

Summary
Aging increases oxidative stress and osteoblast apoptosis and decreases bone mass, whereas forkhead box O (FoxO) transcription factors defend against oxidative stress by activating genes involved in free radical scavenging and apoptosis. Conditional deletion of FoxO1, 3 and 4 in three month-old mice resulted in an increase in oxidative stress in bone and osteoblast apoptosis and a decrease in the number of osteoblasts, the rate of bone formation, and bone mass at cancellous and cortical sites. The effect of the deletion on osteoblast apoptosis was cell autonomous and resulted from oxidative stress. Conversely, overexpression of a FoxO3 transgene in mature osteoblasts decreased oxidative stress and osteoblast apoptosis, and increased osteoblast number, bone formation rate and vertebral bone mass. We conclude that FoxO-dependent oxidative defense provides a mechanism to handle the oxygen free radicals constantly generated by the aerobic metabolism of osteoblasts and is thereby indispensable for bone mass homeostasis.
Introduction
Reactive oxygen species (ROS) are continuously generated as normal by-products of aerobic metabolism and can cause lipid peroxidation, protein damage, and DNA lesions leading to cell death, but also serve as signaling molecules at lower concentrations to control cell proliferation and differentiation (Balaban et al., 2005; Giorgio et al., 2007). Age- or gonadectomy-related bone loss in mice is associated with increased oxidative stress (OS) (Lean et al., 2003; Almeida et al., 2007b; Almeida et al., 2009a; Almeida et al., 2009b; Manolagas, 2009). Likewise, murine models of premature aging and signs of oxidative damage exhibit osteoporotic features (Tyner et al., 2002; De Boer et al., 2002). Moreover, antioxidants prevent the increased osteoclastogenesis and increased osteoblast and osteocyte apoptosis as well as the loss of bone caused by gonadectomy, supporting the view that OS plays an important role in skeletal homeostasis (Lean et al., 2003; Almeida et al., 2007b).
To counteract the adverse consequences of OS, cells possess several oxidative defense mechanisms including those governed by the FoxO family of transcription factors. In mammals, this family comprises four members: FoxO1/FKHR, FoxO3/FKHRL1, FoxO4/AFX and FoxO6 (Greer and Brunet, 2005). FoxO1, 3, and 4 show broad, overlapping patterns of expression in developing and adult tissues, whereas FoxO6 is restricted to specific structures of the developing brain. In the setting of growth factor stimulation, FoxO1, 3 and 4 are subject to Akt-mediated phosphorylation which results in their nuclear export and inhibition of FoxO-mediated transcription (Brunet et al., 1999; Kops et al., 1999; Biggs, III et al., 1999; Tang et al., 1999; Takaishi et al., 1999). OS, on the other hand, causes the retention of FoxOs in the nucleus and activation of transcription, by promoting FoxO post-translational modifications, including phosphorylation (on amino-acid residues distinct from those phosphorylated by Akt), ubiquitylation and acetylation (Calnan and Brunet, 2008; van der Horst and Burgering, 2007). Stress stimuli override the sequestration of FoxOs in the cytoplasm by growth factors, both in mammalian cells and in Drosophila (Brunet et al., 2004; Wang et al., 2005) suggesting that protection of cells from stress is one of the crucial roles of these evolutionarily conserved proteins. FoxOs mediate OS responses by regulating the transcription of antioxidant enzymes such as MnSOD and catalase as well as genes involved in cell cycle, DNA repair, and lifespan (Salih and Brunet, 2008; van der Horst and Burgering, 2007). The importance of the antioxidant properties of FoxOs in mammalian biology has been recently highlighted by the profound effects of the deletion of FoxO3 alone or the combined deletion of FoxO1, 3, and, 4 in mice. Indeed, these studies have revealed that FoxOs are indispensable for normal erythropoiesis and hematopoietic stem cell quiescence and survival, because of their ability to prevent excessive levels of ROS (Marinkovic et al., 2007; Tothova et al., 2007).
Beta-catenin – a factor required for osteoblast differentiation (Glass et al., 2005) – is essential for ROS-induced FoxO activation (Essers et al., 2005). In osteoblastic cell models, OS induces the association of FoxOs with β-catenin and promotes FoxO-mediated transcription at the expense of β-catenin/T-cell specific transcription factor (TCF)-mediated transcription and osteoblast differentiation (Almeida et al., 2007a; Almeida et al., 2009a). The negative effect of ROS on TCF transcription is abrogated by raising the level of β-catenin, suggesting that a limited pool of active β-catenin is diverted from TCF to FoxO transcription under stress conditions (Manolagas and Almeida, 2007; Hoogeboom and Burgering, 2009). FoxOs are also known to suppress the expression and transcriptional activity of PPARγ, which is a potent repressor of osteoblastogenesis (Dowell et al., 2003; Armoni et al., 2006; Akune et al., 2004). Together, these observations prompt speculation that FoxOs play an important role in bone biology by enabling the maintenance of a physiologically appropriate lifespan of mature osteoblasts through their oxidative defense activities. In addition, FoxOs may control the generation of new osteoblasts from their mesenchymal stem cell progenitors by modulating their proliferation and/or differentiation through their antioxidant properties or via modulating the activity of other transcription factors such as β-catenin or PPARγ.
Results
FoxO3 is the predominant FoxO in bone and bone cells
Quantitative (q)RT-PCR analysis of whole tissue RNA revealed that all three FoxO genes were expressed in calvaria and vertebrae, with FoxO3 showing the most abundant expression (2-3 fold higher than FoxO1 and FoxO4) (Figure S1A). FoxO1, 3 and 4 expression remained constant between 4 and 26 month of age, as determined in two independent experiments (Figure S1B). Consistent with the whole bone findings, FoxO3 expression was 2 fold higher than FoxO1 and 4 in bone marrow-derived osteoclasts (Figure S1C), or in calvaria- or bone marrow-derived osteoblastic cells (Figure S1D). Thus, all three FoxOs are expressed in bone cells and FoxO3 is the predominant FoxO family member.
Loss of FoxO signaling causes loss of bone mass
To elucidate the role of FoxOs in bone, we characterized the skeletal phenotype of mice in which the three main FoxO genes FoxO1, 3, and 4 were conditionally deleted. For this purpose, we used 12 wk-old Mx-Cre+;FoxO1,3,4L/L mice harboring the interferon-inducible transgene Mx1-Cre and conditional alleles for each of the FoxO genes and Mx-Cre-;FoxO1,3,4L/L littermate controls, hereafter designated Mx-Cre+ and Mx-Cre-, respectively (Paik et al., 2007). Five wk after injection of polyinosine-polycytidylic acid (pI-pC), used to activate the transgene and consequently the deletion of the three FoxO genes, the levels of expression of FoxO1, 3, and 4 were decreased by 60-75% in calvaria and vertebrae of Mx-Cre+ mice (Figure 1A). The efficiency of deletion of FoxOs in bone was comparable to that of spleen but less than in liver (Figure 1A). Body weight was not affected by FoxO deletion in Mx-Cre+ females or males (Figure S2A). Five wk after FoxO deletion, however, male and female Mx-Cre+ mice exhibited a significantly lower bone mass, as documented by a decrease in total and femoral bone mineral density (BMD) by dual energy X-ray absorptiometry (DEXA) (Figure 1B and Table S1). Spinal BMD also decreased in male animals. Deletion of FoxOs led to a decrease in femoral cortical width in female and male Mx-Cre+ mice as determined by compartment-specific analysis by micro-CT (Figure 1C). Femoral cancellous bone volume was also decreased but to a lesser extent and consistent with this Mx-Cre+ mice exhibited decreased trabecular number and increased trabecular separation (Figure S2B). Histomorphometric analysis of the vertebrae of male mice confirmed the effect of the deletion on bone mass by revealing a significant reduction in cancellous bone area as well as a decrease in trabecular width and number and increased trabecular separation (Figure 1D). In agreement with the decreased cancellous bone mass, osteoblast number, osteoid perimeter as well as bone formation rate (BFR) were also reduced (Figure 2A). In contrast, the number of osteoclasts in cancellous bone was not affected by deletion of FoxOs (Figure 2A).
Figure 1. Bone Mass is Decreased Following FoxOs Deletion.

(A-D) 12 wk-old female and male mice were injected with pI-pC and sacrificed 5 wk later.
(A) mRNA levels of FoxOs in calvaria (calv), vertebra (vert), liver or spleen from male mice determined by qRT-PCR (n=8 mice/group).
