

Abstract
Targeting guanine (G) quadruplex structures is an exciting new strategy with potential for controlling gene expression and designing anticancer agents. Guanine-rich peptide nucleic acid (PNA) oligomers bind to homologous DNA and RNA to form hetero-G-quadruplexes but can also bind to complementary cytosine-rich sequences to form heteroduplexes. In this study, we incorporated backbone modifications into G-rich PNAs to improve the selectivity for quadruplex vs duplex formation. Incorporation of abasic sites as well as chiral modifications to the backbone were found to be effective strategies for improving selectivity as shown by UV-melting and surface plasmon resonance measurements. The enhanced selectivity is due primarily to decreased affinity for complementary sequences, since binding to the homologous DNA to form PNA-DNA heteroquadruplexes retains high affinity. The improved selectivity of these PNAs is an important step toward using PNAs for regulating gene expression by G quadruplex formation.
Introduction
Among the four natural DNA bases, guanine has been of widespread interest because of its strong tendency to form tetrameric planar structures in different contexts. Less than 10 years after the proposal that guanine (G) hydrogen bonds with cytosine in the double-helical DNA structure1, Davies and coworkers2 proposed that guanine can also assemble into tetrameric arrangements called G-quartets, in which the guanines are arranged in a coplanar structure stabilized by eight hydrogen bonds. It was later discovered that G-rich DNA sequences were able to fold into secondary structures where two or more G-quartets stack on top of each other.3 Interest in the biological significance of G-quartets was first stimulated by the discovery of repetitive sequences of guanines at the ends of the chromosomes4–8 with more recent work demonstrating the presence of G-quadruplex forming sequences in other parts of the chromosomes, particularly in promoters.9–12 The preference of quadruplex-forming sequences for genomic regions implicated in regulating gene expression has led to the hypothesis that quadruplexes are themselves transcriptional regulatory elements and are likely to be important in both cancer and the aging process.13–17 The biological interest in G quadruplexes extends to RNA, where their possible presence in introns18 and 5′-untranslated regions19,20 suggests roles in regulating alternative splicing and translation.
The growing awareness of G quadruplexes as potential biological regulators of gene expression has driven great effort to design and synthesize molecules that can bind to these motifs.21,22 A large majority of papers in this area describe small molecules that exhibit shape recognition of G quadruplexes. The planar tetrads, concave grooves and loop nucleotides in quadruplexes offer distinctive surfaces to which small molecules can make hydrogen bonds, salt bridges or van der Waals interactions. The wealth of experience in the medicinal chemistry and pharmaceutical sciences fields with developing small-molecule drugs makes this approach appealing for in vivo applications.
An alternative strategy for quadruplex binding relies on sequence recognition and requires the quadruplex structure to be disrupted in order to access the sequence information encoded within the nucleobases. Inspired by work on antisense regulation of translation, researchers have successfully used high affinity DNA analogues such as locked nucleic acid (LNA)23 and peptide nucleic acid (PNA)24–26 to form complementary heteroduplexes with the quadruplex target. The high percentage of G-C pairs in these duplexes should translate into high affinity.
Our groups have taken a different approach to sequence recognition of quadruplexes: G-rich PNAs can invade DNA and RNA quadruplexes to form PNA-DNA27,28 or PNA-RNA29,30 heteroquadruplexes. The PNAs exhibit low nanomolar Kd values for both model quadruplexes and biologically relevant targets. The ability to form PNA-DNA heteroquadruplexes has been further supported by reports from Appella31 and Ladame32. Recent advances in the intracellular delivery of PNAs and demonstrations of their biological effects33–36 have motivated us to continue developing quadruplex-targeted PNAs.
Any compound designed for targeting a specific G quadruplex, regardless of whether the 13 binding mode relies on shape-or sequence-recognition, faces a multitude of undesirable targets 14 within a cell, including RNAs, double-helical genomic DNA and other, structurally similar quadruplexes (both DNA and RNA). Small molecules that recognize specific structural features of quadruplexes might easily evade most cellular RNA, whereas sequence-recognizing molecules such as PNA are more likely to bind to such RNAs. In our case, G-rich PNAs targeted to a given DNA quadruplex are at risk of binding to a C-rich complementary RNA. Given the high G-C content of the resulting duplex, it is likely that mismatches would be tolerated at physiological conditions, significantly expanding the range of possible off-target effects.
In this report, we describe two strategies for improving selectivity of a G-rich PNA for binding to homologous, quadruplex targets versus complementary sequences. These approaches involve modification of the PNA structure in ways that destabilize Watson-Crick pairing to cytosine-rich sequences while retaining high affinity for the Hoogsteen hydrogen bonding needed to assemble G quadruplexes. In the first approach, bases that are not involved in G-tetrad formation (i.e. loop bases) are removed from the PNA, thereby eliminating potential Watson-Crick base pairs from forming with a C-rich target. The second strategy relies on a chiral backbone modification that induces a left-handed helical structure in the PNA. This selectively destabilizes Watson-Crick pairing versus G-tetrad formation, providing additional selectivity. The ability to independently tune quadruplex and duplex stability is a significant advantage for quadruplex-forming PNAs relative to traditional antisense agents that recognize quadruplexes by complementary base pairing.
Experimental Procedures
Materials
DNA oligonucleotides were purchased from Integrated DNA Technologies (www.idtdna.com) and used after standard desalting by gel filtration chromatography, except for biotinylated oligonucleotides for surface plasmon resonance experiments, which were purified by HPLC. Sequences employed include: rComp (RNA, 5′-AUUACCCCUCCCAUUA-3′), Comp (DNA, 5′-CCCACCC-3′). Myc19 (DNA, 5′-AGGGTGGGAGGGTGGGGA-3′) and Myc19-comp (DNA, 5′-TCCCCACCCTCCCACCCT-3′). γ-modified-t-Boc-protected peptide nucleic acid monomers were synthesized as previously described37, unmodified monomers were purchased from Applied Biosystems (PNA monomers are no longer available from this company) or ASM Research Chemicals. Peptide nucleic acids were synthesized using standard solid phase synthesis,38,39 purified using reverse phase HPLC and verified by MALDI-TOF mass spectrometry (Applied Biosystems, Voyager DE sSTR) using sinapinic acid as the matrix. Samples were run with linear detection and positive ionization. PNAs synthesized for these studies are shown in Table 1. The yield for coupling multiple guanines in a row can be low. We used two coupling rounds for all unmodified monomers on a PNA after the second guanine was added, but only one coupling for the γ-modified guanines. In the case of coupling γ-modified monomers, 2.5 eq were used instead of 5 eq in order to conserve material.
