

Abstract
Accumulating evidence indicates that ovarian hormones regulate a wide variety of non-reproductive functions in the central nervous system by interacting with several molecular and cellular processes. A growing animal literature using both adult and aged rodent models indicates that 17β-estradiol, the most potent of the biologically relevant estrogens, facilitates some forms of learning and memory, in particular those that involve hippocampal-dependent tasks. A recently developed triple-transgenic mouse (3xTg-AD) has been widely used as an animal model of Alzheimer's disease, as this mouse exhibits an age-related and progressive neuropathological phenotype that includes both plaque and tangle pathology mainly restricted to hippocampus, amygdala and cerebral cortex. In this report, we examine recent studies that compare the effects of ovarian hormones on synaptic transmission and synaptic plasticity in adult and aged rodents. A better understanding of the non-reproductive functions of ovarian hormones has far-reaching implications for hormone therapy to maintain health and function within the nervous system throughout aging.
Keywords: 3xTg AD, aging, estrogen, hippocampus, LTP, mouse, synaptic plasticity
Introduction
Within the past decade, there has been increasing interest among neuroscientists in studying the effects of estrogen on neural function. This effort is driven, in part, by the results of clinical studies suggesting that estrogen therapy administered after the menopause may delay or prevent the onset of Alzheimer's disease (AD) in older women. Other, still controversial, research indicates that estrogen may enhance certain forms of memory in postmenopausal women. Much of the most current research related to estrogen and brain function is focused in two primary directions. First, clinical studies have examined the potential effect estrogen might offer in protecting against cognitive decline during normal aging and against AD (neuroprotection). Second, laboratory studies have examined the mechanisms by which estrogen can modify the structure of nerve cells and alter the way neurons communicate with other cells in the brain (neuroplasticity). Rodent models have been used extensively to study a variety of behavioral, cognitive and anatomical changes linked to important features of AD found in humans. In this article, we examine recent evidence from experimental research using rodent models on the effects of estrogen on neuroplasticity and neuroprotection in both adult and aged rats, as well as information regarding the recent triple transgenic mouse model of AD.
AD is an age-related, irreversible and neurodegenerative disorder that causes a progressive deterioration of cognitive function, including a profound loss of memory [50]. Neuropathologically, AD is characterized by an accumulation of amyloid-β (Aβ) deposits in the brain, neurofibrillary tangles which consist of hyperphosphorylated tau aggregates [90], and progressive neuron loss. Aβ has been found to be the primary component of amyloid plaques and is generated from the amyloid-β protein precursor (AβPP) by sequential proteolytic cleavage at the β and γ sites on the peptide [50]. Neurofibrillary tangles are composed of hyperphosphorylated tau, a microtubule-associated protein found in the brain. Tau promotes the assembly of microtubles; hyperphosphorylation of tau interferes with the normal biological functions of tau by reducing its ability to bind to and stabilize microtubules [109]. The occurrence of both amyloid deposits and neurofibrillary tangles are necessary to confirm a diagnosis of AD. Epidemiological studies indicate that women develop a higher risk of AD even after adjusting for age [23,56], suggesting a genetic or hormonal cause. However, the exact cause of higher AD risk in women is unknown. The depletion of the sex steroid hormone estrogen (and progesterone) at menopause appears to be a significant risk factor for the development of AD in women [60,82,83,105,125]. Prospective and case-control studies have demonstrated that hormone therapy (HT) can reduce the risk of AD in women [60,83,105]. However, the relationship between the therapeutic benefits of estrogen- and progesterone-based HT, and both normal cognitive decline and development of AD have been the recent topics of heated debate and controversy (see [29]). Furthermore, clinical findings from the massive Women's Health Initiative Memory Study (WHIMS) demonstrating a higher incidence of dementia in subjects receiving estrogen-based HT [87,93] was quite unexpected, given the early background of estrogen and AD. The WHIMS findings raised many important points, perhaps the most important being the need to better understand the role of estrogen (and progesterone) in AD pathogenesis, and to optimize HT.
Estrogen and Cognition
In humans, the issue of hormone therapy has yielded much debate with many clinical studies supporting a protective role of estrogen, along with those including the use of progesterone to counteract estrogen-induced proliferation of the endometrium [57,91]. The depletion of estrogen that occurs after the menopause increases the susceptibility of women to AD [53,83], whereas estrogen replacement in postmenopausal women improves verbal memory [2,88]. Healthy postmenopausal women with estrogen replacement scored significantly higher on tests of immediate and delayed paragraph recall compared with healthy postmenopausal women not taking estrogen replacement [59]. Other evaluations of estrogen-replacement therapy in AD patients indicate that estrogen does not alleviate cognitive impairment associated with the disease [73], but it does seem to have a beneficial effect as a preventive treatment [105], which is most apparent in younger, postmenopausal women [54]. The WHIMS study [93] was designed to address this seemingly contradictory literature. A possible confound in many of the earlier estrogen studies of cognition was the use of combinations of estrogens and progestins. The WHIMS reported a non-significant increase in the number of women with mild cognitive impairment who were using estrogen (conjugated equine estrogens), but a highly significant increase in the number of women with mild cognitive impairment who were using combined estrogens and progestins.
In animal models, ovarian hormones have been shown to influence memory via actions on neurons, particularly in the hippocampus [71,74,104,115]. A neuroprotective role of estrogen has been established in both in vivo and in vitro animal models of neurodegeneration [39,40,99,101]. Additionally, 17β-estradiol has been found to facilitate some forms of learning and memory function in rodents, particularly for hippocampal-dependent tasks [7,8]. Post-training injection of 17β-estradiol facilitates retention in the Morris water maze [97], and a cholinergic agonist enhance this effect [81]. In other studies, the effects of 17β-estradiol and raloxifene, a selective estrogen-receptor modulator, have been evaluated on the acquisition of a delayed matching to position in a T-maze task and on hippocampal acetylcholine release in ovariectomized rats. 17β-estradiol, but not raloxifene, enhanced the T-maze task performance, and 17β-estradiol and a high dose of raloxifene increased potassium-stimulated acetylcholine release in hippocampus [47]. By contrast, some studies found little or no effect of the estrous cycle, and thereby of endogenous levels of 17β-estradiol, on tasks involving spatial memory [11,43,114,122].
A recent review of sex differences in rodent models of learning and memory function suggests that male rats have advantages in some forms of memory, but this finding was found not to be as strong in mouse models of memory [58]. Most research suggests that 17β-estradiol, and perhaps progesterone, influence learning and memory, but do so in a task-dependent manner [32]. In a series of rat studies, progesterone was found to impair cognitive function [16,17]. Gonadally-intact aged female rats performed much more poorly on a reference memory task (radial arm maze) than gonadally-intact young adult female rats. Interestingly, if the aged animals were ovariectomized, they performed as well as the young animals. It appears that in the gonadally-intact aged animals, 17β-estradiol levels are virtually the same as in young animals, but progesterone levels are much higher in the aged animals. Indeed, if aged ovariectomized animals were chronically implanted with progesterone pellets, they performed the radial arm maze task as poorly as gonadally-intact aged female rats. In this cognitive task, progesterone supplementation appears to reverse the somewhat cognitive enhancing effects of ovariectomy. Despite a vast literature on ovarian hormones and cognition, the effects of ovarian hormones on learning and memory functions still remain unclear. What emerges from many of these reports is that 17β-estradiol and progesterone enhance learning and memory in some instances, but impair, or have no effect on learning in others.
Estrogen and Hippocampus
For more than 30 years, electrophysiological investigations have found estrogen to promote changes in synaptic plasticity within the nervous system. In a pioneering study, decreased hippocampal seizure thresholds were found in animals primed with estrogen and also during proestrus, the time of the estrous cycle when estrogen levels are at their highest levels [106]. In humans, changes in electrical activity of nervous system tissue correlate with hormonal factors that appear to play a role in catamenial epilepsy, a form of epilepsy in which the likelihood of seizures varies during the menstrual cycle. Many women with catamenial epilepsy experience a sharp increase in seizure frequency immediately before menstruation, when estrogen concentrations relative to those of progesterone are also at their highest levels [3]. Changes in hippocampal responsiveness correlate with estrogen activity, as induction of long-term potentiation (LTP), an induced form of synaptic plasticity (see below), is maximal in female rats during the afternoon of proestrus, when endogenous estrogen concentrations are highest [113]. Furthermore, induction of hippocampal LTP is facilitated in ovariectomized rats treated with estrogen as compared to untreated ovariectomized rats [28].
The development of in vitro models to study the mechanisms of neuronal plasticity have provided researchers better tools to investigate how estrogen regulates synaptic excitability in the nervous system, and, in particular, in hippocampus. It should be stressed, however, that the binding of 3H-estradiol in hippocampus does not approach that seen in hypothalamus and related diencephalic structures [69,70]. Nonetheless, studies by Teyler and colleagues using in vitro hippocampal slice preparations have shown that gonadal steroids dramatically affect neuronal excitability in specific pathways of the rodent hippocampus [107,110]. In the initial series of experiments, extracellular monosynaptic population field responses recorded from area CA1 of hippocampal slices from male and female rats were monitored before and after the addition of 17β-estradiol (100 pM) to the slice incubation medium (artificial cerebrospinal fluid; aCSF). In male rats, 17β-estradiol produced a rapid (< 10 min) enhancement of population field responses evoked by stimulation of the afferents to CA1 pyramidal cells (Fig. 1). This was the first published report demonstrating that picomolar concentrations of the gonadal steroid 17β-estradiol directly enhanced glutamatergic synaptic transmission in hippocampus [107].
Figure 1.