(B) BMDs determined by DEXA 1 d before and 5 wk following pI-pC injections (n=10-17 mice/group). *p<0.05 vs respective Mx-Cre- by Student's t-test
(C) Cortical width measured by micro-CT in femurs (n=10-17 mice/group). *p<0.05 vs respective Mx-Cre- by Student's t-test
(D) Histomorphometric analysis of longitudinal undecalcified sections of L1-L3 vertebrae of male mice (n=6-7 mice/group). BA/TA=bone area per tissue area; Tb=trabecular.
*p<0.05 by Student's t-test.
Figure 2. Deletion of FoxOs Increases Oxidative Stress and Decreases Bone Formation.
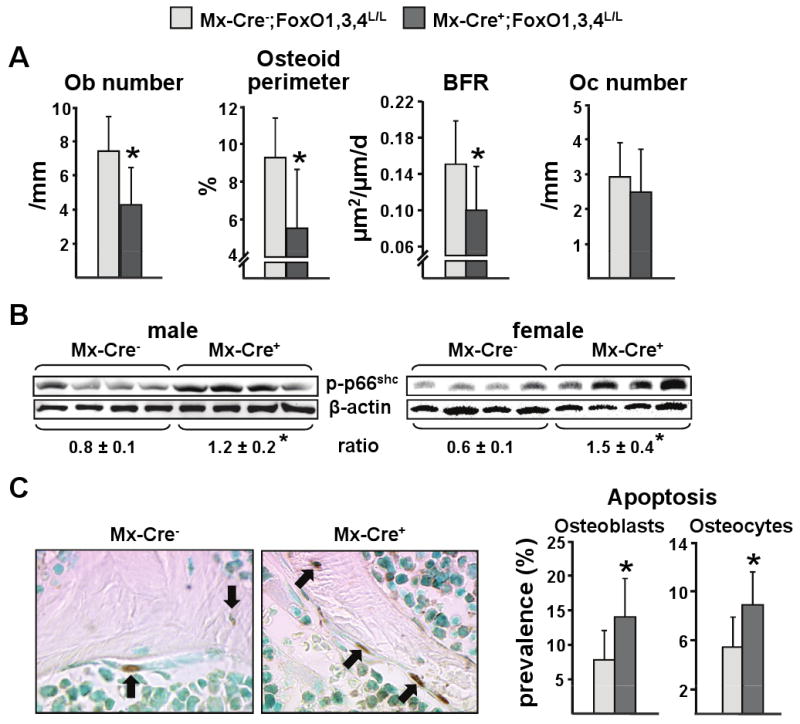
(A-C) Measurements were made using bone from the animals described in Figure 2.
(A) Histomorphometric analysis of longitudinal undecalcified sections of L1-L3 vertebrae of male mice (n=6-7 mice/group). Ob=osteoblast; Oc=osteoclast.
(B) p66shc phosphorylation determined by Western blot in vertebral lysates; each lane represents one animal. The mean ratio ± SD of phosphorylated p66shc to β-actin is depicted numerically at the bottom of the corresponding blots.
(C) Osteoblasts and osteocyte apoptosis in sections of the same mice as in A. In the photomicrographs apoptotic osteocytes and osteoblasts (stained brown by ISEL, original magnification ×630) are indicated by the arrows (n=7-8 mice/group).
*p<0.05 by Student's t-test.
OS and osteoblast apoptosis are increased in FoxO-deficient mice
Because of the well known anti-oxidant role of FoxOs in other tissues (Marinkovic et al., 2007; Tothova et al., 2007), we examined the levels of OS in the bone of the FoxO deleted mice. FoxO deletion resulted in increased phosphorylation of p66shc – a robust marker of OS (Migliaccio et al., 1999; Giorgio et al., 2005) – in vertebral lysates from male and female Mx-Cre+ mice (Figure 2B). In agreement with our earlier findings that increased OS in bone promotes osteoblast and osteocyte apoptosis (Almeida et al., 2007b), the prevalence of apoptosis of both these cell types was increased in vertebral cancellous bone from Mx-Cre+ mice as a consequence of the loss of FoxO function (Figure 2C). This result suggests that decreased osteoblast survival is responsible, at least in part, for the decreased osteoblast number, bone formation, and bone mass.
To investigate whether the protective actions of FoxOs against OS and osteoblast apoptosis in bone were cell autonomous, we cultured calvaria- or bone marrow-derived osteoblastic cells from Mx-Cre+ or Mx-Cre- mice. The efficiency of deletion of FoxOs in calvaria- or bone marrow-derived cells from Mx-Cre+ mice was about 50% and 80%, respectively, as measured by qRT-PCR. The decrease of the three FoxOs was confirmed by Western blotting (Figure 3A and S3A). Similar to the in vivo data, apoptosis of either calvaria- or bone marrow-derived osteoblastic cells from Mx-Cre+ mice was increased as compared to cells from control mice (Figure 3B and S3B). The higher prevalence of apoptosis of FoxO-deleted cells was due to increased OS, as the anti-oxidant N-acetyl-cysteine (NAC) abrogated the pro-apoptotic effect of FoxO deletion in calvaria-derived cells (Figure 3B). In agreement with this finding, OS was increased in calvaria-derived osteoblasts from Mx-Cre+ mice as evidenced by a 2-fold higher basal p66shc phosphorylation (Figure 3C). As with apoptosis, addition of NAC abrogated the increased p66shc phosphorylation, a finding consistent with the view that FoxOs attenuate osteoblastic cell apoptosis through a cell autonomous mechanism that minimizes OS. There was no difference between the Mx-Cre+ and Mx-Cre- mice in the rate of the proliferation of cells in these cultures (data not shown).
Figure 3. ROS-Induced Apoptosis is Increased in Osteoblastic Cells from FoxO-Deleted Mice.
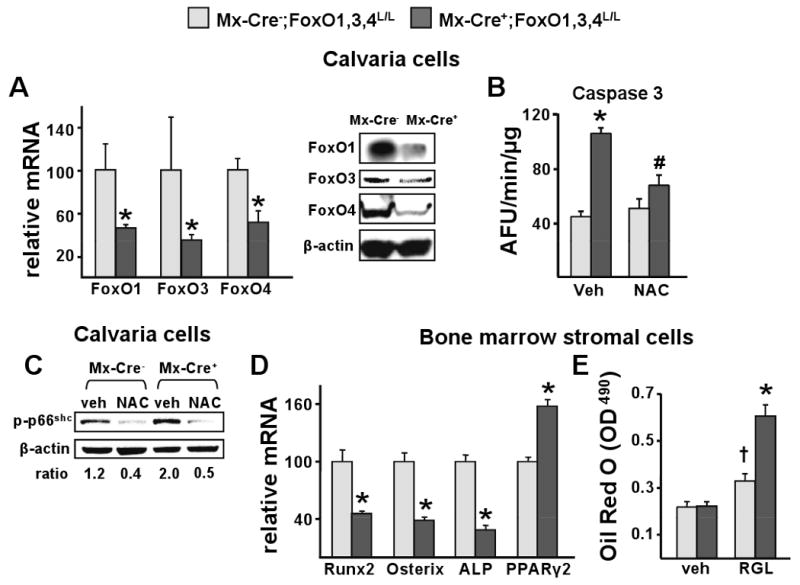
(A-D) Cells were obtained from the mice described in figure 2.
(A) mRNA levels of FoxOs by qRT-PCR and protein levels of FoxOs by Western Blot in calvaria-derived osteoblastic cells cultured for 8 d. *p<0.05 by Student's t-test.
(B) Caspase 3 activity in cells described in A, cultured in the presence of vehicle or NAC (1 mM) for 24 h. *p<0.05 vs veh in Mx-Cre-, # p<0.05 vs veh in Mx-Cre+ by ANOVA.
(C) p66shc phosphorylation determined by western blot in lysates from calvaria cells described in B. The ratio of phosphorylated p66shc to β-actin is depicted numerically at the bottom of the corresponding blots.
(D) mRNA levels of the indicated genes in bone marrow-derived stromal cells cultured for 8 d.
*p<0.05 by Student's t-test
(E) Bone marrow-derived stromal cells were treated with vehicle or rosiglitazone (RGL; 5 nM/ml) for 11 days. Lipid was visualized with Oil Red O and quantified as described in Methods. *p<0.05 vs RGL in Mx-Cre-, †p<0.05 vs vehicle in Mx-Cre- by ANOVA.