Table 1.
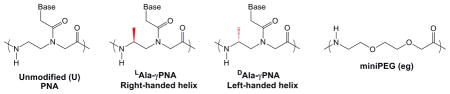
Sequence of the different synthesized PNAs and their calculated and experimental m/z. Structures of backbone modified and abasic resides are shown below.
| PNA | Sequence | Calc. M/Z | Exp. M/Z |
|---|---|---|---|
| Peg1 | H- G G G – (eg) - G G G - LLys - NH2 | 2039.24 | 2039.85 |
| Peg2 | H- G G G – (eg)2 - G G G - LLys - NH2 | 2184.40 | 2188.72 |
| Peg3 | H- G G G – (eg)3 - G G G - LLys - NH2 | 2329.55 | 2332.06 |
| Pmyc | H- G G G - A G – G G G – LLys-NH2 | 2460.20 | 2462.52 |
| Peg2-TO† | TO-G G G – (eg)2 - G G G - LLys - NH2 | 2572.91 | 2571.12 |
| PT | H- G G G - T - G G G - LLys - NH2 | 2160.98 | 2161.00 |
| L-Ala-PT | H- L-alaG L-alaG L-alaG - L-alaT - L-alaG L-alaG L-alaG - LLys – NH2 | 2259.16 | 2259.31 |
| D-Ala-PT | H- D-alaG D-alaG D-alaG - D-alaT - D-alaG D-alaG D-alaG - LLys - NH2 | 2259.16 | 2258.92 |
| D-Ala-PEG2 | H- D-alaG D-alaG D-alaG - (eg)2 - D-alaG D-alaG D-alaG - LLys – NH2 | 2268.28 | 2269.77 |
TO = Thiazole Orange
All DNA, RNA and PNA concentrations were determined by measuring the absorbance at 260nm at 85°C on a Cary 3 Bio spectrophotometer. At high temperature the bases are presumably unstacked, and the extinction coefficient can be calculated as the sum of the individual bases. The DNA base extinction coefficients were obtained from the literature40, whereas the PNA extinction coefficients were obtained from Applied Biosystems.
UV Melting Experiments
UV measurements were performed on a Varian Cary 3 spectrophotometer equipped with a thermoelectrically controlled multicell holder. Samples were prepared in a buffer containing 10mM Tris-HCl (pH 7) and 0.1mM Na2EDTA. For LiCl-containing samples, 0.1mM Li2EDTA was used, instead of the sodium salt. Various concentrations of KCl, NaCl or LiCl were used. The solutions were heated to 95 °C and equilibrated for 5 min. Then a cooling gradient was applied at a rate of 1 °C/min up to 15 °C, when samples were equilibrated for 5 min before starting a heating gradient at the same rate up to 95 °C. The absorbance at 275 nm for complementary binding or 295 nm for homologous binding41 was recorded as a function of temperature every 0.5 °C.
Circular Dichroism (CD) Spectropolarimetry
CD measurements were performed on a Jasco J-715 CD spectropolarimeter equipped with a water-circulating temperature controller. Samples containing 10 mM Tris-HCl (pH 7) and 0.1 mM Na2EDTA (or Li2EDTA), and various concentrations of KCl or LiCl were prepared in microcentrifuge tubes, heated to 95 °C for 5 min, and slowly annealed to room temperature. All spectra were collected at 25 °C or 37 °C. Each spectrum is the average of 3 different samples (unless specified), baseline corrected. For CD melting temperature experiments samples were annealed first and then the CD signal was measured as a function of temperature every 0.5 °C while applying a heating gradient of 1 °C/min and acquiring full-spectrum scans every 10 °C.
Surface Plasmon Resonance Experiments
Surface plasmon resonance (SPR) experiments to determine equilibrium binding constants in solution were performed by competition42 or direct43 binding methods. Measurements were made using a Biacore 2000 system with a 4 channel sensor chip containing covalently bound streptavidin linked by a carboxymethylated dextran layer to a gold surface. For the competition assays, biotinylated Myc19 was immobilized at >1000RU to maximize mass transfer in the determination of binding constants. The assays were performed in a Tris buffer (pH = 7). A range of PNA from 0–30 nM was used to obtain a standard curve. Following that, a pre-incubated mixture of free DNA (competitor, non-biotinylated) and PNA was injected over the immobilized DNA surface. The amount of response was used to determine the concentration of free PNA in the presence of the competitor DNA. In the direct binding experiments biotinylated Myc19 and Myc19-comp were immobilized on separate flow cells to a total RU of 250–300 to minimize mass transfer effects. All PNA samples were prepared in filtered and degassed buffer by serial dilutions from stock solutions. PNA samples were injected over the DNA surface at a flow rate of 50 μl/min followed by flow of running buffer. The sensor chip was regenerated by injection of 10 μl of 50 mM NaOH. Double referencing subtractions were used for data analysis. The first reference subtraction eliminates the bulk refractive index change and injection noise whereas the second subtraction of a blank buffer injection eliminates any systematic changes that are characteristic of a particular flow cell.43
RESULTS
The goal of the following experiments was to improve the selectivity of a G-rich PNA probe for binding to a DNA target of homologous sequence over the corresponding complementary sequence. Based on our previous findings that hybridization of homologous PNA and DNA (or RNA) oligomers yielded high affinity. 2:1 heteroquadruplexes (Kd < 10 nM),28 a strategy that selectively destabilized the competing Watson-Crick heteroduplex was appealing. The two approaches we used to achieve this objective are described below.
Incorporation of Abasic Sites
Rationale
An important difference between heteroduplexes and heteroquadruplexes is the presence of unpaired nucleobases in the latter. These bases form the loops linking adjacent tracts of guanines that form the G-tetrads which stabilize heteroquadruplexes, whereas they are used to make Watson-Crick base pairs in heteroduplexes. Thus, replacing the loop bases in the PNA probe with an abasic linker should destabilize the heteroduplex while having little or no impact on the heteroquadruplex stability (Scheme 1).
Scheme 1.