(A) Diagram of a transverse hippocampal slice. Stimulating electrodes were located in the afferent pathway, which contains the Schaffer (Sch.) collaterals. Recording micropipettes were situated in the pyramidal cell body layer in CA1. Cells of this subfield receive monosynaptic input from the CA3 pyramids via the Schaffer collateral system.
(B) Representative field potentials from slice preparations in the various experimental conditions. Extracellular population spike responses to a given stimulus intensity are shown from the control period (before steroid administration) and after the administration of 1 × 10-10 M 17β-estradiol (E) or 1 × 10-10 M testosterone (T). Potentials from slices obtained from males and from proestrus and diestrus females are shown for purposes of comparison. All potentials are single sweeps recorded at the same voltage and time scales.
(C) Bar graph summarizing the major experimental outcomes. Values on the ordinate are mean percentages of spike amplitudes after steroid administration. Data for each condition are from 6 to 10 animals, each contributing one slice. Cursors representing magnitude of variability (standard error of the mean) are shown for each bar.
Reprinted with permission from [107]. Copyright 2008 American Association for the Advancement of Science.
Although the mechanism of action of gonadal steroids in hippocampus is not entirely understood, it is likely to be receptor-mediated. There was no facilitation of field responses when the inactive estrogen, 17α-estradiol, was added to hippocampal slice medium [38], and the further addition of 17β-estradiol no longer resulted in an increased response, as observed in the presence of 17β-estradiol alone [38,116,117]. Similar results were found when the estrogen receptor antagonist tamoxifen was applied to hippocampal slices before addition of 17β-estradiol [37]. The ability of 17α-estradiol and tamoxifen to block the effects of 17β-estradiol on hippocampal excitability provides strong evidence that the rapid physiological modulation of gonadal hormones is most likely due to the activation of a plasma membrane receptor.
Synaptic Plasticity and Long-Term Potentiation
Long-term potentiation of synaptic transmission in hippocampus and neocortex is considered to be a cellular model of memory trace formation in the brain, at least for certain forms of memory [8,18,64]. Although there is a large body of work regarding the molecular and synaptic mechanisms underlying LTP [9,46], there is a relative paucity of studies demonstrating the critical role of LTP in behavioral learning and memory [92]. Nonetheless, whether LTP is or not the substrate of the synaptic modifications which occur during learning in forebrain structures of vertebrates, studies of its mechanisms have revealed the existence of a number of processes that undoubtedly play critical roles in memory formation [12]. In area CA1 of hippocampus, the most widely studied form of LTP requires NMDA receptor activation for its induction, and an increase in α-amino-3-hydroxy-5-methyl-4-isoxazoleproprianate (AMPA) receptor function for its expression and maintenance. In addition, Teyler and associates have demonstrated a second form of tetanus-induced LTP in CA1 that is independent of NMDA receptors, and involves voltage-dependent calcium channels [48].
Estrogen and Non-Genomic Mechanism of Action
The genomic mechanism of action of estrogen has been the traditional framework for interpreting the effect of estrogen on cell function, but an increasing number of reports document the effects of acute applications of estrogenic steroids that are too rapid (occurring ≤ 10 min) to be accounted for exclusively by a genomic pathway. In particular, the existence of rapid estrogenic steroid-induced changes in neuronal excitability suggests other, non-genomic mechanisms involving direct interactions with sites on the plasma membrane that alter or regulate a variety of ion channels and neurotransmitter transporters [84,119].
In vitro intracellular recordings of CA1 neurons from adult ovariectomized female rats have shown that the addition of 17β-estradiol increases synaptic excitability in part by enhancing the magnitude of AMPA receptor-mediated responses [117]. The rapid onset of the increased excitability, and its blockade by 6-cyano-7-nitroquinaxaline (CNQX, an AMPA receptor antagonist) but not by D-2-amino-5-phosphonovalerate (D-APV a competitive NMDA receptor antagonist), supported a postsynaptic membrane site of action resulting in enhanced non-NMDA glutamate receptor function. Later studies using whole cell recordings found that acute 17β-estradiol application potentiated kainate-induced currents in a subpopulation of CA1 cells [49], although a direct interaction between 17β-estradiol and the receptor channel was not indicated [118].
Estrogen and Nmda Receptor Regulation
A large body of evidence demonstrates that 17β-estradiol-mediated regulation of synapse formation is dependent on NMDA receptor activation. Morphological studies during the course of neuronal development conducted in cultured neurons prepared from embryonic day 18 rat fetuses have shown that estrogenic steroids exert a growth-promoting, neurotrophic effect on hippocampal and cortical neurons via a mechanism that requires NMDA receptor activation [21,22]. In vivo studies using adult ovariectomized female rats have also revealed a proliferation of dendritic spines in hippocampal CA1 pyramidal cells after 17β-estradiol treatment that could be prevented by blockade of NMDA receptors, but not by AMPA or muscarinic receptor antagonists [120]. Other reports using adult ovariectomized female rats provided evidence that chronic 17β-estradiol treatment increased the number of NMDA receptor binding sites and NMDA receptor-mediated responses [44,121]. These studies indicate that estrogen and NMDA receptors are heavily involved in synapse formation.
The possibility of a direct regulation of NMDA receptor-mediated synaptic transmission by 17β-estradiol may not have been detected previously (e.g., [117]) because tests of this hypothesis had not been conducted under optimal conditions. Because of the voltage-dependent blockade of the NMDA receptor channel by Mg2+ and the slow kinetics of the channel opening relative to that of the AMPA receptor, there is only a minor NMDA receptor-mediated component of the excitatory postsynaptic potential (EPSP) evoked by low-frequency stimulation of glutamatergic afferents. This NMDA receptor component can be enhanced with low Mg2+ concentrations or high-frequency stimulation patterns used to induce the depolarization accompanying the summation of overlapping EPSPs [124]. In experiments using low Mg2+ concentrations and in the presence of the AMPA receptor antagonist 6,7-dinitroquinoxaline-2,3-dione (DNQX), an acute application of 17β-estradiol in adult male rat hippocampal slices resulted in a rapid increase in the amplitude of NMDA receptor-mediated EPSPs evoked by stimulation of the Schaffer collaterals [39]. The effect of 17β-estradiol on pharmacologically isolated NMDA receptor-mediated synaptic responses was such that concentrations of 17β-estradiol greater than 10 nM induced seizure activity in hippocampal neurons, and lower concentrations (1 nM) markedly increased the amplitude of NMDA receptor-mediated EPSPs.
Estrogen and Hippocampal LTP in Male Rats
To investigate the effect of estrogen on synaptic plasticity associated with learning and memory function, estrogen was applied to hippocampal slices from adult male rats before the slices were exposed to high-frequency stimulation designed to induce LTP. When LTP was assessed after high-frequency stimulation, fEPSP values were increased significantly for 17β-estradiol-treated slices compared to control-aCSF slices (Fig. 2). fEPSP mean increases in slope was 192% (experimental) vs. 154% (control). Thus, hippocampal slices from adult male rats treated with 17β-estradiol exhibited a pronounced, persisting and significant increase in LTP as measured by both population fEPSP slope and fEPSP amplitude recordings [39,42].
Figure 2.
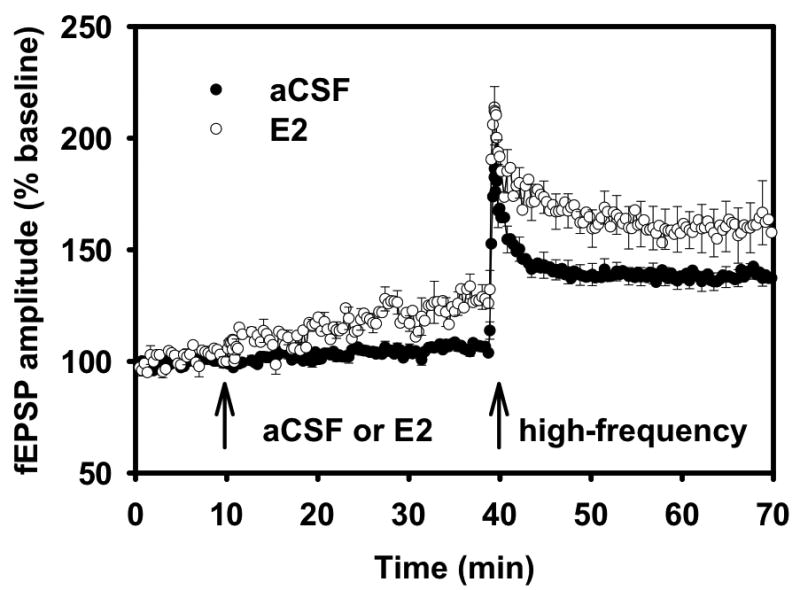
Field EPSP (f-EPSP) recordings in area CA1.
All hippocampal slices were perfused with aCSF for 10 min to obtain fEPSP slope and amplitude percentage baseline data. After 10 min of baseline recording, experimental slices were perfused with 100 pM 17β-estradiol (E2). Control slices continued to be perfused with aCSF. After 30 min of either E2 or aCSF perfusion, all slices received high-frequency stimulation, designed to induce long-term potentiation. Data points represent averaged fEPSP slope ± SE (taken at each 20 sec sweep) for experimental (E2-treated) and control (aCSF) hippocampal slices.
Reprinted with permission from [42]. Copyright 2008 American Physiology Society.