The number of osteoblasts present in bone is determined by their rate of generation and their lifespan. To examine whether the lower osteoblast number in Mx-Cre+ mice could result in part from a decrease in osteoblastogenesis, we examined the expression of Runx2 and Osterix – two transcription factors required for the commitment and differentiation of mesenchymal progenitors to cells of the osteoblastic lineage (Karsenty, 2008). Runx2 and Osterix mRNA levels were decreased as a result of FoxO deletion in bone marrow-derived stromal cells, as well as the levels of the osteoblast marker alkaline phosphatase (Figure 3D). In addition, the same cells displayed increased expression of PPARγ2, a nuclear receptor that downregulates osteoblastogenesis (Akune et al., 2004) (Figure 3D). Consistent with the increased PPARγ2 expression in the Mx-Cre+ mice, the pro-adipogenic ligand rosiglitazone (RGL) caused a greater increased in the number of adipocytes in bone marrow-derived stromal cell cultures from these mice as compared to the controls (Figure 3E).
Deletion of FoxOs alters the number of hematopoietic lineage cells (Tothova et al., 2007). Prompted by this evidence, we quantified the number of osteoclast progenitors present in the bone marrow of Mx-Cre+ mice. As shown in figure 4A, the mRNA and protein levels of the three FoxOs were dramatically diminished in cultured osteoclasts from Mx-Cre+ mice, similar to the case of hematopoietic stem cells (Tothova et al., 2007). Osteoclasts from Mx-Cre+ mice exhibited an increase in the rate of apoptosis (Figure 4B). This was confirmed by the lower expression of the osteoclast-specific markers like the calcitonin receptor, tartrate-resistant acid phosphatase (TRAP), and cathepsin K in parallel cultures (Figure 4C). In line with evidence that FoxO3 binds to and suppresses the FasL promoter (Jonsson et al., 2005), the increased osteoclast apoptosis was associated with a 4-fold increase in FasL mRNA. Osteoclast progenitor number was increased as a result of FoxO deletion (Figure 4D)
Figure 4. Osteoclast Progenitor Number and Mature Osteoclast Apoptosis are Increased Following FoxOs Deletion.
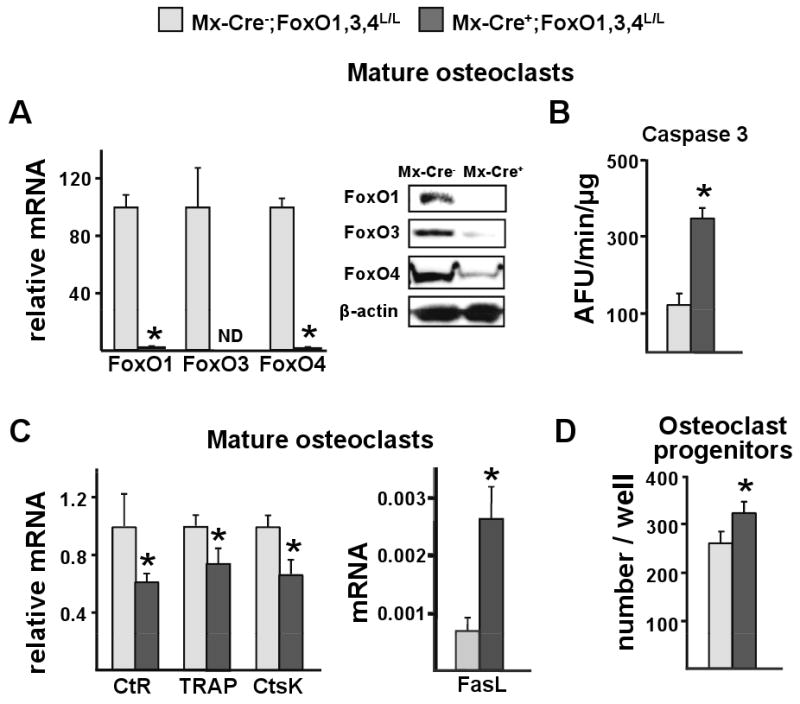
(A-D) Cells were obtained from the mice described in figure 2.
(A) mRNA levels of FoxOs by qRT-PCR and protein levels of FoxOs by Western Blot in mature osteoclasts generated from non-adherent bone marrow cells cultured with M-CSF and RANKL for 5 d; ND=not detected.
(B) Caspase 3 activity and (C) mRNA levels of the indicated genes in cells described in A. CtR=calcitonin receptor; CtsK=cathepsin K.
(D) Number of TRAP positive cells generated from bone marrow cells cultured with M-CSF and RANKL for 6 d. TRAP=tartrate-resistant acid phosphatase.
*p<0.05 by Student's t-test.
Gain of FoxO3 function in osteoblasts causes an increase in bone mass
To seek independent confirmation of the role of FoxOs in bone formation and homeostasis, we generated mice that conditionally overexpress FoxO3 in cells of the osteoblast lineage (Figure 5A). This was accomplished by crossing mice with a conditionally activated WT FoxO3 transgene (FoxO3C) with mice expressing Cre recombinase under the control of the osteocalcin (OCN) promoter. This promoter targets the expression of Cre to osteoblast-lineage cells at later stages of differentiation. Consistent with this, the FoxO3 transgene was highly expressed in calvaria or vertebra from 1 month-old OCN-Cre;FoxO3C mice but undetectable in liver or spleen. As expected, the transgene was also undetectable in all tissues from littermate controls harboring only the OCN-Cre transgene (Figure 5B). The expression of FoxO-target genes, like Gadd45 and catalase, was increased in calvaria of OCN-Cre;FoxO3C mice confirming the functionality of the transgene (Figure 5C). To further examine the functionality of the transgene, we used adenovirus (Ad)-Cre infection to activate it in calvaria cells from FoxO3C mice. Transfection of Ad-Cre infected cells with a FoxO-responsive luciferase (luc) reporter construct led to a 2-fold enhancement of FoxO directed transcriptional activity (Figure 5D).
Figure 5. Generation of FoxO3C Transgenic Mice.

(A) Mice harboring a transgene consisting of a transcriptional/translational stop cassette, flanked by loxP sites, inserted between the broadly-expressed hybrid CMV-chicken β-actin promoter and a cDNA encoding an HA-tagged human FoxO3 WT, designated FoxO3C mice were crossed with mice expressing the Cre recombinase under the control of the OCN promoter. The loxP-flanked stop cassette is removed only in cells expressing Cre resulting in FoxO3 expression in that cell type. PA=polyadenilation site.
(B) mRNA levels of the FoxO3 transgene in calvaria, vertebra (L5), liver and spleen of 4 wk-old mice determined by qRT-PCR (n=3 mice/group); ND=not detected.
(C) mRNA levels of the indicated FoxO-target genes in calvaria from 4 wk-old mice (n=3 mice/group).
(D) Calvaria-derived osteoblastic cells isolated from newborn FoxO3C and WT littermates infected with Ad-Cre for FoxO3 overexpression and transfected with a FoxO-luc reporter construct. Luc activity was measured 24 h later.
*p<0.05 by Student's t-test.
Overexpression of FoxO3 in osteoblasts led to a significant increase in bone mass in the vertebrae, as determined by BMD at 16 wk of age in the female, but not in the male, mice (Figure 6A). Nonetheless, an increase in cancellous bone mass (as determined by the measurement of bone volume over total volume) could be detected by micro-CT in both sexes, most likely reflecting the three dimensional advantage of the micro-CT measurement (Figure 6B). Female or male OCN-Cre;FoxO3C mice also exhibited increased trabecular number, as compared to littermate controls, while trabecular separation was decreased but this latter change was significant only in the male (Figure 6C). Vertebral BMD in female mice harboring the FoxO3C transgene without Cre activation was similar to wild-type littermates, demonstrating that the insertion site of the FoxO3C transgene has no effect on bone mass (Figure S4A). In contrast to the lumbar spine BMD, there was no difference in femoral bone mass between the experimental and the control mice by BMD (data not shown) or micro-CT (Figure S4B). Histomorphometric analysis of vertebral bone independently confirmed that FoxO3 gain-of-function in osteoblasts led to an increase in bone mass as demonstrated by an increase in cancellous bone area (BA/TA) and trabecular number, as well as a decrease in trabecular separation (Figure 6D).
Figure 6. Overexpression of FoxO3 in Osteoblasts Increases Vertebral Bone Mass.