We previously reported that an 8-mer G-rich PNA sequence (Pmyc, Table 1) formed an extremely stable PNA2-DNA heteroquadruplex with a 19-mer target derived from the MYC proto-oncogene promoter region (Myc19, Table 1).28 Given the PNA and DNA sequences, the quadruplex domains should consist of three stacked G-tetrads, implying that the central two bases on the PNA should be expendable. Since the loop lengths and sequences can affect the stability and folding topology of DNA quadruplexes,44,45 it was difficult to predict the impact of abasic loop residues on the stability of a PNA2-DNA heteroquadruplex. However, it seemed unlikely that abasic residues would destabilize the heteroquadruplex by as much as the loss of two base pairs from a heteroduplex, since no inter-base hydrogen bonds are lost in the case of the quadruplex (Scheme 1).
To test this hypothesis, we synthesized a set of PNA oligomers featuring two G3 tracts separated by abasic linkers consisting of one, two, or three commercially available mini-polyethylene glycol (miniPEG) monomers (Pegn, n = 1, 2 or 3; Table 1). As with our previous reports, these G-rich PNAs were readily prepared by solid-phase synthesis, purified by HPLC and gave satisfactory results by mass spectrometry.
Complementary Hybridization
We first compared the binding of the PNAs to an RNA sequence (rComp) that is complementary to Pmyc. The RNA also contained overhanging regions to simulate natural RNA, which would be longer than the PNA probe. Prior reports showed that overhanging bases can significantly stabilize PNA-DNA24 and PNA-RNA46 duplexes. UV melting experiments were used to characterize the PNA-RNA duplexes that should result from hybridization. UV absorbances of solutions containing 1 μM Pegn or Pmyc and 1 μM rComp were monitored at 275 nm while the temperature was increased at 1°C/min from 15°C to 95°C (Figure 1 for Peg2; data for Peg1 and Peg3 shown in Figure S1). Under the conditions of the experiment, the transition of the Pmyc-rComp hybrid was greater than 90°C, based on the beginning of a transition at the highest temperature. As expected, the Pegn-rComp hybrids yielded much lower melting temperatures (ΔTm > 40°C), consistent with the loss of the central two base pairs in the duplex.
Figure 1.

UV melting curves monitored at 275 nm for rComp + PNA. Buffer = 10 mM Tris-HCl (pH 7), 150 mM KCl and 0.1 mM EDTA. [RNA] = [PNA] = 1 μM.
Homologous Hybridization
The 2:1 binding stoichiometry of Pmyc to Myc19 was previously determined through a fluorescence Job plot analysis that relied on thiazole orange (TO), an environmentally sensitive dye that was attached to the N-terminus of the PNA to act as a reporter. Similarly, Peg2 was synthesized with an N-terminal TO label and a Job plot was constructed to determine the binding stoichiometry of Peg2-TO to Myc19. The total strand concentration was held constant at 100nM and the individual PNA and DNA strand concentrations were varied. Figure 2 represents a Job plot demonstrating a turnover between 2:1 and 3:1 (PNA:DNA) binding stoichiometry. The presence of only four G tracts in the DNA target makes a 3:1 binding ratio unlikely, whereas a 2:1 binding stoichiometry is consistent with previous observations for short PNAs. However, it is possible that the TO chromophore allows binding of a third PNA via an end-stacking mode to the expected 2:1 PNA-DNA heteroquadruplex. Alternatively, a minor error (ca. 10%) in the extinction coefficient used to determine the PNA concentration could account for the ambiguity, since the concentrations at 2:1 and 3:1 correspond to 67 and 75 nM, respectively.
Figure 2.

Fluorescence Job Plot binding of Peg2-TO to Myc19. Total strand concentration (PNA + DNA) was 100 nM. Buffer contained 10 mM Tris-HCl (pH 7), 100 mM KCl, and 0.1 mM EDTA.
Subsequently, UV melting experiments were performed to monitor the binding of the abasic PNAs to Myc19. G-quadruplexes typically demonstrate hypochromic transitions at 295 nm upon heating through the Tm. Figure 3 compares the melting curves recorded at this wavelength for samples containing 1 μM Myc19 in the presence or absence of 2 μM Peg2 or 2 μM Pmyc. Although the shapes of the curves differ, nearly identical melting temperatures were obtained from first derivative plots for the two hybrid quadruplexes (76.1 ± 0.3 °C for Pmyc and 75.6 ± 2.3 °C for Peg2). Moreover, the PNAs having one or three miniPEG units in the linker yielded similar melting curves when combined with Myc19 (Figure S2). These results indicate that substituting the central GA bases in the PNA with abasic miniPEG residues has little or no effect on the thermal stability of the hybrid PNA-DNA quadruplex formed with the Myc19 target.
Figure 3.

UV melting curves monitored at 295nm for 1μM Myc19 and 2μM of either Pmyc or Peg2 in buffer containing 10mM Tris-HCl (pH 7), 1mM KCl, and 0.1mM EDTA.
Thermodynamic parameters for hybrid quadruplex formation were estimated by curve-fitting of the melting data (Table S1). While the magnitude of the parameters should be treated with caution due to the likelihood that the melting does not follow a simple two-state pathway,49 the relative values can be compared for the different quadruplexes. The ΔG of formation was found to be strongly exergonic (ca. −31 kcal/mol) and invariant, within the limits of error, for the four PNAs, verifying that replacement of the loop bases with abasic residues did not have an effect on the stability of the hybrid quadruplex. Based on the similar results obtained for the three abasic PNAs, further experiments were restricted to Peg2.
G-quadruplexes are stabilized by coordination of cations within the electronegative interior of the structure created by the lone pair of electrons on the O6 atom of each guanine that participates in tetrad formation. A strong preference for potassium over sodium and lithium has been reported for DNA50 and RNA homoquadruplexes20,51 as well as PNA-containing heteroquadruplexes27,30,42. Melting curves of Peg2-Myc19 (2:1) in the presence of KCl, NaCl and LiCl are shown in Figure 4. The results clearly demonstrate the preference for K+. Interestingly, while the quadruplex is clearly stabilized in KCl, the substantially lower hypochromicity suggests that the tetrads are not as well stacked as in the other two salts.
Figure 4.

UV melting curves monitored at 295nm for 1μM Myc19 + 2μM Peg2. Buffer contained 10mM Tris-HCl (pH7), and 1mM indicated ionic salt.