To further evaluate the effects of 17β-estradiol on the magnitude of hippocampal LTP, the intensity of afferent stimulation to Schaffer collaterals in slices perfused with 17β-estradiol was decreased in order to produce baseline values to pre-17β-estradiol levels immediately before the delivery of the high-frequency stimulation train used to elicit LTP [13]. Under these conditions, 17β-estradiol still produced an increase in the amplitude of LTP from adult male rat hippocampal slices compared to that obtained in control (aCSF) slices (Fig. 3). These findings indicate that 17β-estradiol-induced enhancement of hippocampal LTP is not due to simply a change in basal EPSP level, but is more likely due to biochemical activation of an intracellular cascade, presumably mediated by activation of a src tyrosine pathway that enhances NMDA receptor function.
Figure 3.

Changes in LTP in field CA1 of hippocampal slices from female rats in proestrus and diestrus. Hippocampal slices from female rats in proestrus or diestrus were prepared as described. fEPSP amplitude and slope values were obtained for each slice and averaged across slices to produce one average before and after the train of hfs. fEPSP amplitudes and slopes were normalized for the 10 min pre-hfs period for each slice. Separate ANOVAs and planned two-tailed t tests for the pre-hfs and post-hfs periods were used to evaluate the effects of estrous cycle on fEPSP slope and amplitude.
(A) Representative waveforms from female rats in proestrus and diestrus for pre-hfs (1) and post-hfs (2) periods.
(B) Means ± SEM of fEPSP slopes recorded in slices from female rats in proestrus (filled circles; n=6) and diestrus (open circles; n=5).
Reprinted with permission from [14]. Copyright 2008 National Academy of Sciences, USA.
Estrogen and Hippocampal LTP in Female Rats
In another series of animal studies, estrous cycle changes in rats were correlated with changes in synaptic plasticity. Hippocampal slices from cycling female rats in diestrus (low estrogen concentration) and proestrus (high estrogen concentration) were prepared in aCSF, and LTP was elicited by high-frequency stimulation. The difference in LTP magnitude between these groups following high-frequency stimulation was dramatic: slices from rats in proestrus exhibited LTP representing about a 50% increase over baseline, whereas slices from rats in diestrus had LTP values representing about a 25% increase over baseline [14] (Fig. 4). These findings support the original interpretation of Teyler et al (1980) who identified changes in baseline synaptic transmission that were correlated with the phase of the estrus cycle in female rats at the time of hippocampal slice preparation.
Figure 4.

Changes in LTP in field CA1 of hippocampal slices from female rats in proestrus and diestrus, and with 17β-estradiol (experimental) or aCSF (control) treatment. Hippocampal slices from female rats in proestrus or diestrus were prepared as described.
(A) Means ± SEM of fEPSP amplitudes recorded following tetanus in slices from female rats in diestrus. 17β-estradiol (filled circles) enhanced LTP relative to control aCSF (open circles).
(B) Means ± SEM of fEPSP amplitudes recorded following tetanus in slices from female rats in proestrus. 17β-estradiol (filled circles) impaired LTP relative to control aCSF (open circles).
Reprinted with permission from [36]. Copyright 2008 Cambridge University Press.
Since the electrophysiological study above has shown that female rats in proestrus exhibited an increased magnitude of hippocampal LTP as compared to female in diestrus, results of a current study examining the effect of 17β-estradiol on hippocampal LTP during the two critical time periods in the rat estrous cycle, proestrus and diestrus, are reported here. Estrous cycles of adult (3-5 mo) Sprague-Dawley rats were monitored for 10 days prior to any physiological experiments, and hippocampal slices were prepared from rats that were either in proestrus or diestrus. Recording and stimulating electrodes were positioned in the dendrites of area CA1 and Schaffer collaterals, respectively. Baseline stimulation (0.05 Hz, 100 μsec) was adjusted to elicit 50% of the maximum fEPSP amplitude. After 10 min of stable baseline stimulation, aCSF or 17β-estradiol at a concentration of 100 pM (experimental group) was perfused to the slices for 30 min, and LTP was induced by a brief period of high-frequency stimulation (5 trains of 20 pulses at 100 Hz). Subsequent synaptic responses were monitored for 30 min post-LTP induction. The magnitude of LTP induced in area CA1 was larger in slices from proestrus rats, compared to slices from diestrus rats, as previously reported [14]. However, addition of 17β-estradiol increased LTP in slices from diestrus rats, while it decreased LTP in slices from proestrus rats (Fig. 5). These observations suggest that 17β-estradiol alters hippocampal LTP in female rats, depending on the state of their estrous cycle (i.e., on the levels of circulating 17β-estradiol). In cycling female rats, when endogenous circulating levels of 17β-estradiol are at their highest levels (i.e., proestrus), LTP magnitude is increased, and exogenously applied 17β-estradiol during proestrus decreases LTP magnitude, possibly through the activation of inhibitory or ceiling effect. When endogenous circulating levels of 17β-estradiol are at their lowest levels (i.e., diestrus), the situation is completely reversed from that observed in the proestrus state. Here, LTP magnitude is decreased, and exogenously applied 17β-estradiol increases LTP magnitude under this condition.
Figure 5.


Long-term depression.
(A) LTD Adult aCSF versus E2. In this figure, following baseline and drug/aCSF periods, slices received low-frequency stimulation (low-frequency) to elicit long-term depression. LTD that was initially induced in the aCSF adult males quickly diminished. The arrows indicate the time at which aCSF/E2 was applied, and when low-frequency stimulation (900 pulses at 1 Hz) was delivered.
(B) LTD Aged aCSF versus E2. LTD was examined in slices from aged rats, with fEPSP comparisons between aCSF versus E2. LFS delivered to aged rat slices perfused with 17β-estradiol failed to induce robust LTD. Reprinted with permission from [42].
Copyright 2008 American Psychological Association.
These results suggest that cyclic changes in estrogen levels occurring during the estrous cycle in female rats are associated with changes in the magnitude of LTP recorded from hippocampal CA1 cells. They also corroborate work mentioned earlier indicating the facilitation of LTP induction by estrogen in ovariectomized female rats [28], increased LTP in the afternoon of proestrus of female rats [113], and support a study showing improved memory performance with high estrogen levels in female rats [66].
Estrogen, Synaptic Plasticity and Aging in Rats
It has been reported that during aging, when memory function declines, the processes of synaptic plasticity in hippocampus are altered. Specifically, LTP is impaired and the opposite process of long-term depression (LTD) is enhanced [4-6,34,35,45,62,63,78,79]. We recently replicated this effect of aging on LTD and discovered a profound action of estrogen on this process in aged male rats [42,111]. LTD was induced in CA1 region of hippocampal slices using standard conditions (stimulation of Schaffer collaterals at 1 Hz for 15 min) in adult (3-5 mo) and aged (18-24 mo) Sprague-Dawley rats. In agreement with earlier studies, the standard protocol for inducing LTD resulted in little or no LTD in slices from adult animals, but in marked LTD in slices from aged animals (Fig. 6A) [34,35]. Infusion of 17β-estradiol in slices caused a slight increase in synaptic transmission (baseline), as in previous studies. It had little effect on LTD in slices from adult animals, but markedly attenuated LTD in slices from aged animals (Fig. 6B). Thus, the prevention by 17β-estradiol of age-related LTD enhancement may account, in part, for the protective effects of estrogen on memory functions in aged organisms reported in some studies (see below).
Figure 6.

Schematic representation of the general hypotheses linking estrogen/testosterone with the MAPK/ERK pathway, NMDA receptors and synaptic plasticity in brain. Abbreviations: AMPAR = α-amino-3-hydroxy-5-methyl-4-isoxazoleproprianate receptor; bax = protein of bcl-2 family (pro-apoptotic); bcl-2 = integral membrane protein (anti-apoptotic); Erα = estrogen receptor alpha; Erβ = estrogen receptor beta; ERK = extracellular signal regulated kinase; Erm = estrogen receptor membrane; GFAP = glial fibrillary acidic protein; LTP = long-term potentiation; MAPK = mitogen-activated protein kinase; MEK = MAPK kinase; NMDAR = N-methyl-D-aspartate receptor; Src = family of intracellular tyrosine kinases; Src-1 = member of src kinase family.
Reprinted with permission from [36]. Copyright 2008 Cambridge University Press
Estrogen and Two Forms of LTP
As we noted earlier, Teyler and associates discovered a form of LTP in CA1 pyramidal neurons that is independent of NMDA receptors and involves voltage-dependent calcium channels [48]. This form of LTP is optimally induced by very high frequency stimulation of Schaffer collaterals (e.g., 200 Hz for 1 sec) and is blocked by nifedipine (L-type calcium channel blocker), but not by the NMDA receptor antagonist D-APV. In contrast, the NMDA receptor-dependent form of LTP in CA1 is best induced by lower frequencies of stimulation (e.g., 25 Hz) and is blocked by D-APV but not by nifedipine. The standard stimulation paradigm used for LTP induction (i.e., 100 Hz for 1 sec) therefore induces both forms of LTP [26,72].
We evaluated the effects of acute application of 17β-estradiol (CA1 slice) on both forms of LTP in hippocampal slices from male rats. Using 25 Hz tetanus of Schaffer collaterals, 17β-estradiol and nifedipine were infused and extracellular field EPSPs recorded. 17β-estradiol caused the expected increase in synaptic transmission and pronounced enhancement of LTP whereas nifedipine had no effect on either process, implying that under this condition, 17β-estradiol was acting only on NMDA receptor-dependent LTP [126].