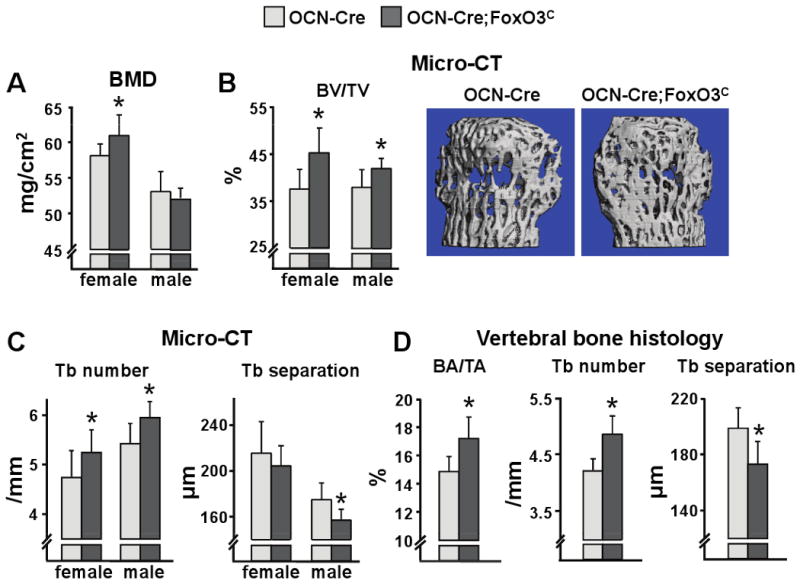
(A) Spine BMD by DEXA and (B-C) micro-CT measurements in the vertebra (L5) of 16 wk-old mice (n=7-14 mice/group). BV/TV=bone volume per total volume; Tb=trabecular. Representative images of vertebral cancellous bone are shown. *p<0.05 vs respective OCN-Cre by Student's t-test
(D) Histomorphometric analysis of longitudinal undecalcified sections of L1-L3 vertebrae of 16 wk-old male mice (n=6 mice/group). *p<0.05 by Student's t-test. BA/TA=bone area per tissue area.
Consistent with the gain in bone mass, the transgenic mice exhibited higher number of osteoblasts and bone formation rate in cancellous bone (Figure 7A). Moreover, in a diametrically opposite manner to the deletion model, FoxO3 overexpression in mature osteoblasts resulted in decreased phosphorylation of p66shc in vertebral lysates from the OCN-Cre;FoxO3C mice (Figure 7B). In line with the evidence for the decrease in OS in the bones of the OCN-Cre;FoxO3C mice, the prevalence of osteoblast apoptosis was diminished; whereas osteocyte apoptosis was not affected (Figure 7C).
Figure 7. Overexpression of FoxO3 in Osteoblasts Protects Against Oxidative Stress-Induced Apoptosis.
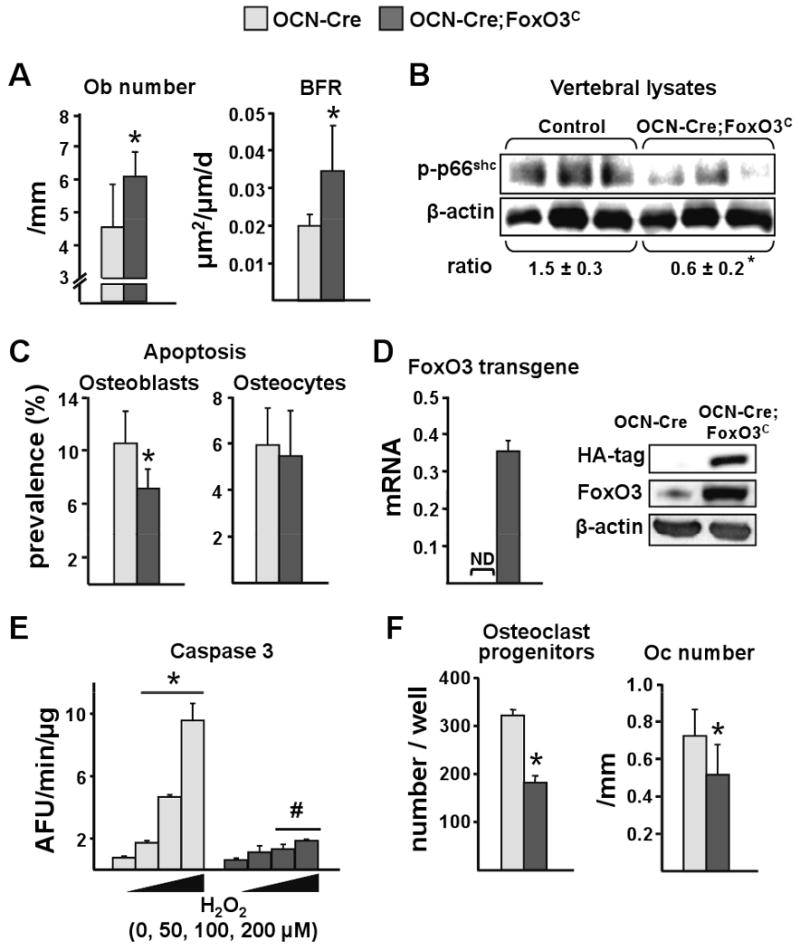
(A-C) Measurements were made using bones from the animals described in Figure 6.
(A) Histomorphometric analysis of longitudinal undecalcified sections of L1-L3 vertebrae of male mice (n=7 mice/group). Ob=osteoblasts; BFR=bone formation rate.
(B) p66shc phosphorylation determined by Western blot in vertebral lysates; each lane represents one animal. The mean ratio ± SD of phosphorylated p66shc to β-actin is depicted numerically at the bottom of the corresponding blots.
(C) Osteoblasts and osteocyte apoptosis in sections of the same mice as in A.
(D) mRNA levels of FoxO3 transgene by qRT-PCR and protein levels of HA-FoxO3 and Foxo3 by Western blot in bone marrow-derived osteoblastic cells cultured for 18 d with ascorbic acid.
(E) Caspase 3 activity in cells described in D, cultured in the presence of vehicle or H2O2 at the indicated concentrations for 16 h. *p<0.05 vs no H2O2 by ANOVA. #p<0.05 vs respective H2O2 dose in OCN-Cre by Student's t-test.
(F) Left panel: number of TRAP positive cells generated from bone marrow cells cultured with M-CSF and RANKL for 6 d. Right panel: osteoclast number was determined in the same sections as in A. TRAP=tartrate-resistant acid phosphatase; Oc=osteoclast.
*p<0.05 by Student's t-test.
To demonstrate that the overexpression of FoxO3 in osteoblasts did actually enhance defense against OS and thereby protected osteoblasts from OS-induced apoptosis we cultured bone marrow-derived osteoblastic cells from the control and the OCN-Cre;FoxO3C mice for 18 days in ascorbic acid (Figure 7D-E). As expected, progression of osteoblast differentiation under these culture conditions (and thereby an increase in the number of osteocalcin expressing cells) led to the expression of the FoxO3 transgene in cells from the OCN-Cre;FoxO3C, but not in cells from control mice as shown in Figure 7D by qRT-PCR and Western Blot for HA and FoxO3. For the Western blot of Figure 7D we have utilized an anti-FoxO3 antibody that recognizes both the human protein produced by the transgene as well as the endogenous mouse protein. Thus, the increased signal intensity observed in the transgene lane reflects the sum of both proteins and thereby an increase in overall FoxO3 expression in the cells from the transgenic mice. As shown in Figure 7E, osteoblasts from the OCN-Cre;FoxO3C mice, exhibited resistance to apoptosis induced by a range of H2O2 concentrations.
Finally, based on our previous observations that FoxOs can divert β-catenin from TCF- to FoxO-mediated transcription and by this mechanism decrease the expression of the anti-osteoclastogenic factor osteoprotegerin (OPG) and consequently increase osteoclast numbers (Glass et al., 2005), we examined the number of osteoclast progenitors in the bone marrow and mature osteoclasts in vertebral bone sections of the FoxO3 overexpressing mice. Unexpectedly, these mice displayed lower progenitors and lower osteoclast numbers (Figure 7F). This decrease could not be explained by changes in the production of osteoclastogenic cytokines by osteoblastic cells because we observed no changes in the mRNA levels of RANKL, OPG or M-CSF in vertebral bone from OCN-Cre;FoxO3C mice (data not shown).