Surface plasmon resonance experiments were next carried out to determine the affinity of Peg2 for Myc19. Briefly, a series of solutions with varying concentration of Peg2 were flowed over a chip with a high density of immobilized Myc19. The initial slope of the sensorgrams under these mass transport limited conditions is linearly dependent on the concentration of PNA in solution.28 These slopes were used to build a calibration curve to quantify free Peg2. Subsequently, solutions containing 15 nM PEG2 and varying amounts of Myc19 were mixed, allowed to equilibrate at room temperature and then flowed over the same chip (Figure 5, left). As the soluble Myc19 concentration increases, there is less free Peg2 available to bind to the immobilized Myc19, leading to the progressively smaller signals in the SPR. The initial slopes obtained from the resulting sensorgrams yielded the free Peg2 concentrations enabling the construction of a binding isotherm. Separate control experiments demonstrated that free Myc19 in solution does not interact with surface bound Myc19 (data not shown). The binding isotherm was fitted to a two equivalent site binding model (Figure 5, right) yielding an average binding constant of 2.5 ± 0.1 × 108 M−1 per site (Kd = 4.0 nM). Note that this value corresponds to quadruplex-formation in solution since the SPR is used only to report the unbound PNA concentration. For comparison, we previously found the association constant of Pmyc to Myc19 to be 2.0 ± 0.2 × 108 M−1 (Kd = 5.0 nM).28 The similar binding constants for the two PNAs further demonstrates that, in spite of replacing the linking bases with abasic miniPEG units, the high affinity of G-rich PNA for the homologous Myc19 DNA target is preserved.
Figure 5.

SPR analysis of Peg2 binding to Myc19. (Left) Sensorgrams obtained upon flowing solutions containing 15nM Peg2 and varying amounts of Myc19 over a chip containing immobilized Myc19. Increasing Myc19 concentration leads to decreased free Peg2 and smaller changes in RU. (Right) Fitting to a 2:1 model with two equivalent sites gives an average Ka = 2.5 ± 0.1 × 108 M−1.
Circular dichroism (CD) spectropolarimetry is often used to obtain structural information about intramolecular and hybrid G-quadruplexes. CD spectra were recorded for Myc19 alone as well as Myc19 + Peg2 or Pmyc. The CD spectrum for Myc19 alone shows a peak at 260 nm and a minimum at 240 nm, consistent with a parallel G-quadruplex structure. As observed previously for Pmyc,28 addition of the homologous PNA to Myc19 did not cause a significant change in the spectrum (Figure 6), even though the SPR, Tm and fluorescence results clearly demonstrate that the PNAs can bind to Myc19 under these conditions. These results indicate that the Pmyc-Myc19 and Peg2-Myc19 heteroquadruplexes are structurally similar to each other and to the Myc19 homoquadruplex.
Figure 6.

CD spectra of 2.5 μM Myc19 in the presence and absence of 5 μM Peg2 or Pmyc. Buffer contained 150 mM KCl, 10 mM Tris (pH 7), and 0.1 mM EDTA. Spectra were recorded at 37°C
Modifications of the PNA Backbone
Rationale
With the aim of improving even further the properties of the PNAs, modifications in the original backbone structures have been performed. While modifications at the alpha carbon of the PNA monomer have only modest effects on thermal stability,52,53 modifications that introduce chirality at the gamma carbon can have profound effects on hybridization. Ly and coworkers37 demonstrated that modification in the gamma position induces a helical structure in the PNA: an L-modified backbone results in a right-handed helix, whereas a D-modified backbone gives rise to a left-handed helix. Since the L-modified structure matches the preferred right-handed helicity of DNA and RNA duplexes, binding of γ-PNA to complementary DNA and RNA is favored thermodynamically and results in a stronger binding than unmodified PNA37. Appella and coworkers also reported enhanced thermal stability for γ-PNAs based on an L-lysine side chain.54 Based on this we predicted that the corresponding D-modified PNAs would show weak binding towards the complementary sequence due to their left-handed helical conformation. The impact on quadruplex formation was difficult to predict, although the fact that quadruplexes are typically underwound relative to duplexes suggested that heteroquadruplexes involving D-γ-PNA might suffer lower destabilization than heteroduplexes. In addition, results from the Appella group showing that a PNA backbone constrained by a trans-cyclopentane ring enhanced PNA-DNA quadruplex stability31 indicated that the PNA could tolerate considerable modifications without compromising quadruplex recognition.
To test our hypotheses, we synthesized a series of PNAs having D-or L-alanine modified backbones and compared them to an unmodified PNA (Table 1). In addition, we synthesized a PNA having both a D-alanine-modified backbone and two miniPEG residues in the loop to combine the two types of modifications under investigation in this study.
PNA Characterization
The structures of the γ-PNAs in the absence of DNA were studied by CD spectroscopy. As shown in Figure 7A, the two fully modified PNAs exhibit exciton coupling between 250–290 nm in their CD spectra, indicating helical stacking of the nucleobases. The positive peak at 280 nm, the negative peak at 260 nm, and the strong negative peak at 210 nm for L-Ala-PT are consistent with earlier findings from the Ly group, which established the right-handed helicity of single-stranded L-γ-PNA oligomers by correlating the CD spectrum with 2D-NMR experiments.55 The near-mirror image spectrum for D-Ala-PT is then assigned to the desired left-handed helix. That the intensities of the respective peaks are not equal for the two PNAs is likely due to the fact that both PNAs have L-lysines at the C-terminus, i.e. the two PNAs are not true enantiomers.
Figure 7.

CD spectra of L-and D-Ala-PT samples. Samples contained 2μM PNA, 10mM Tris HCl (pH 7), 0.1mM Na2EDTA and either 1 mM KCl (A) or 150 mM KCl (B).
When the KCl concentration is increased from 1 mM to 150 mM, the intensities of the peaks increase and the spectral profiles change, with four clearly resolved peaks (Figure 7B). The most significant change to the spectra is the inversion of the 260 nm peak, from positive-to-negative for D-Ala-PT and negative-to-positive for L-Ala-PT. However, repeating the experiment using 150 mM LiCl instead of KCl results in spectra that look like the low potassium spectra shown in Figure 7A (see supporting information, Figure S3). This result indicates that the spectra shown in Figure 7B are due to a PNA homoquadruplex, which is stabilized by higher potassium concentration but destabilized by lithium ions. Further characterization of these quadruplexes will be reported elsewhere. Other examples of PNA homoquadruplexes have been reported previously.56
Complementary Hybridization
The destabilizing effect of the D-γ-modified PNAs was demonstrated by UV melting experiments with the complementary DNA oligonucleotide 5′-CCCACCC-3′. The shorter sequence and choice of DNA (rather than RNA) backbone for this oligonucleotide allowed us to observe the melting transition of even the most stable duplex, formed by L-Ala-PT (Figure 8). Table 2 collects the Tm data and shows that the right-handed L-modification improves hybridization by 16.3°C, while the left-handed D-modification destabilizes hybridization by −26°C, consistent with earlier findings.37 The difference in magnitude of the ΔTm values for the L-and D-γ-PNAs reflects the fact that the two types of PNA-DNA duplexes are diastereomeric, rather than enantiomeric. The PNA that combines both D-γ and abasic residues, D-Ala-Peg2, does not give an observable transition, indicating that complementary hybridization is completely eliminated under these conditions.
