Northrop S, Choy J, Rocchini A, Matthes L, Chavis J, Diab A, Drew PD, Racke MK. PPARα agonists as therapy for autoimmune disease. J Immunol. 2004;172:5790–8. doi: 10.4049/jimmunol.172.9.5790. [DOI] [PubMed] [Google Scholar]
- 15.Kennedy MK, Torrance DS, Picha KS, Mohler KM. Analysis of cytokine mRNA expression in the central nervous system of mice with experimental autoimmune encephalomyelitis reveals that IL-10 mRNA expression correlates with recovery. J Immunol. 1992;149:2496–505. [PubMed] [Google Scholar]
- 16.Khoruts A, Miller SD, Jenkins MK. Neuroantigen-specific Th2 cells are inefficient suppressors of experimental autoimmune encephalomyelitis induced by effector Th1 cells. J Immunol. 1995;155:5011–7. [PubMed] [Google Scholar]
- 17.Correale J, Gilmore W, McMillan M, Li S, McCarthy K, Le T, Weiner LP. Patterns of cytokine secretion by autoreactive proteolipid protein-specific T cell clones during the course of multiple sclerosis. J Immunol. 1995;154:2959–68. [PubMed] [Google Scholar]
- 18.Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman GC, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–69. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- 19.Szabo SJ, Sullivan BM, Stemmann C, Satoskar AR, Sleckman BP, Glimcher LH. Distinct effects of T-bet in Th1 lineage commitment and IFN-γ production in CD4 and CD8 T cells. Science. 2002;295:338–42. doi: 10.1126/science.1065543. [DOI] [PubMed] [Google Scholar]
- 20.Thierfelder WE, van Deursen JM, Yamamoto K, Tripp RA, Sarawar SR, Carson RT, Sangster MY, Vignali DA, Doherty PC, et al. Requirement for Stat4 in interleukin-12-mediated responses of natural killer and T cells. Nature. 1996;382:171–4. doi: 10.1038/382171a0. [DOI] [PubMed] [Google Scholar]
- 21.Trinchieri G, Pflanz S, Kastelein RA. The IL-12 family of heterodimeric cytokines: New players in the regulation of T cell responses. Immunity. 2003;19:641–4. doi: 10.1016/s1074-7613(03)00296-6. [DOI] [PubMed] [Google Scholar]
- 22.Zhang GX, Gran B, Yu S, Li J, Siglienti I, Chen X, Kamoun M, Rostami A. Induction of experimental autoimmune encephalomyelitis in IL-12 receptor-beta 2-deficient mice: IL-12 responsiveness is not required in the pathogenesis of inflammatory demyelination in the central nervous system. J Immunol. 2003;170:2153–60. doi: 10.4049/jimmunol.170.4.2153. [DOI] [PubMed] [Google Scholar]
- 23.Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–8. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 24.Lovett-Racke AE, Rocchini AE, Choy J, Northrop SC, Hussain RZ, Ratts RB, Sikder D, Racke MK. Silencing T-bet defines a critical role in the differentiation of autoreactive T lymphocytes. Immunity. 2004;21:719–31. doi: 10.1016/j.immuni.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 25.Ouyang W, Ranganath SH, Weindel K, Bhattacharya D, Murphy TL, Sha WC, Murphy KM. Inhibition of Th1 developmental mediated by GATA-3 through an IL-4 independent mechanism. Immunity. 1998;9:745–55. doi: 10.1016/s1074-7613(00)80671-8. [DOI] [PubMed] [Google Scholar]
- 26.Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev. 1999;20:649–88. doi: 10.1210/edrv.20.5.0380. [DOI] [PubMed] [Google Scholar]
- 27.Diab A, Deng C, Smith JD, Hussain RZ, Phanavanh B, Lovett-Racke AE, Drew PD, Racke MK. Peroxisome proliferator-activated receptor-γ agonist 15-deoxy-Δ12,14-PGJ2 ameliorates experimental autoimmune encephalomyelitis. J Immunol. 2002;168:2508–15. doi: 10.4049/jimmunol.168.5.2508. [DOI] [PubMed] [Google Scholar]
- 28.Jiang C, Ting AT, Seed B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82–6. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- 29.Chawla A, Barak Y, Nagy L, Liao D, Tontonoz P, Evans RM. PPAR-γ dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat Med. 2001;7:48–52. doi: 10.1038/83336. [DOI] [PubMed] [Google Scholar]
- 30.Cunard R, Ricote M, DiCampli D, Archer DC, Kahn DA, Glass CK, Kelly CJ. Regulation of cytokine expression by ligands of peroxisome proliferator activated receptors. J Immunol. 2002;168:2795–802. doi: 10.4049/jimmunol.168.6.2795. [DOI] [PubMed] [Google Scholar]
- 31.Marx N, Kehrle B, Kohlhammer K, Grub M, Koenig W, Hombach V, Libby P, Plutzky J. PPAR activators as antiinflammatory mediators in human T lymphocytes: Implications for atherosclerosis and transplantation-associated arteriosclerosis. Circ Res. 2002;90:703–10. doi: 10.1161/01.res.0000014225.20727.8f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang XY, Wang LH, Chen T, Hodge DR, Resau JH, DaSilva L, Farrar WL. Activation of human T lymphocytes is inhibited by peroxisome proliferator-activated receptor gamma (PPARgamma) agonists. PPARgamma co-association with transcription factor NFAT. J Biol Chem. 2000;275:4541–4. doi: 10.1074/jbc.275.7.4541. [DOI] [PubMed] [Google Scholar]