Discussion
FoxOs translate environmental stimuli, such as OS and hormonal changes, into dynamic gene expression programs involved in many physiological as well as pathological processes (Salih and Brunet, 2008; Dansen and Burgering, 2008; van der Horst and Burgering, 2007). Based on this and evidence that decreased defense against OS is responsible, at least in part, for the adverse effects of aging and sex steroid deficiency on the murine skeleton, we have genetically manipulated FoxOs in mice and investigated the impact of such manipulation on skeletal homeostasis. Specifically, we hypothesized, that if one were to remove an important defense mechanism against OS from bone cells, one may recapitulate, at least some of, the adverse effects of aging on bone in young mice. Our results demonstrate that conditional deletion of the three broadly expressed genes, FoxO1, 3, and 4, in young adult mice or targeted overexpression of FoxO3 in osteoblasts leads to significant changes in bone mass. Specifically, loss of FoxO function in 3 mo old mice for a period of 5 wk results in increased OS in bone as well as increased osteoblast and osteocyte apoptosis and an osteoporotic phenotype characterized by decreased bone mass at both cancellous and cortical sites. We also show that the increased osteoblast apoptosis following the triple FoxO deletion is cell autonomous and the result of increased OS, as the rate of osteoblast apoptosis of the FoxO-deficient osteoblasts in ex-vivo cultures can be reverted to normal by the addition of the antioxidant NAC.
In sharp contrast to the increased OS and osteoblast apoptosis, decreased osteoblast number and bone formation, and the loss of bone mass following the loss of FoxO function, overexpression of FoxO3 in mature osteoblasts led to opposite effects: decreased OS and osteoblast apoptosis, increased osteoblast number and bone formation and increased bone mass. Albeit, increased bone mass was detectable in the vertebrae but not in the femur, perhaps because of the high variability of the measurement at the latter site during growth. An alternative explanation for the manifestation of the gain of FoxO3 function only in vertebrae may be the dramatically higher rate of turnover and the basal rate of osteoblast apoptosis in cancellous versus the periosteal compartment (Jilka et al., 2009) and the fact that cancellous bone makes larger contribution to BMD in the vertebrae than in the femur.
Collectively, the findings of this paper suggest that FoxOs are constantly defending against increased OS in osteoblasts, thereby assuring a normal healthy lifespan. In support of this conclusion, earlier work from our group and others over the years has established that apoptosis is the fate of the majority (60-80%) of mature osteoblasts (Jilka et al., 2007); and that this process is regulated by all major factors that influence skeletal homeostasis including systemic hormones, glucocorticoid administration, locally produced factors like IL-6 type cytokines, IGF-I, Wnts and their antagonists, parathyroid related peptide, mechanical forces, as well as OS (Jilka et al., 2008). Consistent with the contention that loss of FoxOs increases OS and thereby decreases the lifespan of osteoblasts, the number of mature osteoblasts in bone sections from the FoxO-deficient mice was decreased, and as a consequence their work output (i.e. the synthesis and mineralization of the bone matrix) was compromised.
Similar to our findings with osteoblasts, FoxOs promote mammalian cell survival by attenuating OS in other cell types (Tothova and Gilliland, 2007). Nonetheless, this effect is highly dependent on the cell type and even the tissue in which a particular cell type resides. For example, loss of FoxO function promotes murine hematopoietic stem cell apoptosis but decreases the apoptosis of thymocytes in the same animals (Paik et al., 2007; Tothova et al., 2007). More strikingly, loss of FoxOs decreases the apoptosis of endothelial cells from the liver but has no effect on endothelial cells residing in the lungs (Paik et al., 2007).
Osteoblast generation (determined by the expression of several lineage specific markers) in ex-vivo cultures of osteoblastic progenitors derived from the bone marrow of FoxO-deficient mice was attenuated as compared to the control mice. Specifically, Runx2 and Osterix expression as well as alkaline phosphatase, markers of osteoblast differentiation, were decreased. This could result from either an increase in the apoptosis of these progenitors or attenuation of their differentiation. In support of the latter explanation, deletion of FoxOs caused an increase of PPARγ — a nuclear receptor that stimulates adipogenesis (Tontonoz and Spiegelman, 2008) while represses osteoblastogenesis (Akune et al., 2004) and is tonically suppressed by FoxOs (Dowell et al., 2003; Armoni et al., 2006). Moreover cells from the FoxO deficient mice exhibited increased adipogenic potential in response to the PPARγ ligand rosiglitazone. Hence, increased adipocyte differentiation at the expense of osteoblast differentiation from their common mesenchymal progenitors, may have contributed to the decreased osteoblast number and bone formation in these mice.
We and others have demonstrated that both old age and acute loss of sex steroids in mice leads to increased OS and an increase in the prevalence of osteoblast and osteocyte apoptosis; and that antioxidants prevent the increased osteoblast and osteocyte apoptosis, as well as the loss of bone caused by gonadectomy (Almeida et al., 2007b; Lean et al., 2003). In addition, FoxO transcriptional activity, as measured by the expression of the Gadd45 gene expression, was increased in the aged mice of our earlier studies (Almeida et al., 2007a). The most likely explanation for the inability of FoxOs to prevent osteoblast apoptosis in old age is that the defense provided by these factors is overwhelmed by the high level of OS or that OS activates other pathways which interfere with the activity of FoxOs, or both. Extensive evidence indicates that ROS-induced p66shc activation leads to FoxO inactivation via Akt-mediated FoxO phosphorylation (Nemoto and Finkel, 2002; Smith et al., 2005). Moreover, ROS-induced stimulation of NF-kB may lead to IKK-mediated phosphorylation and inhibition of Foxo3 activity, at least in part by targeting it to ubiquitin-dependent degradation (Hu et al., 2004). Increased phosphorylation of p66shc as well as increased activity of NF-kB are common features of old age and sex steroid deficiency (Almeida et al., 2007b; Biswas et al., 2005; Adler et al., 2007; De Bosscher et al., 2006). Hence, several counteracting mechanisms can account for the overwhelming of FoxOs ability to thwart osteoblast apoptosis in old age.
Deletion of FoxOs also caused an increase in mature osteoclast apoptosis, perhaps secondary to the observed increase in FasL production. This latter finding is in agreement with the observation that FoxO3 can bind and suppress the FasL promoter in neutrophils; whereas deletion of FoxO3 results in elevated expression of FasL and increased neutrophil apoptosis (Jonsson et al., 2005). In support of this contention, FasL is an important pro-apoptotic signal for osteoclasts, and mice lacking Fas have increased osteoclast numbers and decreased bone mass (Wu et al., 2003). Moreover, up-regulation of FasL expression by estrogens may represent an important mechanism of the pro-apoptotic effect of these hormones on osteoclasts (Manolagas, 2000; Nakamura et al., 2007). The stimulatory effect of the FoxO deletion on osteoclast apoptosis was accompanied by an increase in osteoclast progenitors, most likely due to the expansion of myeloid progenitors, described earlier by Tothova et al. (Tothova et al., 2007). In spite of these changes the number of osteoclasts in cancellous bone of the Mx-Cre+ mice was unaltered, suggesting that the two changes may have cancelled each other. Genetic manipulation of FoxOs exclusively in cells of the osteoclast lineage in future work should help to explain these phenomena and their biologic relevance.
Lastly, overexpression of FoxO3 in osteoblasts led to a decrease in osteoclast numbers, which was unexpected, considering the inhibitory effect of FoxOs on canonical Wnt signaling and evidence that β-catenin reduces the levels of OPG (a direct target of β-catenin/TCF transcriptional activity), and increases RANKL and consequently osteoclast numbers in bone (Glass et al., 2005; Holmen et al., 2005). Yet, OPG or RANKL expression was not altered in the bone of our OCN-Cre;FoxO3C mice. This latter finding suggests that increased FoxO3 transcriptional activity in mature osteoblasts controls osteoclastogenesis independently of the canonical Wnt signaling. Instead, FoxO3 overexpression may decrease osteoclast number by decreasing ROS in the bone microenviroment. This suggestion is supported by mounting evidence that mitochondria biogenesis and ROS are indeed required for osteoclast generation (Garrett et al., 1990; Levasseur et al., 2003; Bai et al., 2005; Lean et al., 2005; Lee et al., 2005).
In conclusion, the results reported in this paper strongly support the working hypothesis that ROS are seminal signals for the fate of osteoblasts and that inappropriate increase of ROS adversely affects bone formation. The FoxO family of transcription factors defends against such an increase by constantly up-regulating free radical scavenging and DNA-repair enzymes, thereby representing an indispensable homeostatic mechanism for skeletal health.