Figure 8.

UV melting experiments for the different PNAs with the complementary DNA. Samples contained 2 μM PNA and DNA, 10 mM Tris-HCl (pH 7), 0.1 mM Na2EDTA and 150 mM KCl. The lack of a transition for D-egPNA is due to the failure of this PNA to hybridize to the complementary DNA.
Table 2.
Melting temperature data for the different PNAs and the complementary DNA 5′-CCCACCC-3′ (Figure 8).
| PNA | Tm (°C) | ΔTm (°C) |
|---|---|---|
| PT | 62.6 ± 0.4 | - |
| L-Ala-PT | 78.9 ± 0.5 | +16.3 |
| D-Ala-PT | 36.6 ± 0.1 | −26.0 |
| D-Ala-Peg2 | - | - |
Homologous Hybridization
We next investigated heteroquadruplex formation between the γ-PNAs and Myc19. UV melting experiments were performed at a 2:1 PNA:DNA stoichiometry (Figure 9; actual melting curves are shown in Figure S4). There are several interesting results in this experiment. First, under the experimental conditions of 1 mM KCl, the Myc19 homoquadruplex melted at 61 °C while all PNA-DNA heteroquadruplexes were significantly more stable (ΔTm > 10 °C). Second, the fact that the unmodified PT, which has only a single thymine between the two G3 tracts rather than the GA segment of Pmyc, still forms a stable heteroquadruplex provides further evidence that the linker between the G tracts in the PNA does not significantly affect heteroquadruplex stability. Third, while PT and L-Ala-PT show similar melting temperatures at the same KCl concentration, D-Ala-PT and D-Ala-Peg2 show small decreases (2–6 °C), indicating that the left-handed helical structure of these PNAs has a modest destabilizing effect on the resulting heteroquadruplexes. Nevertheless, these heteroquadruplexes are still more stable than the Myc19 homoquadruplex, consistent with results presented by our group42. In addition, the relatively weak effect of the change in chirality on heteroquadruplex formation contrasts favorably with heteroduplex formation, which was virtually eliminated for D-Ala-Peg2 (Figure 8).
Figure 9.

Histogram showing melting temperature for the Myc19 DNA quadruplex alone or in a 2:1 heteroquadruplex with the indicated PNA. Samples contained 2 μM PNA and 1 μM DNA, 10 mM Tris-HCl (pH 7), 0.1 mM Na2EDTA and 1.0 mM KCl.
CD spectropolarimetry experiments were performed to obtain information about the secondary structure of the complexes. As observed previously for Pmyc and demonstrated above for the abasic PNAs PEGn, hybridization of the γ-PNAs to Myc19 resulted in minimal perturbation of the CD spectrum, indicating minimal reorganization of the DNA structure upon heteroquadruplex formation (Figure S5).42
Hybridization Selectivity
We next used SPR to further compare the ability of our PNAs to discriminate between complementary and homologous targets. In this experiment, Myc19 was immobilized on the SPR chip to provide a homologous target for heteroquadruplex formation. On an independent flow cell, we immobilized the complement to Myc19, i.e. 5′-TCCCCACCCTCCCACCCT-3′ (Myc19-comp), where the underlined sequences are perfectly complementary to the unmodified PNA and the γ-PNAs, except for D-Ala-Peg2. (We were unable to perform this experiment using immobilized RNA, which would be degraded during the denaturation wash with NaOH.) Sensorgrams for hybridization of 20 nM PNA to Myc19 and Myc19-comp are shown in Figure 10A for the unmodified PNA and three of the γ-PNAs. All four of the PNAs bind reasonably well to the homologous target, consistent with the UV melting results described above. The incomplete association and dissociation phases complicate extraction of the kinetic parameters from the sensorgrams. However, there is less than a factor of two difference in the amount of PNA bound during the association phase for each PNA. These results further support the conclusion that γ-modifications to the PNA backbone are well tolerated when forming heteroquadruplexes with DNA.
Figure 10.

SPR data for γ-PNAs with homologous (Myc19; A) or complementary (Myc19-comp; B) DNA. Samples contain 20nM PNA, 10mM Tris HCl (pH 7), 0.1mM Na2EDTA and 100mM KCl.
Strikingly different results are obtained for hybridization to the complementary DNA target (Figure 10B). Hybridization of PT is rapid and saturates the immobilized DNA whereas dissociation is very slow, presumably due to the high GC content of the resulting PNA-DNA duplex. Hybridization of L-Ala-PT is somewhat slower than for the unmodified PNA, but once formed, the duplex also strongly resists dissociation. Inverting the chirality of the backbone to D-Ala-PT significantly retards hybridization, as expected due to the left-handed helicity of this PNA. This PNA dissociates markedly faster than L-Ala-PT. Most importantly, D-Ala-Peg2 exhibits very little hybridization to the immobilized complementary target followed by rapid and complete dissociation. These results illustrate the high quadruplex versus duplex selectivity that can be achieved by appropriately modified PNAs.
Finally, the promising results for D-Ala-Peg2 motivated us to determine its binding constant for Myc19 using the SPR competition method. Figure 11 illustrates that free Myc19 efficiently competes with surface-immobilized Myc19 for hybridization to the PNA: there is essentially no free PNA once the concentration of Myc19 in the solution reaches 55 nM. The binding plot shown in Figure 11 reveals an unexpected result: the value of r (i.e. ratio of bound PNA to total DNA) is less than 1 even when the PNA is completely bound, indicating that this 57 PNA preferentially forms a 1:1 complex with Myc19. (Future experiments will be directed to understanding the apparent change in binding stoichiometry.) The data in Figure 11B are fit to a 1:1 binding equation giving a value of 4.1 ± 0.3 × 108 M−1 (Kd = 2.4 nM). While the fit is not ideal, particularly at low PNA concentration, the raw SPR data shown in Figure 11A indicate that 50% of the PNA is bound by <20 nM free Myc19, verifying that even with the D-γ-backbone modifications and abasic residues, heteroquadruplex formation by D-Ala-Peg2 is a favorable process.