We then used a 100 Hz tetanus of Schaffer collaterals (in the CA1 region of hippocampal slices from adult male rats), and both extracellular field EPSPs and intracellular EPSPs were recorded from pyramidal neurons and 17β-estradiol and nifedipine were infused. 17β-estradiol alone caused the expected increase in synaptic transmission and pronounced enhancement of LTP, but both effects of 17β-estradiol were reduced in magnitude by nifedipine. Therefore, under this condition, it would seem that 17β-estradiol is acting by modulating both L-type voltage gated calcium channels and NMDA receptors. Intracellularly recorded EPSPs in response to paired subthreshold stimuli with a short interstimulus interval (50 ms) in the presence of 17β-estradiol indicated an increase in EPSP amplitude to both stimuli without changes in the paired-pulse ratio, strongly supporting a postsynaptic origin of the effects of 17β-estradiol [1]. The possibility that 17β-estradiol may modulate calcium influx through L-type calcium channels is consistent with the effects of aging on synaptic transmission and plasticity in hippocampus. Thus, it has been reported that aging is associated with enhanced activity of voltage-gated calcium channels in hippocampal CA1 neurons [24], and that blocking calcium influx through L-type calcium channels inhibits LTD induction and enhances LTP in aged animals in the CA1 region of hippocampal slices [79]. Blocking L-type calcium channels in hippocampus has been reported to enhance memory in several paradigms and particularly to enhance learning and memory processes in aged animals [31,85,86].
Development of 3xTg-AD Mouse (Alzheimer's Disease)
A recently generated triple-transgenic mouse model of AD (called 3xTg-AD) was developed in LaFerla's laboratory at the University of California, Irvine [80]. Mutations in three genes linked to AD and frontotemporal dementia were utilized. Human AβPPSWE and tauP301L transgenes were co-microinjected into single-cell embryos harvested from homozygous mutant PS1M146V knock in mice. There are several advantages in using this model. First, the reported tight AβPP and tau linkage paired with the ‘knock in’ PS1 approach yielded homozygous mice that breed readily, thus facilitating rapid, straightforward and cost-effective generation of a study colony. Second, and more importantly, the 3xTg-AD mouse exhibits age-related neuropathological phenotype that includes both Aβ and hyperphosphorylated tau pathologies that develop with a regional pattern similar to AD. Specifically, intracellular Aβ accumulates first in cortical regions (around 4 mo) and later in hippocampus, while tau hyperphosphorylation develops after Aβ accumulation (between 12-15 mo), beginning in limbic structures and progressing to cortical regions [15,80]. An in vitro study examining synaptic dysfunction in 3xTg-AD mice at 6 months found lowered levels of hippocampal basal synaptic transmission and reduced levels of LTP compared to non-transgenic mice [80]. In this report, it was suggested that the synaptic dysfunction found in transgenic mice might represent an early change preceding the accumulation of the hallmark pathological lesions that accompany AD. Another study using 3xTg-AD mice found that in ovariectomized transgenic mice, progesterone blocked the beneficial effect on Aβ peptide accumulation provided by estrogen treatment alone, but did not affect spontaneous alternation behavior in a Y-maze, a rodent model of working memory [25]. Significantly higher amounts of Aβ peptide pathology was also found in female 3xTg-AD mice compared to male 3xTg-AD mice [55]. Research utilizing the 3xTg-AD mouse model has indicated that this mouse also shows hypothalamic-pituitary-adrenal (HPA) axis hyperactivity in both an age- and sex-dependent fashion [108]. 3xTg-AD mice exhibit HPA hyperactivity in response to stress, which is more pronounced in 9 mo old female 3xTg-AD mice compared to age-matched non-transgenic female mice and 3xTg-AD male mice [27].
In an in vitro electrophysiological study, synaptic plasticity was examined comparing the effects of 17β-estradiol in hippocampal slices prepared from both gonadally-intact and gonadectomized 6 month-old male 3xTg-AD and wild-type (wt: 129/C57BL/6 F1 hybrid) mice [41]. 17β-estradiol induced an increase in LTP in both 3xTg-AD groups (intact and gonadectomized) compared to their respective vehicle controls [41]. However, in the wild-type groups, 17β-estradiol produced a smaller enhancement of LTP. These findings suggest a differential effect of 17β-estradiol on hippocampal synaptic plasticity in 3xTg-AD and wild-type mice.
In a related behavioral study, the impact of allopregnanolone, a metabolite of progesterone promoting proliferation of neural progenitor cells derived from rat hippocampus and cerebral cortex [112], was evaluated on the hippocampal-dependent trace eyeblink conditioning task in 3xTg-AD mice [96]. In delay eyeblink conditioning, the conditioned stimulus (CS; e.g., auditory tone) onset precedes the unconditioned stimulus (US; e.g., an airpuff to the cornea of the eye) onset, and the two CS and US stimuli overlap and coterminate with one another. In trace eyeblink conditioning, the CS precedes the US, and there is a short stimulus free period (trace interval) between the CS offset and the US onset. Research has found that both delay and trace eyeblink conditioning require the cerebellum, but the trace procedure also requires involvement of the hippocampus [100,103]. Mice were injected with 10 mg/kg allopregnanolone (s.c.) and 100 mg/kg BrdU. Seven days later, mice were trained in the trace eyeblink-conditioning paradigm (250 ms tone followed by 100 ms, 60 Hz shock, 30 trials, 2 sessions/day for 5 days, trace interval of 250 ms). Following the learning trials, mice were returned to their home cages for another seven days, and subsequently tested for memory of the learned association. Allopregnanolone treatment enhanced the rate of learning in 3xTg-AD mice, increased the magnitude of the learning performance, and reversed the memory deficit of 3xTg-AD [96]. These results also suggest that allopregnanolone is a potent cognitive enhancer in a mouse model of AD, and that allopregnanolone could be a potential therapeutic to prevent or delay cognitive deficits associated with AD.
In summary, the neuropathology of AD mimicked in the 3xTg-AD mouse (development of Aβ and tau pathology) provides an exciting model with which to the study the molecular, cellular and behavioral interactions between the many processes underlying learning and memory function, and their modifications during the post-menopausal period and aging-related disorders.
Estrogen and Cellular Neuroprotection
In a well-established model of estrogen-induced neuroprotection, primary cultures of hippocampal neurons were exposed to the excitatory amino acid, glutamic acid, and neuronal injury was assessed by measuring lactate dehydrogenase (LDH) release in the culture medium. A 5 min treatment with 100 μM glutamate caused significant cell death compared to control conditions; neuronal cell death was significantly decreased following pre-treatment with 17β-estradiol [75]. Maximal neuroprotection of approximately 18% was provided by a concentration of 10 ng/ml 17β-estradiol [75].
To investigate the mechanism underlying E2-mediated neuroprotection, and in particular, the contributions of changes in intracellular calcium concentration, calcium concentration was determined by imaging techniques and microfluorescence in cultured hippocampal neurons. Surprisingly, in neurons pretreated with 17β-estradiol, changes in intracellular calcium concentration elicited by glutamate application were increased by about 70% [76]. 17β-estradiol itself induced a rapid increase in intracellular calcium concentration within minutes of exposure, an effect that was blocked by an L-type calcium channel antagonist suggesting that 17β-estradiol directly or indirectly regulates some properties of these voltage-gated calcium channels [123]. 17β-estradiol-induced calcium ion influx was required for 17β-estradiol-mediated activation of a biochemical signaling pathway involving Src, ERK, CREB, and Bcl-2; a schematic diagram of the molecular signaling cascade leading to 17β-estradiol-induced neuroprotection was described in [123]. These results demonstrate that at the single-cell level, a 17β-estradiol membrane-associated receptor mediates rapid 17β-estradiol effects in cultured neurons. Estrogen-induced neuroprotection against excitotoxic glutamate also requires the mitogen-activated protein kinase (MAPK) cascade in primary cortical neuron cultures [77,95]. A similar neuroprotective effect of 17β-estradiol against NMDA-mediated neurotoxicity was reported in cultured hippocampal slices, and this effect also involved the activation of the MAP kinase pathway [14].
Collectively, these reports indicate that although significant progress has been made regarding the identification of the cellular mechanisms involved in 17β-estradiol-mediated neuroprotection, there is a need to further elucidate a number of unresolved issues regarding the complex and mostly indirect ways in which estrogen interacts with numerous cellular signaling pathways regulating cell survival and death.
Molecular Mechanisms of Estrogen Effects in Brain
Recent results from several laboratories have provided a general framework to understand the mechanisms underlying the multiple effects of 17β-estradiol on synaptic structure and function (Fig. 9) [65]. Physiological concentrations of 17β-estradiol (10 pM-1 nM) interact with Erα and Erβ receptors to produce both direct and indirect genomic effects. The direct genomic effects are due to interactions between 17β-estradiol and traditional cytoplasmic receptors followed by translocation of the hormone-receptor complex to the nucleus and the regulation of transcription of specific genes, through interactions with ERE regulatory elements present in these genes. In neurons, these include anti-apoptotic genes of the bcl-2 family, which are probably responsible for the neuroprotective effects of 17β-estradiol observed in a number of models of neuronal death. In astrocytes, they include the GFAP (down regulation) and laminin (up regulation) genes, which might be involved in regulating sprouting responses following lesions, as well as in normal astrocyte activation in brains from old animals [61]. The indirect genomic effects of 17β-estradiol might be linked to the stimulation of the phosphoinositol-3 (PI3) kinase/Akt system [30,94] and/or of a G protein, and/or of Src tyrosine kinase and ERK/MAP kinase pathways [98].