Experimental Procedures
Generation of FoxO3 transgenic mice
We generated mice harboring a transgene consisting of a transcriptional/translational stop cassette, flanked by loxP sites, inserted between the broadly-expressed hybrid CMV-chicken β-actin promoter (Matsuda and Cepko, 2007) and a cDNA encoding an HA-tagged wild-type human FoxO3 (Fig. 5A), designated FoxO3C mice. HA-FoxO3 (Brunet et al., 1999) was provided by M. Greenberg, Harvard Medical School (Addgene plasmid 1787). When crossed with mice expressing the Cre recombinase in a specific cell type, the loxP-flanked stop cassette is removed only in the cells expressing Cre resulting in FoxO3 expression in that cell type (Fig. 5A). We generated 10 independent FoxO3C lines using this construct in a C57BL/6 background and tested the ability of the transgene to be activated in a cell type-specific manner by crossing each line with transgenic mice expressing Cre under the control of the human osteocalcin promoter (OCN-Cre) (Zhang et al., 2002). A single FoxO3C line demonstrating bone-specific expression (Fig. 5A) was utilized for all the studies described herein. Experimental mice were generated by crossing homozygous OCN-Cre mice, which are in a FVB/N background, with hemizygous FoxO3C mice to generate offspring that were all hemizygous for the OCN-Cre transgene and 50% of these were also hemizygous for the FoxO3C transgene. Thus all experimental mice were littermates in an F1 C57BL/6-FVB/N genetic background. Transgenic offspring were identified by PCR using the following primer sequences: forward (101bp) 5′-GAG TCG CTC GGT ACG ATT TA-3′ reverse (101bp) 5′-CGG GAG GCT AGC ATA ATC AG-3′.
Animal experimentation
Twelve week-old Mx-Cre+;FoxO1,3,4L/L and Mx-Cre-;FoxO1,3,4L/L littermate control mice, generated as previously described (Paik et al., 2007), received a total of three intraperitoneal injections of 300 μg of pI-pC (Sigma) administered every other day to induce Cre activation. Five wk later the animals were sacrificed and the various tissues and cells collected. BMD and body weight measurements were performed before the first pI-pC injection and before sacrifice, as previously described (O'Brien et al., 2005). Mice were injected with tetracycline 6 and 2 days before harvesting. Protocols involving genetically modified mice and their wild-type littermates were approved by the Institutional Animal Care and Use Committees of the University of Arkansas for Medical Sciences and the Central Arkansas Veterans Healthcare System.
Bone analysis
Femur and vertebra micro-CT scans were performed on isolated bones with a μCT40 scanning system (Scanco). Histology was performed in undecalcified vertebral bone sections as previously described (Weinstein et al., 1998; Weinstein et al., 2002). Details of the bone analysis can be found in Supplemental Experimental Procedures.
Cell culture
Osteoblastic cells derived from mouse calvaria were obtained and cultured as described previously (Jilka et al., 1998) for 8 days. Adherent bone marrow cells were obtained from long bones as described previously (DiGregorio et al., 2001) and cultured in α-MEM supplemented with 10% FBS and 1% PSG. Bone marrow-derived cells cultured in the absence of ascorbic acid are designated stromal cells (Fig. 3D and 3E). Cells cultured in the presence of 1% ascorbic acid to promote osteoblast differentiation are designated bone marrow osteoblastic cells (Fig. 7D, 7E, S1D and S4). To quantify adipogenesis, bone marrow-derived stromal cells were cultured to 70% confluence, and the media supplemented with rosiglitazone (5 nM/ml) or with 3.3% BSA in PBS as vehicle control. Medium was changed every 2 d for 11 d. Oil Red O staining and quantification were performed as described previously (Almeida et al., 2009a). Osteoclast progenitors were derived from bone marrow cells cultured in α-MEM supplemented with 10% FBS, 1% PSG, 30 ng/ml M-CSF and 30 ng/ml soluble RANKL (R&D Systems). Osteoclasts were enumerated after staining for TRAP; only multinucleated cells (more than three nuclei) were counted. Mature osteoclasts were derived from non-adherent bone marrow cells maintained for 5 d in same medium as for osteoclast progenitors. Apoptotic cells were quantified by measuring caspase 3 activity as described previously (Plotkin et al., 1999).
Western blot and qRT-PCR analysis
Phosphorylated p66shc was assayed by immunoblotting in fifth lumbar vertebra or cultured cell lysates, as previously described (Kousteni et al., 2003) using a mouse monoclonal antibody recognizing Ser36 phosphorylated p66shc (Calbiochem). FoxO1, 3 and 4 levels in cultured cell lysates were determined using a rabbit monoclonal antibody for Foxo1 (Cell Signaling), a rabbit polyclonal antibody for FoxO3 (Santa Cruz Biotechnology; GenWay Biotech, Inc), and a rabbit polyclonal antibody for Foxo4 (Cell Signaling). Protein levels of HA-FoxO3 or β-actin were analyzed using mouse monoclonal antibodies (Cell Signaling and Sigma, respectively). Quantification of the intensity of the bands in the autoradiograms was performed using a VersaDocTM imaging system (Bio-Rad). Total RNA was extracted from tissues or cultured cells and reverse-transcribed as previously described (Almeida et al., 2007a). Primers and probes for the different genes were manufactured by the TaqMan® Gene Expression Assays service (Applied Biosystems). Primers and probe for HA-FoxO3 were manufactured by Assays-on-demand service (Applied Biosystems): forward primer sequence 5′-CTCTGACTGACCGCGTTACTC-3′; reverse primer sequence 5′-GCCAAAATGATGAGACAGCACAATA-3′. The mRNA levels in Figure 4C, 5B, 5C, 7D and S1 were calculated by normalizing to the house-keeping gene ribosomal protein S2 using the ΔCt method (Livak and Schmittgen, 2001). The relative mRNA levels in Figure 1A, as well as in Figures 3A, 3D, 4A, 4C, S4A were calculated by the same method with the additional step of assigning an arbitrary value of 100 to the control sample.
Adenovirus infection and luciferase assay
Newborn calvaria cells from Foxo3C or wild-type mice were infected twice (with 24 h interval) with adenovirus encoding Cre recombinase (Ad-Cre) (Vector Biolabs) at a titer of 100 multiplicity of infection. Twenty four h after the last infection cells were plated on a 48-well plate and 16 h later transfected with a reporter plasmid containing 6 copies of daf-16 family protein binding element (FoxO-luc) provided by B. Burgering (University Medical Center, Utrecht, Netherlands) using Lipofectamine Plus (Invitrogen). Luciferase activity was determined as previously described (Kousteni et al., 2003).