Figure 11.

SPR competition experiment for measuring equilibrium constant for D-Ala-Peg2 + Myc19 hybridization in solution. All samples contained 15 nM PNA, 10 mM Tris HCl (pH 7), 0.1 mM Na2EDTA and 100 mM KCl with varying concentrations of Myc19. Left: SPR data for individual samples. Right: Binding isotherm where r values (r = [PNA bound]/[DNA total]) were calculated based on the amount of free PNA available to hybridize to the SPR surface. Line corresponds to fit based on 1:1 binding equation.
DISCUSSION
Any G-quadruplex targeted compound faces challenges in achieving selective binding in cells. Structure-targeted compounds such as small molecules that recognize quadruplexes by end-stacking or groove binding must exhibit significantly higher affinity for quadruplexes than for double-helical DNA, which, because of its relatively high concentration, could be a formidable competitor. Additionally, the small molecule must avoid binding to cellular RNA, which can fold into complex three-dimensional structures that might provide suitable binding sites. Finally, if the goal is to target a single quadruplex, then the molecule must discriminate among several structurally similar DNA and RNA quadruplexes. The development of G-quadruplex-binding proteins57,58 using powerful selection schemes promises to yield higher selectivity, but face the typical challenges of protein reagents, e.g. higher cost, lower stability and inefficient cell uptake.
Sequence-targeted approaches to G-quadruplex recognition face different obstacles. Complementary oligonucleotides such as PNA and LNA must exhibit a high degree of sequence selectivity in order to avoid binding to unintended DNA or RNA. This is particularly difficult when the target is highly enriched in guanine, since duplexes containing single mismatches might still be quite stable at physiological temperatures. In the homologous hybridization strategy, the probe must still avoid binding to a complementary competitor. The results presented above illustrate two ways in which this can be accomplished.
Our overall strategy was to modify the PNA in a way that would discourage binding to complementary sites without compromising the affinity for homologous sites. We first replaced the PNA loop residues with abasic, miniPEG units, since two potential Watson-Crick base pairs with a complementary competitor would be eliminated. This is similar to a report from Risitano and Fox, which demonstrated that similar ethylene glycol-based units inserted into a G-rich DNA oligonucleotide led to a strong preference for intramolecular folding into a quadruplex over intermolecular hybridization to a C-rich DNA.59 As shown, Pmyc binds tightly to a complementary RNA (Tm > 90 °C) but replacing the central GA with two miniPEG units depressed the melting temperature by over 40 °C without compromising the affinity for the Myc19 DNA quadruplex target.
We next used γ-modified PNAs to further weaken the affinity for complementary targets. γ-PNAs derived from L-or D-alanine feature methyl groups attached to the γ-carbon of the PNA backbone, which induce either right-or left-handed helicity, respectively. As shown in Table 2, the left-handed D-γ-PNA significantly weakened binding to a complementary competitor DNA, an observation that was reinforced by SPR experiments. Combining abasic and D-γ residues into the same PNA (D-Ala-Peg2) almost completely eliminated hybridization to the complementary DNA (Figures 8 and 10), which was the main goal of this project.
While it was unsurprising to learn that abasic and D-γ-modified residues weakened hybridization of PNA to complementary RNA and DNA, it was less obvious what the impact would be on binding to a homologous DNA target. Ethylene glycol (EG)-like residues have been inserted into loops in various DNA hairpin60,61 and dumbbell61 constructs, but their use in quadruplex motifs has been less common. Risitano and Fox published two reports showing that such modifications were tolerated in motifs such as G3-(X)-G3-(X)-G3-(X)-G3.45,59 The resulting quadruplexes were less stable than a Myc-model quadruplex, but of similar or higher stability compared with a quadruplex modeled on the human telomere sequence. Meanwhile, Cevec and Plavec showed that EG residues inserted between two G4 tracts accelerated formation of intermolecular dimeric quadruplexes, although the stability of the resulting quadruplex decreased.62
To determine the impact of the modifications on the affinity for quadruplex DNA, we tested hybridization of the PNAs to Myc19, which was previously targeted with the unmodified PNA Pmyc. Because of the high stability of the PNA-DNA heteroquadruplex structures, melting temperature experiments could only be performed at low KCl concentrations. Unlike the experiments with the complementary RNA, the right-handed L-γ-PNAs did not affect the stability of the heteroquadruplex, compared with the unmodified PNA (Figure 9). This suggests that the favorable entropic contribution of a pre-organized helical PNA strand is offset by an unfavorable enthalpic contribution if the helicity of the PNA does not match that of the final heteroquadruplex structure. (The methyl group on the modified PNA backbone is also likely to alter the effect of hydration on the PNA hybridization thermodynamics.) Meanwhile, the D-γ-PNAs exhibit a modest destabilization (ΔTm = 4–9 °C), but stable heteroquadruplexes are formed in all three cases.
CD experiments were performed to evaluate the structure of the PNA-DNA heteroquadruplexes. Interestingly, in contrast to the inverted CD spectra exhibited by the chiral PNAs (Figure 7), all PNAs show similar CD spectra when bound to the Myc19 DNA, with a maximum at 260nm and minimum at 240nm, similar to the characteristic CD signal of a parallel quadruplex. These results indicate that the helical secondary structures and intermolecular quadruplex tertiary structures adopted by the γ-PNAs are readily disrupted in order to form stable heteroquadruplexes with Myc19.
Final confirmation that the modified PNAs exhibit improved selectivity for homologous vs complementary targets was obtained from SPR experiments. The best PNA, D-Ala-Peg2, combines abasic and D-γ-modified residues, leading to low nanomolar affinity for Myc19 with minimal binding to a complementary target. Such PNAs should be able to access their cellular quadruplex targets without significant off-target binding to complementary RNA or DNA.
CONCLUSIONS
The results presented above illustrate the ease with which quadruplex-targeted PNAs can be modified to avoid binding to complementary targets, addressing a major concern with using G-rich PNAs as gene-targeted agents. The success of the modified PNAs exploits the different binding modes for the PNAs with their homologous targets, i.e. heteroquadruplex formation based on G-tetrads, versus complementary competitors, i.e. heteroduplex formation based on Watson-Crick pairs. It would be much more difficult to avoid off-target effects for C-rich probes that targeted quadruplexes by duplex formation.