- 33.Marx N, Mach F, Sauty A, Leung JH, Sarafi MN, Ransohoff RM, Libby P, Plutzky J, Luster AD. Peroxisome proliferator-activated receptor-gamma activators inhibit IFN- gamma-induced expression of the T cell-active CXC chemokines IP-10, Mig, and I-TAC in human endothelial cells. J Immunol. 2000;164:6503–8. doi: 10.4049/jimmunol.164.12.6503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kawahito Y, Kondo M, Tsubouchi Y, Hashiramoto A, Bishop-Bailey D, Inoue K, Kohno M, Yamada R, Hla T, Sano H. 15-deoxy-delta(12,14)-PGJ(2) induces synoviocyte apoptosis and suppresses adjuvant-induced arthritis in rats. J Clin Invest. 2000;106:189–97. doi: 10.1172/JCI9652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li AC, Brown KK, Silvestre MJ, Willson TM, Palinski W, Glass CK. Peroxisome proliferator-activated receptor gamma ligands inhibit development of atherosclerosis in LDL receptor-deficient mice. J Clin Invest. 2000;106:523–31. doi: 10.1172/JCI10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Su CG, Wen X, Bailey ST, Jiang W, Rangwala SM, Keilbaugh SA, Flanigan A, Murthy S, Lazar MA, Wu GD. A novel therapy for colitis utilizing PPAR-gamma ligands to inhibit the epithelial inflammatory response. J Clin Invest. 1999;104:383–9. doi: 10.1172/JCI7145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Niino M, Iwabuchi K, Kikuchi S, Ato M, Morohashi T, Ogata A, Tashiro K, Onoe K. Amelioration of experimental autoimmune encephalomyelitis in C57BL/6 mice by an agonist of peroxisome proliferator-activated receptor-γ. J Neuroimmunol. 2001;116:40–8. doi: 10.1016/s0165-5728(01)00285-5. [DOI] [PubMed] [Google Scholar]
- 38.Natarajan C, Bright JJ. Peroxisome proliferator-activated receptor-gamma agonists inhibit experimental allergic encephalomyelitis by blocking IL-12 production, IL-12 signaling, and Th1 differentiation. Genes Immun. 2002;3:59–70. doi: 10.1038/sj.gene.6363832. [DOI] [PubMed] [Google Scholar]
- 39.Feinstein DL, Galea E, Gavrilyuk V, Brosnan CF, Whitacre CC, Dumitrescu-Ozimek L, Landreth GE, Pershadsingh HA, Weinberg G, Heneka MT. Peroxisome proliferator-activated receptor-gamma agonists prevent experimental auotimmune encephalomyelitis. Ann Neurol. 2002;51:694–702. doi: 10.1002/ana.10206. [DOI] [PubMed] [Google Scholar]
- 40.Devchand PR, Keller H, Peters JM, Vazquez M, Gonzalez FJ, Wahli W. The PPARα-leukotriene B4 pathway to inflammation control. Nature. 1996;384:39–42. doi: 10.1038/384039a0. [DOI] [PubMed] [Google Scholar]
- 41.Delerive P, De Bosscher K, Besnard S, Vanden Berghe W, Peters JM, Gonzalez FJ, Fruchart JC, Tedgui A, Haegeman G, Staels B. Peroxisome proliferator-activated receptor a negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-κB and AP-1. J Biol Chem. 1999;274:32048–54. doi: 10.1074/jbc.274.45.32048. [DOI] [PubMed] [Google Scholar]
- 42.Spencer NF, Poynter ME, Im SY, Daynes RA. Constitutive activation of NF-κB in an animal model of aging. Int Immunol. 1997;9:1581–8. doi: 10.1093/intimm/9.10.1581. [DOI] [PubMed] [Google Scholar]
- 43.Delerive P, Gervois P, Fruchart JC, Staels B. Induction of IkappaBalpha expression as a mechanism contributing to the anti-inflammatory activities of peroxisome proliferator-activated receptor-α activators. J Biol Chem. 2000;275:36703–7. doi: 10.1074/jbc.M004045200. [DOI] [PubMed] [Google Scholar]
- 44.Cunard R, DiCampli D, Archer DC, Stevenson JL, Ricote M, Glass CK, Kelly CJ. WY14,642, a PPAR alpha ligand, has profound effects on immune responses in vivo. J Immunol. 2002;169:6806–12. doi: 10.4049/jimmunol.169.12.6806. [DOI] [PubMed] [Google Scholar]
- 45.Cornu-Chagnon MC, Dupont H, Edgar A. Fenofibrate: metabolism and species differences for peroxisome proliferation in cultured hepatocytes. Fundam Appl Toxicol. 1995;26:63–74. doi: 10.1006/faat.1995.1075. [DOI] [PubMed] [Google Scholar]
- 46.Hill MR, Clarke S, Rodgers K, Thornhill B, Peters JM, Gonzalez FJ, Gimble JM. Effect of peroxisome proliferator-activated receptor alpha activators on tumor necrosis factor expression in mice during endotoxemia. Infect Immun. 1999;67:3488–93. doi: 10.1128/iai.67.7.3488-3493.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- 48.Chinetti G, Griglio S, Antonucci M, Torra IP, Delerive P, Majd Z, Fruchart JC, Chapman J, Najib J, Staels B. Activation of proliferator-activated receptors α and γ induces apoptosis of human monocyte-derived macrophages. J Biol Chem. 1998;273:25573–80. doi: 10.1074/jbc.273.40.25573. [DOI] [PubMed] [Google Scholar]
- 49.Jones DC, Ding X, Zhang TY, Daynes RA. Peroxisome proliferator-activated receptor a negatively regulates T-bet transcription through suppression of p38 mitogen-activated protein kinase activation. J Immunol. 2003;171:196–203. doi: 10.4049/jimmunol.171.1.196. [DOI] [PubMed] [Google Scholar]