The MAP kinase pathway occupies a central place in the regulation of synaptic plasticity [68,102]. Pharmacological manipulations directed at blocking this pathway have consistently produced impairments in synaptic plasticity and learning and memory, and this pathway is activated with LTP-inducing tetanus or in different learning paradigms [10,19,20,89]. We have shown that endogenous estrogen levels in cycling female rats produce a tonic phosphorylation/activation of extracellular signal-regulated kinase 2 (ERK2)/MAP kinase [14]. In addition, we have shown that this activation of the MAP kinase pathway is also linked to the regulation of glutamate ionotropic receptors and might be involved in the “cognitive enhancing” effects of 17β-estradiol. Indeed, the acute estrogen-mediated enhancement of LTP is mediated by activation of a src tyrosine kinase pathway [13]. Thus, acute application of the src inhibitor PP2 in the perfusing medium of hippocampal slices from adult male rats abolished 17β-estradiol-mediated enhancement of both synaptic transmission and LTP, but had no effect on LTP itself (see Fig. 5). Similarly, this pathway might also be involved in the neuroprotective effects of 17β-estradiol, as MAP kinase inhibitors have consistently been shown to block the neuroprotective effects of 17β-estradiol in a variety of models of neurodegeneration. Moreover, growth factors and other factors providing neuroprotection, such as PDGF and BDNF, also use the MAP kinase pathway for their neuroprotective effects.
Interestingly, it appears that Erα stimulation is critically involved in the neuroprotective effect of estrogen, as Erα knock-out mice are not protected by 17β-estradiol against ischemia-induced neuronal damage [33]. Furthermore, recent results obtained from the same knock-out mice suggest the possible existence of novel 17β-estradiol receptors responsible for the activation of the ERK/MAP kinase pathway [98]. These results indicate that several steps described in Figure 9 remain to be elucidated.
Summary of Estrogen's Effects on Synaptic Plasticity
The studies mentioned in this report establish several fundamental characteristics of the effects of estrogen on synaptic transmission in the mammalian central nervous system (CNS). Estrogen acts rapidly via presumed membrane mechanisms to enhance both NMDA and AMPA receptor/channel responses elicited by glutamate released from excitatory presynaptic terminals.
17β-estradiol can also markedly enhance hippocampal LTP in CA1 neurons of adult, male rats. The enhancement of LTP after acute 17β-estradiol application is due to an increase in NMDA receptor and AMPA receptor functions. Both possibilities are consistent with intracellular data. Changes in estrogen levels in cycling female rats have also been correlated with changes in synaptic plasticity, as measured by changes in LTP magnitude. Furthermore, 17β-estradiol has also been found to enhance LTP from male hippocampal slices prepared from 3xTg-AD mice. These findings suggest a mechanism by which naturally fluctuating endogenous hormone levels can impact a cellular model associated with important aspects of learning and/or memory in mammalian CNS.
To the extent that LTP is a mechanism involved in processes of coding and storage of information, i.e., in memory formation, 17β-estradiol appears to enhance these processes. Indeed, the 17β-estradiol enhancement of LTP suggests a possible mechanism by which 17β-estradiol can exert its facilitatory effects on memory processes in humans. Clinical evidence indicates that estrogenic steroids can enhance cognitive functions in humans, particularly in postmenopausal women [51,52,60], however, some prospective observational studies have yet to find a protective effect of estrogen on either cognition, or the incidence of dementia [7,67]. Understanding the mechanisms underlying the changes in some of the effects of estrogen associated with aging will represent a significant advance in understanding the mechanisms involved in decline in cognitive function with aging.
Acknowledgments
This work was supported by the National Institute of Health Grants NIA P01 AG014751 (Caleb E. Finch), Project 3 (RFT); P01 AG026572 (RDB), Project 2 (RFT); and R01 AG023742 (Diana Woodruff-Pak & RFT).
References
- 1.Akopian G, Foy MR, Thompson RF. 17B-estradiol enhancement of LTP involves activation of voltage-dependent calcium channels. Soc Neurosci Abstr. 2003 Program No. 255.252. [Google Scholar]
- 2.Asthana S, Baker LD, Craft S, Stanczyk FZ, Veith RC, Raskind MA, Plymate SR. High-dose estradiol improves cognition for women with AD: results of a randomized study. Neurology. 2001;57:605–612. doi: 10.1212/wnl.57.4.605. [DOI] [PubMed] [Google Scholar]
- 3.Backstrom T. Epileptic seizures in women related to plasma estrogen and progesterone during the menstrual cycle. Acta Neurol Scand. 1976;54:321–347. doi: 10.1111/j.1600-0404.1976.tb04363.x. [DOI] [PubMed] [Google Scholar]
- 4.Barnes CA. Memory deficits associated with senescence: a neurophysiological and behavioral study in the rat. Journal of Comparative Physiological Psychology. 1979;93:74–104. doi: 10.1037/h0077579. [DOI] [PubMed] [Google Scholar]
- 5.Barnes CA, Rao G, Foster TC, McNaugton BL. Region-specific age effects on AMPA sensitivity: electrophysiological evidence for loss of synaptic contacts in hippocampal field CA1. Hippocampus. 1992;2:457–468. doi: 10.1002/hipo.450020413. [DOI] [PubMed] [Google Scholar]
- 6.Barnes CA. Normal aging: regionally specific changes in hippocampal synaptic transmission. Trends in Neuroscience. 1994;17:13–18. doi: 10.1016/0166-2236(94)90029-9. [DOI] [PubMed] [Google Scholar]
- 7.Barrett-Conner E, Kritz-Silverstein D. Estrogen replacement therapy and cognitive function in older women. Journal of the American Medical Association. 1993;269:2637–2641. [PubMed] [Google Scholar]
- 8.Baudry M, Davis JL, Thompson RF. Advances in Synaptic Plasticity. The MIT Press; Cambridge, MA: 2000. [Google Scholar]
- 9.Bear MF, Malenka RC. Synaptic plasticity: LTP and LTD. Current Opinion in Neurobiology. 1994;4:389–399. doi: 10.1016/0959-4388(94)90101-5. [DOI] [PubMed] [Google Scholar]
- 10.Berman DE, Hazvi S, Rosenblum K, Seger RYD. Specific and differential activation of mitogen-activated protein kinase cascades by unfamiliar taste in the insular cortex of the behaving rat. Journal of Neuroscience. 1998;18:10037–10044. doi: 10.1523/JNEUROSCI.18-23-10037.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berry B, McMahan R, Gallagher M. Spatial learning and memory at defined points of the estrous cycle: effects on performance of a hippocampal-dependent task. Behav Neurosci. 1997;111:267–274. doi: 10.1037//0735-7044.111.2.267. [DOI] [PubMed] [Google Scholar]
- 12.Bi GQ, Poo MM. Synaptic modification by correlated activity: Hebb's postulate revisited. Annual Review of Neuroscience. 2001;24:139–166. doi: 10.1146/annurev.neuro.24.1.139. [DOI] [PubMed] [Google Scholar]
- 13.Bi R, Broutman G, Foy M, Thompson RF, Baudry M. The tyrosine kinase and MAP kinase pathways mediate multiple effects of estrogen in hippocampus. Proceedings of the National Academy of Sciences (USA) 2000;97:3602–3607. doi: 10.1073/pnas.060034497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bi R, Foy MR, Vouimba RM, Thompson RF, Baudry M. Cyclic changes in estradiol regulate synaptic plasticity through the MAP kinase pathway. Proc Natl Acad Sci U S A. 2001;98:13391–13395. doi: 10.1073/pnas.241507698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM. Intraneuronal Abeta causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron. 2005;45:675–688. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- 16.Bimonte-Nelson HA, Singleton RS, Hunter CL, Price KL, Moore AB, Granholm AC. Ovarian hormones and cognition in the aged female rat: I. Long-term, but not short-term, ovariectomy enhances spatial performance. Behav Neurosci. 2003;117:1395–1406. doi: 10.1037/0735-7044.117.6.1395. [DOI] [PubMed] [Google Scholar]
- 17.Bimonte-Nelson HA, Singleton RS, Williams BJ, Granholm AC. Ovarian hormones and cognition in the aged female rat: II. progesterone supplementation reverses the cognitive enhancing effects of ovariectomy. Behav Neurosci. 2004;118:707–714. doi: 10.1037/0735-7044.118.4.707. [DOI] [PubMed] [Google Scholar]