Statistical analysis
All data are reported as the mean ± standard deviation. Group mean values were compared, as appropriate, by Student's two-tailed t test or one-way ANOVA with Bonferroni's multiple comparison test. A p value ≤0.05 was considered significant.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (P01 AG13918, R01 AR49794); the Department of Veterans Affairs (Merit Review grants to S.C.M., R.S.W. and R.L.J. and a VA Research Enhancement Award Program); and Tobacco Settlement funds provided by the University of Arkansas for Medical Sciences. E.A. is supported by a Ph.D fellowship from the University of Pisa, Italy. M.M.M is recipient of an award from Marques de Valdecilla Foundation, Santander, Spain. J-H.P is Damon Runyon Fellow (DRG 1900-06). R.A.D. is an American Cancer Society Research Professor and supported by the Robert A. and Renee E. Belfer Foundation Institute for Innovative Cancer Science. We thank A. Warren, K. Vyas, A. DeLoose, X. Qiu, W. Webb, C. Wicker III, S. Berryhill, R. Shelton, and T. Chambers for technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Adler AS, Sinha S, Kawahara TL, Zhang JY, Segal E, Chang HY. Motif module map reveals enforcement of aging by continual NF-kappaB activity. Genes Dev. 2007;21:3244–3257. doi: 10.1101/gad.1588507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akune T, Ohba S, Kamekura S, Yamaguchi M, Chung UI, Kubota N, Terauchi Y, Harada Y, Azuma Y, Nakamura K, Kadowaki T, Kawaguchi H. PPARgamma insufficiency enhances osteogenesis through osteoblast formation from bone marrow progenitors. J Clin Invest. 2004;113:846–855. doi: 10.1172/JCI19900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida M, Ambrogini E, Han L, Manolagas SC, Jilka RL. Increased lipid oxidation causes oxidative stress, increased PPAR{gamma} expression and diminished pro-osteogenic Wnt signaling in the skeleton. J Biol Chem. 2009a;284:27438–27448. doi: 10.1074/jbc.M109.023572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida M, Han L, Martin-Millan M, O'Brien CA, Manolagas SC. Oxidative stress antagonizes Wnt signaling in osteoblast precursors by diverting beta-catenin from T cell factor- to forkhead box O-mediated transcription. J Biol Chem. 2007a;282:27298–27305. doi: 10.1074/jbc.M702811200. [DOI] [PubMed] [Google Scholar]
- Almeida M, Han L, Martin-Millan M, Plotkin LI, Stewart SA, Roberson PK, Kousteni S, O'Brien CA, Bellido T, Parfitt AM, Weinstein RS, Jilka RL, Manolagas SC. Skeletal involution by age-associated oxidative stress and its acceleration by loss of sex steroids. J Biol Chem. 2007b;282:27285–27297. doi: 10.1074/jbc.M702810200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida M, Martin-Millan M, Ambrogini E, Han L, Weinstein RS, Jilka RL, O'Brien CA, Manolagas SC. Estrogens attenuate oxidative stress, osteoblast differentiation and apoptosis by DNA binding-independent actions of the ERalpha. J Bone Mineral Res. 2009b doi: 10.1359/jbmr.091017. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armoni M, Harel C, Karni S, Chen H, Bar-Yoseph F, Ver MR, Quon MJ, Karnieli E. FOXO1 represses peroxisome proliferator-activated receptor-gamma1 and -gamma2 gene promoters in primary adipocytes A novel paradigm to increase insulin sensitivity. J Biol Chem. 2006;281:19881–19891. doi: 10.1074/jbc.M600320200. [DOI] [PubMed] [Google Scholar]
- Bai XC, Lu D, Liu AL, Zhang ZM, Li XM, Zou ZP, Zeng WS, Cheng BL, Luo SQ. Reactive oxygen species stimulates receptor activator of NF-kappaB ligand expression in osteoblast. J Biol Chem. 2005;280:17497–17506. doi: 10.1074/jbc.M409332200. [DOI] [PubMed] [Google Scholar]
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Biggs WH, III, Meisenhelder J, Hunter T, Cavenee WK, Arden KC. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc Natl Acad Sci USA. 1999;96:7421–7426. doi: 10.1073/pnas.96.13.7421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas DK, Singh S, Shi Q, Pardee AB, Iglehart JD. Crossroads of estrogen receptor and NF-kappaB signaling. Sci STKE. 2005;2005:e27. doi: 10.1126/stke.2882005pe27. [DOI] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- Calnan DR, Brunet A. The FoxO code. Oncogene. 2008;27:2276–2288. doi: 10.1038/onc.2008.21. [DOI] [PubMed] [Google Scholar]
- Dansen TB, Burgering BM. Unravelling the tumor-suppressive functions of FOXO proteins. Trends Cell Biol. 2008;18:421–429. doi: 10.1016/j.tcb.2008.07.004. [DOI] [PubMed] [Google Scholar]
- De Boer J, Andressoo JO, de Wit J, Huijmans J, Beems RB, van Steeg H, Weeda G, van der Horst GT, van Leeuwen W, Themmen AP, Meradji M, Hoeijmakers JH. Premature aging in mice deficient in DNA repair and transcription. Science. 2002;296:1276–1279. doi: 10.1126/science.1070174. [DOI] [PubMed] [Google Scholar]
- De Bosscher K, Vanden Berghe W, Haegeman G. Cross-talk between nuclear receptors and nuclear factor kappaB. Oncogene. 2006;25:6868–6886. doi: 10.1038/sj.onc.1209935. [DOI] [PubMed] [Google Scholar]
- DiGregorio G, Yamamoto M, Ali A, Abe E, Roberson P, Manolagas SC, Jilka RL. Attenuation of the self-renewal of transit amplifying osteoblast progenitors in the murine bone marrow by 17β-estradiol. J Clin Invest. 2001;107:803–812. doi: 10.1172/JCI11653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowell P, Otto TC, Adi S, Lane MD. Convergence of peroxisome proliferator-activated receptor gamma and Foxo1 signaling pathways. J Biol Chem. 2003;278:45485–45491. doi: 10.1074/jbc.M309069200. [DOI] [PubMed] [Google Scholar]
- Essers MA, Vries-Smits LM, Barker N, Polderman PE, Burgering BM, Korswagen HC. Functional interaction between beta-catenin and FOXO in oxidative stress signaling. Science. 2005;308:1181–1184. doi: 10.1126/science.1109083. [DOI] [PubMed] [Google Scholar]
- Garrett IR, Boyce BF, Oreffo RO, Bonewald L, Poser J, Mundy GR. Oxygen-derived free radicals stimulate osteoclastic bone resorption in rodent bone in vitro and in vivo. J Clin Invest. 1990;85:632–639. doi: 10.1172/JCI114485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, Contursi C, Pelliccia G, Luzi L, Minucci S, Marcaccio M, Pinton P, Rizzuto R, Bernardi P, Paolucci F, Pelicci PG. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell. 2005;122:221–233. doi: 10.1016/j.cell.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Giorgio M, Trinei M, Migliaccio E, Pelicci PG. Hydrogen peroxide: a metabolic byproduct or a common mediator of ageing signals? Nat Rev Mol Cell Biol. 2007;8:722–728. doi: 10.1038/nrm2240. [DOI] [PubMed] [Google Scholar]
- Glass DA, Bialek P, Ahn JD, Starbuck M, Patel MS, Clevers H, Taketo MM, Long F, McMahon AP, Lang RA, Karsenty G. Canonical wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell. 2005;8:751–764. doi: 10.1016/j.devcel.2005.02.017. [DOI] [PubMed] [Google Scholar]
- Greer EL, Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene. 2005;24:7410–7425. doi: 10.1038/sj.onc.1209086. [DOI] [PubMed] [Google Scholar]
- Holmen SL, Zylstra CR, Mukherjee A, Sigler RE, Faugere MC, Bouxsein ML, Deng L, Clemens TL, Williams BO. Essential role of beta-catenin in postnatal bone acquisition. J Biol Chem. 2005;280:21162–21168. doi: 10.1074/jbc.M501900200. [DOI] [PubMed] [Google Scholar]
- Hoogeboom D, Burgering BM. Should I stay or should I go: beta-catenin decides under stress. Biochim Biophys Acta. 2009 doi: 10.1016/j.bbcan.2009.02.002. [DOI] [PubMed] [Google Scholar]
- Hu MC, Lee DF, Xia W, Golfman LS, Ou-Yang F, Yang JY, Zou Y, Bao S, Hanada N, Saso H, Kobayashi R, Hung MC. IkappaB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell. 2004;117:225–237. doi: 10.1016/s0092-8674(04)00302-2. [DOI] [PubMed] [Google Scholar]
- Jilka RL, Bellido T, Almeida M, Plotkin LI, O'Brien CA, Weinstein RS, Manolagas SC. Apoptosis and Bone Cells. In: Bilezikian JP, Raisz LG, Martin T, editors. In Principles of Bone Biology. San Diego: Academic Press; 2008. pp. 235–259. [Google Scholar]
- Jilka RL, O'Brien CA, Ali AA, Roberson PK, Weinstein RS, Manolagas SC. Intermittent PTH stimulates periosteal bone formation by actions on post-mitotic preosteoblasts. Bone. 2009;44:275–286. doi: 10.1016/j.bone.2008.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jilka RL, Weinstein RS, Bellido T, Parfitt AM, Manolagas SC. Osteoblast programmed cell death (apoptosis): modulation by growth factors and cytokines. J Bone Miner Res. 1998;13:793–802. doi: 10.1359/jbmr.1998.13.5.793. [DOI] [PubMed] [Google Scholar]