This report addresses only half of the selectivity problem for quadruplex-targeted PNAs. In order to successfully hybridize to a specific quadruplex, hybridization to all other quadruplexes (or, more generally, all putative quadruplex sequences) must be avoided. Given the large number of such sequences in genomic DNA and mRNA, this is a significant challenge. Successful strategies will likely rely on secondary recognition elements in the PNA that interact with the DNA loop and flanking nucleotides, which are the distinguishing features of similarly folded quadruplexes.
Supplementary Material
Acknowledgments
S.L. synthesized D-γ-PNAs and performed optical spectroscopy and thermal denaturation experiments for γ-PNAs, C.M. synthesized and performed optical spectroscopy and thermal denaturation experiments for miniPEG-containing PNAs, S. R. synthesized and performed optical spectroscopy and thermal denaturation experiments for PEG2, F. T. performed all SPR experiments and I.S. synthesized D-γ-PNA monomers. This research was supported by the National Institutes of Health (R01 GM58547 to B.A.A. and R01 GM61587 to W.D.W.) Mass spectra were recorded in the Center for Molecular Analysis at Carnegie Mellon, supported by NSF CHE-9808188 and DBI-9729351. We thank Dr. Srinivas Rapireddy for synthesis of L-γ-PNA monomers and oligomers.
References
- 1.Watson JD, Crick FHC. Nature. 1953;171:737–738. doi: 10.1038/171737a0. [DOI] [PubMed] [Google Scholar]
- 2.Gellert M, Lipsett MN, Davies DR. Proc Natl Acad Sci U S A. 1962;48:2013–8. doi: 10.1073/pnas.48.12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arnott S, Chandrasekaran R, Marttila CM. Biochem J. 1974;141:537–43. doi: 10.1042/bj1410537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blackburn EH. Nature. 1991;350:569–73. doi: 10.1038/350569a0. [DOI] [PubMed] [Google Scholar]
- 5.Blackburn EH. Science. 1990;249:489–90. doi: 10.1126/science.2200120. [DOI] [PubMed] [Google Scholar]
- 6.Hardin CC, Henderson E, Watson T, Prosser JK. Biochemistry. 1991;30:4460–72. doi: 10.1021/bi00232a013. [DOI] [PubMed] [Google Scholar]
- 7.Oka Y, Thomas CA., Jr Nucleic Acids Res. 1987;15:8877–98. doi: 10.1093/nar/15.21.8877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sundquist WI, Klug A. Nature (London, United Kingdom) 1989;342:825–9. doi: 10.1038/342825a0. [DOI] [PubMed] [Google Scholar]
- 9.Huppert JL, Balasubramanian S. Nucleic Acids Res. 2005;33:2908–2916. doi: 10.1093/nar/gki609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Todd AK, Johnston M, Neidle S. Nucleic Acids Res. 2005;33:2901–2907. doi: 10.1093/nar/gki553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huppert J, Balasubramanian S. Nucleic Acids Res. 2007;35:406–413. doi: 10.1093/nar/gkl1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao Y, Du Z, Li N. FEBS Lett. 2007;581:1951–1956. doi: 10.1016/j.febslet.2007.04.017. [DOI] [PubMed] [Google Scholar]
- 13.Viglasky V. FEBS J. 2009;276:401–409. doi: 10.1111/j.1742-4658.2008.06782.x. [DOI] [PubMed] [Google Scholar]
- 14.Patel DJ, Phan AT, Kuryavyi V. Nucleic Acids Research. 2007;35:7429–7455. doi: 10.1093/nar/gkm711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maizels N. Nat Struct Mol Biol. 2006;13:1055–1059. doi: 10.1038/nsmb1171. [DOI] [PubMed] [Google Scholar]
- 16.Siddiqui-Jain A, Grand CL, Bearss DJ, Hurley LH. Proc Natl Acad Sci USA. 2002;99:11593–11598. doi: 10.1073/pnas.182256799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qin Y, Hurley LH. Biochimie. 2008;90:1149–1171. doi: 10.1016/j.biochi.2008.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gomez D, Lemarteleur T, Lacroix L, Mailliet P, Mergny JL, Riou JF. Nucleic Acids Res. 2004;32:371–379. doi: 10.1093/nar/gkh181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kumari S, Bugaut A, Huppert J, Balasubramanian S. Nature Chem Biol. 2007;3:218–221. doi: 10.1038/nchembio864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arora A, Dutkiewicz M, Scaria V, Hariharan M, Maiti S, Kurreck J. RNA. 2008;14:1290–1296. doi: 10.1261/rna.1001708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huppert J. Chem Soc Rev. 2008;37:1375–1384. doi: 10.1039/b702491f. [DOI] [PubMed] [Google Scholar]
- 22.Monchaud D, Teulade-Fichou MP. Org Biomol Chem. 2008;6:627–636. doi: 10.1039/b714772b. [DOI] [PubMed] [Google Scholar]
- 23.Kumar N, Patowary A, Sivasubbu S, Petersen M, Maiti S. Biochemistry. 2008;47:13179–13188. doi: 10.1021/bi801064j. [DOI] [PubMed] [Google Scholar]
- 24.Datta B, Armitage BA. J Am Chem Soc. 2001;123:9612–9619. doi: 10.1021/ja016204c. [DOI] [PubMed] [Google Scholar]
- 25.Green JJ, Ying L, Klenerman D, Balasubramanian S. J Am Chem Soc. 2003;125:3763–3767. doi: 10.1021/ja029149w. [DOI] [PubMed] [Google Scholar]
- 26.Amato J, Oliviero G, De Pauw E, Gabelica V. Biopolymers. 2009;91:244–255. doi: 10.1002/bip.21124. [DOI] [PubMed] [Google Scholar]
- 27.Datta B, Schmitt C, Armitage BA. J Am Chem Soc. 2003;125:4111–4118. doi: 10.1021/ja028323d. [DOI] [PubMed] [Google Scholar]