- 18.Bliss TVP, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 19.Blum S, Moore AN, Adams F, Dash PK. A mitogen-activated protein kinase cascade in the CA1/CA2 subfield of the dorsal hippocampus is essential for long-term spatial memory. Journal of Neuroscience. 1999;19:3535–3544. doi: 10.1523/JNEUROSCI.19-09-03535.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brambilla R, Gnesutta N, Minichiello L, White G, Roylance AJ, Herron CE, Ramsey M, Wolfer DP, Cestari V, Rossi-Arnaud C, Grant SG, Chapman PF, Lipp HP, Sturani E, Klein R. A role for the Ras signaling pathway in synaptic transmission and long-term memory. Nature. 1997;290:281–286. doi: 10.1038/36849. [DOI] [PubMed] [Google Scholar]
- 21.Brinton RD, Proffitt P, Tran J, Luu R. Equilin a principal component of the estrogen replacement therapy, premarin, increases the growth of cortical neurons via an NMDA receptor dependent mechanism. Experimental Neurology. 1997;147:211–220. doi: 10.1006/exnr.1997.6619. [DOI] [PubMed] [Google Scholar]
- 22.Brinton RD, Tran J, Proffitt P, Kihil M. 17B-estradiol increases the growth and survival of cultured cortical neurons. Neurochemical Research. 1997;22:1339–1351. doi: 10.1023/a:1022015005508. [DOI] [PubMed] [Google Scholar]
- 23.Brookmeyer R, Gray S, Kawas C. Projections of Alzheimer's disease in the United States and the public health impact of delaying disease onset. Am J Public Health. 1998;88:1337–1342. doi: 10.2105/ajph.88.9.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Campbell LW, Hao SY, Thibault O, Blalock EM, Landfield PW. Aging changes in voltage-gated calcium currents in hippocampal CA1 neurons. Journal of Neuroscience. 1996;16:6286–6295. doi: 10.1523/JNEUROSCI.16-19-06286.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carroll JC, Rosario ER, Chang L, Stanczyk FZ, Oddo S, LaFerla FM, Pike CJ. Progesterone and estrogen regulate Alzheimer-like neuropathology in female 3xTg-AD mice. J Neurosci. 2007;27:13357–13365. doi: 10.1523/JNEUROSCI.2718-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cavus I, Teyler TJ. Two forms of long-term potentiation in area CA1 activate different signal transduction cascades. Journal of Neurophysiology. 1996;76:3038–3047. doi: 10.1152/jn.1996.76.5.3038. [DOI] [PubMed] [Google Scholar]
- 27.Clinton LK, Billings LM, Green KN, Caccamo A, Ngo J, Oddo S, McGaugh JL, LaFerla FM. Age-dependent sexual dimorphism in cognition and stress response in the 3xTg-AD mice. Neurobiol Dis. 2007;28:76–82. doi: 10.1016/j.nbd.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cordoba Montoya DA, Carrer HF. Estrogen facilitates induction of long term potentiation in the hippocampus of awake rats. Brain Res. 1997;778:430–438. doi: 10.1016/s0006-8993(97)01206-7. [DOI] [PubMed] [Google Scholar]
- 29.Couzin J. Estrogen research. The great estrogen conundrum. Science. 2003;302:1136–1138. doi: 10.1126/science.302.5648.1136. [DOI] [PubMed] [Google Scholar]
- 30.Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Development. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 31.Disterhoft JF, Wu WW, Ohno M. Biophysical alterations of hippocampal pyramidal neurons in learning, ageing and Alzheimer's disease. Ageing Research Reviews. 2004;3:383–406. doi: 10.1016/j.arr.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 32.Dohanich GP. Gonadal steroids, learning and memory. In: Rubin RT, editor. Hormones, brain and behavior. Academic Press; San Diego: 2002. pp. 265–327. [Google Scholar]
- 33.Dubal DB, Zhu H, Yu J, Rau SW, Shughrue PJ, Merchenthaler I, Kindy MS, Wise PM. Estrogen receptor alpha, not beta, is a critical link in estradiol-mediated protection against brain injury. Proceedings of the National Academy of Sciences (USA) 2001;98:1952–1957. doi: 10.1073/pnas.041483198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Foster TC, Norris CM. Age-associated changes in Ca2+-dependent processes: relation to hippocampal synaptic plasticity. Hippocampus. 1997;7:602–612. doi: 10.1002/(SICI)1098-1063(1997)7:6<602::AID-HIPO3>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 35.Foster TC. Involvement of hippocampal synaptic plasticity in age-related memory decline. Brain Research Reviews. 1999;30:236–249. doi: 10.1016/s0165-0173(99)00017-x. [DOI] [PubMed] [Google Scholar]
- 36.Foy M, Baudry M, Thompson R. Estrogen and hippocampal synaptic plasticity. Neuron Glia Biol. 2004;1:327–338. doi: 10.1017/S1740925X05000165. [DOI] [PubMed] [Google Scholar]
- 37.Foy MR. Doctoral Dissertation. Kent State University; Kent, OH: 1983. Neuromodulation: Effects of estradiol and THC on brain excitability. [Google Scholar]
- 38.Foy MR, Teyler TJ. 17-alpha-Estradiol and 17-beta-estradiol in hippocampus. Brain Res Bull. 1983;10:735–739. doi: 10.1016/0361-9230(83)90206-x. [DOI] [PubMed] [Google Scholar]
- 39.Foy MR, Xu J, Xie X, Brinton RD, Thompson RF, Berger TW. 17beta-estradiol enhances NMDA receptor-mediated EPSPs and long-term potentiation. J Neurophysiol. 1999;81:925–929. doi: 10.1152/jn.1999.81.2.925. [DOI] [PubMed] [Google Scholar]
- 40.Foy MR. 17beta-estradiol: effect on CA1 hippocampal synaptic plasticity. Neurobiol Learn Mem. 2001;76:239–252. doi: 10.1006/nlme.2001.4018. [DOI] [PubMed] [Google Scholar]
- 41.Foy MR, Akopian G, Thompson RF. 17B-estradiol enhances LTP in hippocampal slices from 3xTg-AD mice. Soc Neurosci Abstr. 2006 Program No. 536.515. [Google Scholar]
- 42.Foy MR, Baudry M, Foy JG, Thompson RF. 17beta-estradiol modifies stress-induced and age-related changes in hippocampal synaptic plasticity. Behav Neurosci. 2008;122:301–309. doi: 10.1037/0735-7044.122.2.301. [DOI] [PubMed] [Google Scholar]
- 43.Galea LA, Kavaliers M, Ossenkopp KP, Hampson E. Gonadal hormone levels and spatial learning performance in the Morris water maze in male and female meadow voles, Microtus pennsylvanicus. Horm Behav. 1995;29:106–125. doi: 10.1006/hbeh.1995.1008. [DOI] [PubMed] [Google Scholar]
- 44.Gazzaley AH, Weiland NG, McEwen BS, Morrison JH. Differential regulation of NMDAR1 mRNA and protein by estradiol in the rat hippocampus. Journal of Neuroscience. 1996;16:6830–6838. doi: 10.1523/JNEUROSCI.16-21-06830.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Geinisman Y, Detoledo-Morell F, Heller RE. Hippocampal markers of age-related memory dysfunction: behaivoral, electrophysiological and morphological perspectives. Progress in Neurobiology. 1995;45:223–252. doi: 10.1016/0301-0082(94)00047-l. [DOI] [PubMed] [Google Scholar]
- 46.Geinisman Y. Structural synaptic modifications associated with hippocampal LTP and behavioral learning. Cerebral Cortex. 2000;10:952–962. doi: 10.1093/cercor/10.10.952. [DOI] [PubMed] [Google Scholar]
- 47.Gibbs RB, Wu D, Hersh LB, Pfaff DW. Effects of estrogen replacement on the relative levels of choline acetyltransferase, trkA, and nerve growth factor messenger RNAs in the basal forebrain and hippocampal formation of adult rats. Exp Neurol. 1994;129:70–80. doi: 10.1006/exnr.1994.1148. [DOI] [PubMed] [Google Scholar]
- 48.Grover LM, Teyler TJ. Two components of long-term potentiation induced by different patterns of afferent activation. Nature. 1990;347:477–479. doi: 10.1038/347477a0. [DOI] [PubMed] [Google Scholar]
- 49.Gu Q, Moss RL. 17B-estradiol potentiates kainite-induced currents via activation of the camp cascade. Journal of Neuroscience. 1996;16:3620–3629. doi: 10.1523/JNEUROSCI.16-11-03620.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 51.Henderson VW. Estrogen replacement therapy for the prevention and treatment of Alzheimer's disease. CNS Drugs. 1997;8:343–351. [Google Scholar]
- 52.Henderson VW. Hormone therapy and the brain: a clinical perspective on the role of estrogen. Parthenon Publishing; New York: 2000. [DOI] [PubMed] [Google Scholar]
- 53.Henderson VW, Paganini-Hill A, Miller BL, Elble RJ, Reyes PF, Shoupe D, McCleary CA, Klein RA, Hake AM, Farlow MR. Estrogen for Alzheimer's disease in women: randomized, double-blind, placebo-controlled trial. Neurology. 2000;54:295–301. doi: 10.1212/wnl.54.2.295. [DOI] [PubMed] [Google Scholar]
- 54.Henderson VW, Benke KS, Green RC, Cupples LA, Farrer LA. Postmenopausal hormone therapy and Alzheimer's disease risk: interaction with age. J Neurol Neurosurg Psychiatry. 2005;76:103–105. doi: 10.1136/jnnp.2003.024927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hirata-Fukae C, Li HF, Hoe HS, Gray AJ, Minami SS, Hamada K, Niikura T, Hua F, Tsukagoshi-Nagai H, Horikoshi-Sakuraba Y, Mughal M, Rebeck GW, Laferla FM, Mattson MP, Iwata N, Saido TC, Klein WL, Duff KE, Aisen PS, Matsuoka Y. Females exhibit more extensive amyloid, but not tau, pathology in an Alzheimer transgenic model. Brain Res. 2008;1216:92–103. doi: 10.1016/j.brainres.2008.03.079. [DOI] [PubMed] [Google Scholar]
- 56.Hy LX, Keller DM. Prevalence of AD among whites: a summary by levels of severity. Neurology. 2000;55:198–204. doi: 10.1212/wnl.55.2.198. [DOI] [PubMed] [Google Scholar]