- Jilka RL, Weinstein RS, Parfitt AM, Manolagas SC. Quantifying Osteoblast and Osteocyte Apoptosis: Challenges and Rewards. J Bone Miner Res. 2007;22:1492–1501. doi: 10.1359/jbmr.070518. [DOI] [PubMed] [Google Scholar]
- Jonsson H, Allen P, Peng SL. Inflammatory arthritis requires Foxo3a to prevent Fas ligand-induced neutrophil apoptosis. Nat Med. 2005;11:666–671. doi: 10.1038/nm1248. [DOI] [PubMed] [Google Scholar]
- Karsenty G. Transcriptional control of skeletogenesis. Annu Rev Genomics Hum Genet. 2008;9:183–196. doi: 10.1146/annurev.genom.9.081307.164437. [DOI] [PubMed] [Google Scholar]
- Kops GJ, de Ruiter ND, Vries-Smits AM, Powell DR, Bos JL, Burgering BM. Direct control of the Forkhead transcription factor AFX by protein kinase B. Nature. 1999;398:630–634. doi: 10.1038/19328. [DOI] [PubMed] [Google Scholar]
- Kousteni S, Han L, Chen JR, Almeida M, Plotkin LI, Bellido T, Manolagas SC. Kinase-mediated regulation of common transcription factors accounts for the bone-protective effects of sex steroids. J Clin Invest. 2003;111:1651–1664. doi: 10.1172/JCI17261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lean JM, Davies JT, Fuller K, Jagger CJ, Kirstein B, Partington GA, Urry ZL, Chambers TJ. A crucial role for thiol antioxidants in estrogen-deficiency bone loss. J Clin Invest. 2003;112:915–923. doi: 10.1172/JCI18859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lean JM, Jagger CJ, Kirstein B, Fuller K, Chambers TJ. Hydrogen peroxide is essential for estrogen-deficiency bone loss and osteoclast formation. Endocrinology. 2005;146:728–735. doi: 10.1210/en.2004-1021. [DOI] [PubMed] [Google Scholar]
- Lee NK, Choi YG, Baik JY, Han SY, Jeong DW, Bae YS, Kim N, Lee SY. A crucial role for reactive oxygen species in RANKL-induced osteoclast differentiation. Blood. 2005;106:852–859. doi: 10.1182/blood-2004-09-3662. [DOI] [PubMed] [Google Scholar]
- Levasseur R, Barrios R, Elefteriou F, Glass DA, Lieberman MW, Karsenty G. Reversible skeletal abnormalities in gamma-glutamyl transpeptidase-deficient mice. Endocrinology. 2003;144:2761–2764. doi: 10.1210/en.2002-0071. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Manolagas SC. Birth and death of bone cells: basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr Rev. 2000;21:115–137. doi: 10.1210/edrv.21.2.0395. [DOI] [PubMed] [Google Scholar]
- Manolagas SC. From estrogen-centric to aging and oxidative stress: a revised perspective of the pathogenesis of osteoporosis. Endocrine Reviews. 2009 doi: 10.1210/er.2009-0024. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolagas SC, Almeida M. Gone with the Wnts: beta-catenin, T-cell factor, forkhead box O, and oxidative stress in age-dependent diseases of bone, lipid, and glucose metabolism. Mol Endocrinol. 2007;21:2605–2614. doi: 10.1210/me.2007-0259. [DOI] [PubMed] [Google Scholar]
- Marinkovic D, Zhang X, Yalcin S, Luciano JP, Brugnara C, Huber T, Ghaffari S. Foxo3 is required for the regulation of oxidative stress in erythropoiesis. J Clin Invest. 2007;117:2133–2144. doi: 10.1172/JCI31807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda T, Cepko CL. Controlled expression of transgenes introduced by in vivo electroporation. Proc Natl Acad Sci U S A. 2007;104:1027–1032. doi: 10.1073/pnas.0610155104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliaccio E, Giorgio M, Mele S, Pelicci G, Reboldi P, Pandolfi PP, Lanfrancone L, Pelicci PG. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature. 1999;402:309–313. doi: 10.1038/46311. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Imai Y, Matsumoto T, Sato S, Takeuchi K, Igarashi K, Harada Y, Azuma Y, Krust A, Yamamoto Y, Nishina H, Takeda S, Takayanagi H, Metzger D, Kanno J, Takaoka K, Martin TJ, Chambon P, Kato S. Estrogen prevents bone loss via estrogen receptor alpha and induction of Fas ligand in osteoclasts. Cell. 2007;130:811–823. doi: 10.1016/j.cell.2007.07.025. [DOI] [PubMed] [Google Scholar]
- Nemoto S, Finkel T. Redox regulation of forkhead proteins through a p66shc-dependent signaling pathway. Science. 2002;295:2450–2452. doi: 10.1126/science.1069004. [DOI] [PubMed] [Google Scholar]
- O'Brien CA, Jilka RL, Fu Q, Stewart S, Weinstein RS, Manolagas SC. IL-6 is not required for parathyroid hormone stimulation of RANKL expression, osteoclast formation, and bone loss in mice. Am J Physiol Endocrinol Metab. 2005;289:E784–E793. doi: 10.1152/ajpendo.00029.2005. [DOI] [PubMed] [Google Scholar]
- Paik JH, Kollipara R, Chu G, Ji H, Xiao Y, Ding Z, Miao L, Tothova Z, Horner JW, Carrasco DR, Jiang S, Gilliland DG, Chin L, Wong WH, Castrillon DH, DePinho RA. FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell. 2007;128:309–323. doi: 10.1016/j.cell.2006.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin LI, Weinstein RS, Parfitt AM, Roberson PK, Manolagas SC, Bellido T. Prevention of osteocyte and osteoblast apoptosis by bisphosphonates and calcitonin. J Clin Invest. 1999;104:1363–1374. doi: 10.1172/JCI6800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salih DA, Brunet A. FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr Opin Cell Biol. 2008;20:126–136. doi: 10.1016/j.ceb.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith WW, Norton DD, Gorospe M, Jiang H, Nemoto S, Holbrook NJ, Finkel T, Kusiak JW. Phosphorylation of p66Shc and forkhead proteins mediates Abeta toxicity. J Cell Biol. 2005;169:331–339. doi: 10.1083/jcb.200410041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaishi H, Konishi H, Matsuzaki H, Ono Y, Shirai Y, Saito N, Kitamura T, Ogawa W, Kasuga M, Kikkawa U, Nishizuka Y. Regulation of nuclear translocation of forkhead transcription factor AFX by protein kinase B. Proc Natl Acad Sci U S A. 1999;96:11836–11841. doi: 10.1073/pnas.96.21.11836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang ED, Nunez G, Barr FG, Guan KL. Negative regulation of the forkhead transcription factor FKHR by Akt. J Biol Chem. 1999;274:16741–16746. doi: 10.1074/jbc.274.24.16741. [DOI] [PubMed] [Google Scholar]
- Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem. 2008;77:289–312. doi: 10.1146/annurev.biochem.77.061307.091829. [DOI] [PubMed] [Google Scholar]
- Tothova Z, Gilliland DG. FoxO transcription factors and stem cell homeostasis: insights from the hematopoietic system. Cell Stem Cell. 2007;1:140–152. doi: 10.1016/j.stem.2007.07.017. [DOI] [PubMed] [Google Scholar]
- Tothova Z, Kollipara R, Huntly BJ, Lee BH, Castrillon DH, Cullen DE, McDowell EP, Lazo-Kallanian S, Williams IR, Sears C, Armstrong SA, Passegue E, DePinho RA, Gilliland DG. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007;128:325–339. doi: 10.1016/j.cell.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Tyner SD, Venkatachalam S, Choi J, Jones S, Ghebranious N, Igelmann H, Lu X, Soron G, Cooper B, Brayton C, Hee PS, Thompson T, Karsenty G, Bradley A, Donehower LA. p53 mutant mice that display early ageing-associated phenotypes. Nature. 2002;415:45–53. doi: 10.1038/415045a. [DOI] [PubMed] [Google Scholar]
- van der Horst A, Burgering BM. Stressing the role of FoxO proteins in lifespan and disease. Nat Rev Mol Cell Biol. 2007;8:440–450. doi: 10.1038/nrm2190. [DOI] [PubMed] [Google Scholar]
- Wang MC, Bohmann D, Jasper H. JNK extends life span and limits growth by antagonizing cellular and organism-wide responses to insulin signaling. Cell. 2005;121:115–125. doi: 10.1016/j.cell.2005.02.030. [DOI] [PubMed] [Google Scholar]
- Weinstein RS, Chen JR, Powers CC, Stewart SA, Landes RD, Bellido T, Jilka RL, Parfitt AM, Manolagas SC. Promotion of osteoclast survival and antagonism of bisphosphonate-induced osteoclast apoptosis by glucocorticoids. J Clin Invest. 2002;109:1041–1048. doi: 10.1172/JCI14538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein RS, Jilka RL, Parfitt AM, Manolagas SC. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids: potential mechanisms of their deleterious effects on bone. J Clin Invest. 1998;102:274–282. doi: 10.1172/JCI2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, McKenna MA, Feng X, Nagy TR, McDonald JM. Osteoclast apoptosis: the role of Fas in vivo and in vitro. Endocrinology. 2003;144:5545–5555. doi: 10.1210/en.2003-0296. [DOI] [PubMed] [Google Scholar]
- Zhang M, Xuan S, Bouxsein ML, von Stechow D, Akeno N, Faugere MC, Malluche H, Zhao G, Rosen CJ, Efstratiadis A, Clemens TL. Osteoblast-specific knockout of the insulin-like growth factor (IGF) receptor gene reveals an essential role of IGF signaling in bone matrix mineralization. J Biol Chem. 2002;277:44005–44012. doi: 10.1074/jbc.M208265200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.