- 28.Roy S, Tanious F, Wilson WD, Ly DH, Armitage BA. Biochemistry. 2007;46:10433–10443. doi: 10.1021/bi700854r. [DOI] [PubMed] [Google Scholar]
- 29.Marin VL, Armitage BA. J Am Chem Soc. 2005;127:8032–8033. doi: 10.1021/ja051102y. [DOI] [PubMed] [Google Scholar]
- 30.Marin VL, Armitage BA. Biochemistry. 2006;45:1745–1754. doi: 10.1021/bi051831q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Englund EA, Xu Q, Witschi MA, Appella DH. J Am Chem Soc. 2006;128:16456–16457. doi: 10.1021/ja064317w. [DOI] [PubMed] [Google Scholar]
- 32.Paul A, Sengupta P, Krishnan Y, Ladame S. Chem Eur J. 2008;14:8682–8689. doi: 10.1002/chem.200800605. [DOI] [PubMed] [Google Scholar]
- 33.Ivanova GD, Arzumanov A, Abes R, Yin H, Wood MJA, Lebleu B, Gait MJ. Nucleic Acids Res. 2008;36:6418–6428. doi: 10.1093/nar/gkn671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koppelhus U, Shiraishi T, Zachar V, Pankratova S, Nielsen PE. Bioconjugate Chem. 2008;19:1526–1534. doi: 10.1021/bc800068h. [DOI] [PubMed] [Google Scholar]
- 35.Dragulescu-Andrasi A, Zhou P, He G, Ly DH. Chem Commun. 2005:244–246. doi: 10.1039/b412522c. [DOI] [PubMed] [Google Scholar]
- 36.Zhou P, Wang M, Du L, Fisher GW, Waggoner A, Ly DH. J Am Chem Soc. 2003;125:6878–6879. doi: 10.1021/ja029665m. [DOI] [PubMed] [Google Scholar]
- 37.Dragulescu-Andrasi A, Rapireddy S, Frezza BM, Gayathri C, Gil RR, Ly DH. J Am Chem Soc. 2006;128:10258–10267. doi: 10.1021/ja0625576. [DOI] [PubMed] [Google Scholar]
- 38.Christensen L, Fitzpatrick R, Gildea B, Petersen KH, Hansen HF, Koch T, Egholm M, Buchardt O, Nielsen PE, Coull J, Berg RH. J Peptide Sci. 1995;3:175–183. doi: 10.1002/psc.310010304. [DOI] [PubMed] [Google Scholar]
- 39.Koch T. In: Peptide Nucleic Acids: Protocols and Applications. 2. Nielsen PE, editor. Horizon Bioscience; Norfolk: 2004. pp. 37–60. [Google Scholar]
- 40.Dawson RMC. Data for Biochemical Research. 3. Oxford University Press; New York: 1986. [Google Scholar]
- 41.Mergny JL, Phan AT, Lacroix L. FEBS Lett. 1998;435:74–78. doi: 10.1016/s0014-5793(98)01043-6. [DOI] [PubMed] [Google Scholar]
- 42.Roy S, Tanious FA, Wilson WD, Ly DH, Armitage BA. Biochemistry. 2007;46:10433–10443. doi: 10.1021/bi700854r. [DOI] [PubMed] [Google Scholar]
- 43.Nguyen B, Tanious FA, Wilson WD. Methods. 2007;142:150–161. doi: 10.1016/j.ymeth.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 44.Bugaut A, Balasubramanian S. Biochemistry. 2008;47:689–697. doi: 10.1021/bi701873c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Risitano A, Fox KR. Nucleic Acids Res. 2004;32:2598–2606. doi: 10.1093/nar/gkh598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dias N, Sénamaud-Beaufort C, le Forestier E, Auvin C, Hélène C, Saison-Behmoaras TE. J Mol Biol. 2002;320:489–501. doi: 10.1016/S0022-2836(02)00474-6. [DOI] [PubMed] [Google Scholar]
- 47.Marin VL, Armitage BA. Biochemistry. 2006;45:1745–1754. doi: 10.1021/bi051831q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Svanvik N, Westman G, Wang D, Kubista M. Anal Biochem. 2000;281:26–35. doi: 10.1006/abio.2000.4534. [DOI] [PubMed] [Google Scholar]
- 49.Haq I, Chowdhry BZ, Chaires JB. Eur J Biophys. 1997;26:419–426. doi: 10.1007/s002490050096. [DOI] [PubMed] [Google Scholar]
- 50.Williamson JR. Annu Rev Biophys Biomol Struct. 1994;23:703–730. doi: 10.1146/annurev.bb.23.060194.003415. [DOI] [PubMed] [Google Scholar]
- 51.Ramos A, Hollingworth D, Pastore A. RNA. 2003;9:1198–1207. doi: 10.1261/rna.5960503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Püschl A, Sforza S, Haaima G, Dahl O, Nielsen PE. Tetrahedron Lett. 1998;39:4707–4710. [Google Scholar]
- 53.Haaima G, Lohse A, Buchardt O, Nielsen PE. Angew Chem Int Ed. 1996;35:1939–1942. [Google Scholar]
- 54.Englund EA, Appella DH. Angew Chem Int Ed. 2007;46:1414–1418. doi: 10.1002/anie.200603483. [DOI] [PubMed] [Google Scholar]
- 55.Dragulescu-Andrasi A, Rapireddy S, Frezza BM, Gayathri C, Gil RR, Ly DH. J Am Chem Soc. 2006;128:10258–10267. doi: 10.1021/ja0625576. [DOI] [PubMed] [Google Scholar]
- 56.Datta B, Bier ME, Roy S, Armitage BA. J Am Chem Soc. 2005;127:4199–4207. doi: 10.1021/ja0446202. [DOI] [PubMed] [Google Scholar]
- 57.Fernando H, Rodriguez R, Balasubramanian S. Biochemistry. 2008;47:9365–9371. doi: 10.1021/bi800983u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schaffitzel C, Berger I, Postberg J, Hanes J, Lipps HJ, Plückthun A. Proc Natl Acad Sci USA. 2001;98:8572–8577. doi: 10.1073/pnas.141229498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Risitano A, Fox KR. Biochemistry. 2003;42:6507–6513. doi: 10.1021/bi026997v. [DOI] [PubMed] [Google Scholar]
- 60.Rumney SI, Kool ET. J Am Chem Soc. 1995;117:5635–5646. doi: 10.1021/ja00126a004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McCullagh M, Zhang L, Karaba AH, Zhu H, Schatz GC, Lewis FD. J Phys Chem B. 2008;112:11415–11421. doi: 10.1021/jp802378a. [DOI] [PubMed] [Google Scholar]
- 62.Cevec M, Plavec J. Biochemistry. 2005;44:15238–15246. doi: 10.1021/bi0514414. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.