- 57.Ito K, Utsunomiya H, Yaegashi N, Sasano H. Biological roles of estrogen and progesterone in human endometrial carcinoma--new developments in potential endocrine therapy for endometrial cancer. Endocr J. 2007;54:667–679. doi: 10.1507/endocrj.kr-114. [DOI] [PubMed] [Google Scholar]
- 58.Jonasson Z. Meta-analysis of sex differences in rodent models of learning and memory: a review of behavioral and biological data. Neurosci Biobehav Rev. 2005;28:811–825. doi: 10.1016/j.neubiorev.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 59.Kampen DL, Sherwin BB. Estrogen use and verbal memory in healthy postmenopausal women. Obstet Gynecol. 1994;83:979–983. doi: 10.1097/00006250-199406000-00017. [DOI] [PubMed] [Google Scholar]
- 60.Kawas C, Resnick S, Morrison A, Brookmeyer R, Corrada M, Zonderman A, Bacal C, Lingle DD, Metter E. A prospective study of estrogen replacement therapy and the risk of developing Alzheimer's disease: the Baltimore Longitudinal Study of Aging. Neurology. 1997;48:1517–1521. doi: 10.1212/wnl.48.6.1517. [DOI] [PubMed] [Google Scholar]
- 61.Kohama SG, Goss JR, Finch CE, McNeill TH. Increases of glial fibrillary acidic protein in the aging female mouse brain. Neurobiology of Aging. 1995;16:59–67. doi: 10.1016/0197-4580(95)80008-f. [DOI] [PubMed] [Google Scholar]
- 62.Landfield PW, Lynch G. Impaired monosynaptic potentiation in in vitro hippocampal slices from age, memory-deficient rats. Journal of Gerontology. 1977;32:523–533. doi: 10.1093/geronj/32.5.523. [DOI] [PubMed] [Google Scholar]
- 63.Landfield PW, Pitler TA, Applegate MD. The effects of high Mg2+-Ca2+ ratios on frequency potentiation in hippocampal slices of young and aged rats. Journal of Neurophysiology. 1986;56:797–811. doi: 10.1152/jn.1986.56.3.797. [DOI] [PubMed] [Google Scholar]
- 64.Landfield PW, Deadwyler SA. Long-term potentiation: From biophysics to behavior. Alan R Liss, Inc.; New York: 1988. [Google Scholar]
- 65.Lee SJ, McEwen BS. Neurotrophic and neuroprotective actions of estrogens and their therapeutic implications. Annual Review of Pharmacology and Toxicology. 2001;41:569–591. doi: 10.1146/annurev.pharmtox.41.1.569. [DOI] [PubMed] [Google Scholar]
- 66.Leuner B, Mendolia-Loffredo S, Shors TJ. High levels of estrogen enhance associative memory formation in ovariectomized females. Psychoneuroendocrinology. 2004;29:883–890. doi: 10.1016/j.psyneuen.2003.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Matthews K, Cauley J, Yaffe K, Zmuda JM. Estrogen replacement therapy and cognitive decline in older community women. Journal of the American Geriatric Society. 1999;47:518–523. doi: 10.1111/j.1532-5415.1999.tb02563.x. [DOI] [PubMed] [Google Scholar]
- 68.Mazzucchelli C, Brambilla R. Ras-related and MAPK signaling in neuronal plasticity and memory function. Cell Molecular Life Science. 2000;57:604–611. doi: 10.1007/PL00000722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McEwen B, Gerlach J, Micco D. Putative glucocorticoid receptors in hippocampus and other regions of the rat brain. In: Pribram RIK, editor. The Hippocampus, Vol 2: Neurophysiology and behavior. Plenum; 1975. [Google Scholar]
- 70.McEwen B, Alves SE. Estrogen actions in the central nervous system. Endocr Rev. 1999;20:279–307. doi: 10.1210/edrv.20.3.0365. [DOI] [PubMed] [Google Scholar]
- 71.McEwen B, Akama K, Alves S, Brake WG, Bulloch K, Lee S, Li C, Yuen G, Milner TA. Tracking the estrogen receptor in neurons: implications for estrogen-induced synapse formation. Proc Natl Acad Sci U S A. 2001;98:7093–7100. doi: 10.1073/pnas.121146898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Morgan SL, Coussens CM, Teyler TJ. Depotentiation of vdccLTP requires NMDAR activation. Neurobiology of Learning and Memory. 2001;76:229–238. doi: 10.1006/nlme.2001.4016. [DOI] [PubMed] [Google Scholar]
- 73.Mulnard RA, Cotman CW, Kawas C, van Dyck CH, Sano M, Doody R, Koss E, Pfeiffer E, Jin S, Gamst A, Grundman M, Thomas R, Thal LJ. Estrogen replacement therapy for treatment of mild to moderate Alzheimer disease: a randomized controlled trial. Alzheimer's Disease Cooperative Study. JAMA. 2000;283:1007–1015. doi: 10.1001/jama.283.8.1007. [DOI] [PubMed] [Google Scholar]
- 74.Murphy DD, Cole NB, Greenberger V, Segal M. Estradiol increases dendritic spine density by reducing GABA neurotransmission in hippocampal neurons. J Neurosci. 1998;18:2550–2559. doi: 10.1523/JNEUROSCI.18-07-02550.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nilsen J, Brinton RD. Impact of progestins on estrogen-induced neuroprotection: synergy by progesterone and 19-norprogesterone and antagonism by medroxprogesterone acetate. Endocrinology. 2002;143:205–212. doi: 10.1210/endo.143.1.8582. [DOI] [PubMed] [Google Scholar]
- 76.Nilsen J, Brinton RD. Impact of progestins on estradiol potentiation of the glutamate calcium response. Neuroreport. 2002;13:825–830. doi: 10.1097/00001756-200205070-00018. [DOI] [PubMed] [Google Scholar]
- 77.Nilsen J, Brinton RD. Divergent impact of progesterone and medroxyprogesterone acetate (Provera) on nuclear mitogen-activated protein kinase signaling. Proceedings of the National Academy of Sciences (USA) 2003;100:10506–10511. doi: 10.1073/pnas.1334098100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Norris CM, Korol DL, Foster' Increased susceptibility to induction of long-term depression and long-term potentiation reversal during aging. Journal of Neuroscience. 1996;16:5382–5392. doi: 10.1523/JNEUROSCI.16-17-05382.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Norris CM, Halpain S, Foster TC. Reversal of age-related alterations in synaptic plasticity by blockage of L-type Ca2+ channels. Journal of Neuroscience. 1998;18:3171–3179. doi: 10.1523/JNEUROSCI.18-09-03171.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 81.Packard MG, Teather LA. Posttraining estradiol injections enhance memory in ovariectomized rats: cholinergic blockade and synergism. Neurobiol Learn Mem. 1997;68:172–188. doi: 10.1006/nlme.1997.3785. [DOI] [PubMed] [Google Scholar]
- 82.Paganini-Hill A, Henderson VW. Estrogen deficiency and risk of Alzheimer's disease in women. Am J Epidemiol. 1994;140:256–261. doi: 10.1093/oxfordjournals.aje.a117244. [DOI] [PubMed] [Google Scholar]
- 83.Paganini-Hill A, Henderson VW. Estrogen replacement therapy and risk of Alzheimer disease. Arch Intern Med. 1996;156:2213–2217. [PubMed] [Google Scholar]
- 84.Pfaff DW, McEwen BS. Actions of estrogens and progestins on nerve cells. Science. 1983;219:808–814. doi: 10.1126/science.6297008. [DOI] [PubMed] [Google Scholar]
- 85.Power JM, Wu WW, Sametsky E, Oh MM, Disterhoft JF. Age-related enhancement of the slow outward calcium-activated potassium current in hippocampal CA1 pyramidal neurons in vitro. Journal of Neuroscience. 2002;22:7234–7243. doi: 10.1523/JNEUROSCI.22-16-07234.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Quevedo J, Vianna M, Daroit D, Born AG, Kuyven CR, Roesler R, Quillfeldt JA. L-type voltage-dependent calcium channel blocker nifedipine enhances memory retention when infused into the hippocampus. Neurobiology of Learning and Memory. 1998;69:320–325. doi: 10.1006/nlme.1998.3822. [DOI] [PubMed] [Google Scholar]
- 87.Rapp SR, Espeland MA, Shumaker SA, Henderson VW, Brunner RL, Manson JE, Gass ML, Stefanick ML, Lane DS, Hays J, Johnson KC, Coker LH, Dailey M, Bowen D. Effect of estrogen plus progestin on global cognitive function in postmenopausal women: the Women's Health Initiative Memory Study: a randomized controlled trial. JAMA. 2003;289:2663–2672. doi: 10.1001/jama.289.20.2663. [DOI] [PubMed] [Google Scholar]
- 88.Resnick SM, Maki PM. Effects of hormone replacement therapy on cognitive and brain aging. Ann N Y Acad Sci. 2001;949:203–214. doi: 10.1111/j.1749-6632.2001.tb04023.x. [DOI] [PubMed] [Google Scholar]
- 89.Selcher JC, Atkins CM, Trzaskos JM, Paylor R, Sweatt JD. A necessity for MAP kinase activation in mammalian spatial learning. Learning and Memory. 1999;6:478–490. doi: 10.1101/lm.6.5.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 91.Shabani N, Mylonas I, Jeschke U, Thaqi A, Kuhn C, Puchner T, Friese K. Expression of estrogen receptors alpha and beta, and progesterone receptors A and B in human mucinous carcinoma of the endometrium. Anticancer Res. 2007;27:2027–2033. [PubMed] [Google Scholar]
- 92.Shors TJ, Matzel LD. Long-term potentiation: what's learning got to do with it? Brain and Behavioral Sciences. 1997;20:597–614. doi: 10.1017/s0140525x97001593. [DOI] [PubMed] [Google Scholar]
- 93.Shumaker SA, Legault C, Rapp SR, Thal L, Wallace RB, Ockene JK, Hendrix SL, Jones BN, 3rd, Assaf AR, Jackson RD, Kotchen JM, Wassertheil-Smoller S, Wactawski-Wende J. Estrogen plus progestin and the incidence of dementia and mild cognitive impairment in postmenopausal women: the Women's Health Initiative Memory Study: a randomized controlled trial. Jama. 2003;289:2651–2662. doi: 10.1001/jama.289.20.2651. [DOI] [PubMed] [Google Scholar]
- 94.Simoncini T, Hafezi-Moghadam A, Brazil DP, Ley K, Chin WW, Liao JK. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature. 2000;407:538–541. doi: 10.1038/35035131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Singer CA, Figueroa-Masot XA, Batchelor RH, Dorsa DM. The mitogen-activated protein kinase pathway mediates estrogen neuroprotection after glutamate toxicity in primary cortical neurons. Journal of Neuroscience. 1999;19:2455–2463. doi: 10.1523/JNEUROSCI.19-07-02455.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Singh C, Wang JM, Guerrero B, Brinton RD, Thompson RF. Allopregnanolone reverses the learning and memory deficits in the triple transgenic Alzheimer disease mouse model. Soc Neurosci Abstr. 2007 Program No. 49.10. [Google Scholar]
- 97.Singh M, Meyer EM, Millard WJ, Simpkins JW. Ovarian steroid deprivation results in a reversible learning impairment and compromised cholinergic function in female Sprague-Dawley rats. Brain Res. 1994;644:305–312. doi: 10.1016/0006-8993(94)91694-2. [DOI] [PubMed] [Google Scholar]
- 98.Singh M, Setalo G, Jr, Guan X, Frail DE, Toran-Allerand CD. Estrogen-induced activation of the mitogen-activated protein kinase cascade in the cerebral cortex of estrogen receptor-alpha knock-out mice. Journal of Neuroscience. 2000;20:1694–1700. doi: 10.1523/JNEUROSCI.20-05-01694.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Singh M, Sumien N, Kyser C, Simpkins JW. Estrogens and progesterone as neuroprotectants: what animal models teach us. Front Biosci. 2008;13:1083–1089. doi: 10.2741/2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Squire LR, Stark CE, Clark RE. The medial temporal lobe. Annu Rev Neurosci. 2004;27:279–306. doi: 10.1146/annurev.neuro.27.070203.144130. [DOI] [PubMed] [Google Scholar]
- 101.Suzuki S, Brown CM, Wise PM. Mechanisms of neuroprotection by estrogen. Endocrine. 2006;29:209–215. doi: 10.1385/ENDO:29:2:209. [DOI] [PubMed] [Google Scholar]
- 102.Sweatt JD. The neuronal MAP kinase cascade: a biochemical signal integration system subserving synaptic plasticity and memory. Journal of Neurochemistry. 2001;76:1–10. doi: 10.1046/j.1471-4159.2001.00054.x. [DOI] [PubMed] [Google Scholar]
- 103.Takehara K, Kawahara S, Kirino Y. Time-dependent reorganization of the brain components underlying memory retention in trace eyeblink conditioning. J Neurosci. 2003;23:9897–9905. doi: 10.1523/JNEUROSCI.23-30-09897.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tanapat P, Hastings NB, Gould E. Ovarian steroids influence cell proliferation in the dentate gyrus of the adult female rat in a dose- and time-dependent manner. J Comp Neurol. 2005;481:252–265. doi: 10.1002/cne.20385. [DOI] [PubMed] [Google Scholar]
- 105.Tang MX, Jacobs D, Stern Y, Marder K, Schofield P, Gurland B, Andrews H, Mayeux R. Effect of oestrogen during menopause on risk and age at onset of Alzheimer's disease. Lancet. 1996;348:429–432. doi: 10.1016/S0140-6736(96)03356-9. [DOI] [PubMed] [Google Scholar]
- 106.Terasawa E, Timiras PS. Electrical activity during the estrous cycle of the rat: cyclic changes in limbic structures. Endocrinology. 1968;83:207–216. doi: 10.1210/endo-83-2-207. [DOI] [PubMed] [Google Scholar]
- 107.Teyler TJ, Vardaris RM, Lewis D, Rawitch AB. Gonadal steroids: effects on excitability of hippocampal pyramidal cells. Science. 1980;209:1017–1018. doi: 10.1126/science.7190730. [DOI] [PubMed] [Google Scholar]
- 108.Touma C, Ambree O, Gortz N, Keyvani K, Lewejohann L, Palme R, Paulus W, Schwarze-Eicker K, Sachser N. Age- and sex-dependent development of adrenocortical hyperactivity in a transgenic mouse model of Alzheimer's disease. Neurobiol Aging. 2004;25:893–904. doi: 10.1016/j.neurobiolaging.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 109.Trojanowski JQ, Lee VM. Phosphorylation of paired helical filament tau in Alzheimer's disease neurofibrillary lesions: focusing on phosphatases. FASEB J. 1995;9:1570–1576. doi: 10.1096/fasebj.9.15.8529836. [DOI] [PubMed] [Google Scholar]
- 110.Vardaris RM, Teyler TJ. Sex differences in the response of hippocampal CA1 pyramids to gonadal steroids: Effects of testosterone and estradiol on the in vitro slice preparation. Soc Neurosci Abstr. 1980:A153–159. [Google Scholar]
- 111.Vouimba RM, Foy MR, Thompson RF. 17B-estradiol suppresses expression of long-term depression in aged rats. Brain Research Bulletin. 2000;53:783–787. doi: 10.1016/s0361-9230(00)00377-4. [DOI] [PubMed] [Google Scholar]
- 112.Wang JM, Johnston PB, Ball BG, Brinton RD. The neurosteroid allopregnanolone promotes proliferation of rodent and human neural progenitor cells and regulates cell-cycle gene and protein expression. J Neurosci. 2005;25:4706–4718. doi: 10.1523/JNEUROSCI.4520-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Warren SG, Humphreys AG, Juraska JM, Greenough WT. LTP varies across the estrous cycle: enhanced synaptic plasticity in proestrus rats. Brain Res. 1995;703:26–30. doi: 10.1016/0006-8993(95)01059-9. [DOI] [PubMed] [Google Scholar]
- 114.Warren SG, Juraska JM. Spatial and nonspatial learning across the rat estrous cycle. Behav Neurosci. 1997;111:259–266. doi: 10.1037//0735-7044.111.2.259. [DOI] [PubMed] [Google Scholar]
- 115.Wise PM, Dubal DB, Wilson ME, Rau SW, Bottner M, Rosewell KL. Estradiol is a protective factor in the adult and aging brain: understanding of mechanisms derived from in vivo and in vitro studies. Brain Res Brain Res Rev. 2001;37:313–319. doi: 10.1016/s0165-0173(01)00136-9. [DOI] [PubMed] [Google Scholar]
- 116.Wong M, Moss RL. Electrophysiological evidence for a rapid membrane action of the gonadal steroid, 17 beta-estradiol, on CA1 pyramidal neurons of the rat hippocampus. Brain Res. 1991;543:148–152. doi: 10.1016/0006-8993(91)91057-8. [DOI] [PubMed] [Google Scholar]
- 117.Wong M, Moss RL. Long-term and short-term electrophysiological effects of estrogen on the synaptic properties of hippocampal CA1 neurons. J Neurosci. 1992;12:3217–3225. doi: 10.1523/JNEUROSCI.12-08-03217.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wong M, Moss RL. Patch-clamp analysis of direct steroidal modulation of glutamate receptor-channels. Journal of Neuroendocrinology. 1994;6:347–355. doi: 10.1111/j.1365-2826.1994.tb00592.x. [DOI] [PubMed] [Google Scholar]
- 119.Wong M, Thompson TL, Moss RL. Nongenomic actions of estrogens in the brain: physiological significance and cellular mechanisms. Critical Reviews in Neurobiology. 1996;10:189–203. doi: 10.1615/critrevneurobiol.v10.i2.30. [DOI] [PubMed] [Google Scholar]
- 120.Woolley CS, McEwen BS. Estradiol regulates hippocampal dendritic spine density via an N-methyl-D-aspartate receptor-dependent mechanism. Journal of Neuroscience. 1994;14:7680–7687. doi: 10.1523/JNEUROSCI.14-12-07680.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Woolley CS, Weiland NG, McEwen BS, Schwartzkroin PA. Estradiol increases the sensitivity of hippocampal CA1 pyramidal cells to NMDA receptor-mediated synaptic input: correlation with dendritic spine density. Journal of Neuroscience. 1997;17:1848–1859. doi: 10.1523/JNEUROSCI.17-05-01848.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Woolley CS. Estrogen-mediated structural and functional synaptic plasticity in the female rat hippocampus. Horm Behav. 1998;34:140–148. doi: 10.1006/hbeh.1998.1466. [DOI] [PubMed] [Google Scholar]
- 123.Wu TW, Wang JM, Chen S, Brinton RD. 17Beta-estradiol induced Ca2+ influx via L-type calcium channels activates the Src/ERK/cyclic-AMP response element binding protein signal pathway and BCL-2 expression in rat hippocampal neurons: a potential initiation mechanism for estrogen-induced neuroprotection. Neuroscience. 2005;135:59–72. doi: 10.1016/j.neuroscience.2004.12.027. [DOI] [PubMed] [Google Scholar]
- 124.Xie X, Berger TW, Barrionuevo G. Isolated NMDA receptor-mediated synaptic responses express both LTP and LTD. Journal of Neurophysiology. 1992;67:1009–1013. doi: 10.1152/jn.1992.67.4.1009. [DOI] [PubMed] [Google Scholar]
- 125.Zec RF, Trivedi MA. The effects of estrogen replacement therapy on neuropsychological functioning in postmenopausal women with and without dementia: a critical and theoretical review. Neuropsychol Rev. 2002;12:65–109. doi: 10.1023/a:1016880127635. [DOI] [PubMed] [Google Scholar]
- 126.Zeng Y, Foy MR, Teyler TJ, Thompson RF. 17B-estradiol enhancement of nmda LTP in hippocampus. Soc Neurosci Abstr. 2004 Program No. 972.972. [Google Scholar]