

Abstract
Targeting canarypox (CP)-HIV vaccine to dendritic cells (DCs) elicits anti-HIV-1 immune responses in vitro. We conducted a phase I/II clinical trial to evaluate whether adding DC to a CP-HIV vaccine improved virologic control during analytic treatment interruption (ATI) in HIV-1-infected subjects. Twenty-nine subjects on suppressive antiretroviral therapy were randomized to vaccination with autologous DCs infected with CP-HIV + keyhole limpet hemocyanin (KLH) (arm A, n = 14) or CP-HIV + KLH alone (arm B, n = 15). The mean viral load (VL) setpoint during ATI did not differ between subjects in arms A and B. A higher percentage of subjects in the DC group had a VL setpoint <5000 c/mL during ATI (4/13 or 31% in arm A compared with 0/13 in arm B, p = 0.096), but virologic control was transient. Subjects in arm A had a greater increase in KLH lymphoproliferative response than subjects in arm B; however, summed ELISPOT responses to HIV-1 antigens did not differ by treatment arm. We conclude that a DC-CP-HIV vaccine is well-tolerated in HIV-1-infected patients, but does not lower VL setpoint during ATI compared with CP-HIV alone. New methods to enhance the immunogenicity and antiviral efficacy of DC-based vaccines for HIV-1 infection are needed.
Keywords: HIV-1, Dendritic cells, Canarypox, Therapeutic vaccine, Clinical trial
1. Introduction
The goal of therapeutic vaccination for HIV-1 is to control viral replication by boosting immune responses. Dendritic cells (DCs)are potent antigen presenting cells that can be manipulated ex vivo to serve as powerful adjuvants for vaccine immunogens [1,2]. Canarypox virus vectors carrying HIV-1 genes (CP-HIV) have been used in preventive and therapeutic HIV-1 vaccine trials [3–7], but usually elicit only weak or intermittent immune responses. In studies in HIV-1-infected patients on antiretroviral therapy (ART), vaccination with CP-HIV has not, in general, resulted in control of viremia during analytic treatment interruption (ATI) [8–11]. For example, although one study demonstrated a trend toward virologic control during ATI [12], another trial found an enhancement of viral replication [10]. Targeting of CP-HIV to DCs has been shown to elicit both HIV-1-specific CD4 and CD8 responses in vitro [13]. Because these types of immune responses have been associated with control of viral replication in HIV-1 controllers (reviewed in [14]), we evaluated whether addition of DC to a CP-HIV vaccine enhanced immunogenicity and lowered viral load (VL) setpoint during ATI in HIV-1-infected patients.
2. Methods
2.1. Vaccine and study design
HIV-1-infected subjects on ART with plasma VL ≤400 copies (c)/mL and CD4 cell counts ≥400/mm3 for at least 3 months, and VL ≤50c/mL at screening, were randomized to receive either autologous DCs infected with ALVAC-HIV vCP1452 (arm A) or ALVAC-HIV vCP1452 alone (arm B) (Fig.1). Informed consent was obtained from all subjects and the study was approved by the Institutional Review Boards of participating sites.
Fig. 1.
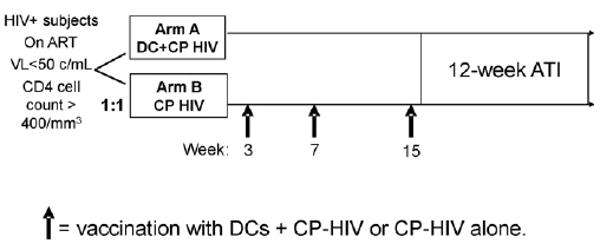
HIV-1-infected subjects on antiretroviral therapy with suppressed viral load were randomized to receive either CP-HIV or DCs + CP-HIV. Subjects were vaccinated at weeks 3, 7 and 15. Following vaccination, subjects underwent analytic treatment interruption. At weeks 3 and 7, subjects also received keyhole limpet hemocyanin (KLH). KLH was a control to determine whether priming of new immune responses occurred. DC, dendritic cells; CP-HIV, canarypox-HIV; ATI, analytic treatment interruption; ART, antiretroviral therapy; VL, viral load.
ALVAC vCP1452 (Sanofi-Pasteur) is a recombinant CP vector that expresses HIV-1 env and gag and a synthetic polypeptide encompassing epitopes from nef and pol [15]. The vector is a plaque-purified isolate of an attenuated canarypox virus, grown on pathogen-free chicken embryo fibroblasts, into which the vaccinia virus E3L and K3L coding sequences are inserted into ALVAC to increase virus-specific gene expression by downregulating PKR activity. The recombinant virus is suspended in a solution of serum-free, antibiotic-free culture medium, and is then lyophilized prior to shipment.
Subjects in group A underwent leukapheresis to obtain peripheral blood mononuclear cells (PBMC) from which DCs were expanded and matured, as previously described [16]. Mature DCs were infected with ALVAC-HIV vCP1452 at a multiplicity of infection (MOI) of 3. Subjects in arm A received 1.5–6 million ALVAC-HIV vCP1452-infected DCs. The DC dose was chosen based on previous studies suggesting that delivery of this range of DCs resulted in an adjuvant effect in vivo [17,18]. The dose and MOI of ALVAC-HIV was chosen based on a previous studies demonstrating immunogenicity in vitro [13,16]. Subjects in arm B received 1 mL of ALVAC vCP1452 (≥107 Cell Culture Infective Dose (CCID)50). This dose was chosen based on the following considerations: (1) in previous clinical trials conducted in HIV-1-negative individuals, this dose induced optimal antibody responses and there was no evidence for a dose–response relationship for CD8 cytotoxic T lymphocytes; (2) increasing the dose beyond 107 CCID50 led to increased reactogenicity [19,20]. Taken together with manufacturing considerations, these findings resulted in selection of a dose of 107 CCID50.
To test the ability to prime immune responses to a neoantigen, keyhole limpet hemocyanin (KLH) (Biosyn Corp.) was included in the first two injections in both treatment arms. In arm A, 10 μg/mL of KLH was pulsed onto DCs in vitro for 24 h prior to injection. In arm B, 0.1 mL of KLH (1 mg/mL) was injected at the same time as ALVAC vCP1452.
Subjects were vaccinated at weeks 3, 7 and 15 by superficial subcutaneous injection in the inner aspect of the arm, 6–12 cm from the axilla. After three injections, subjects underwent a minimum of a 12-week ATI during which time ART was stopped. Subjects reinitiated ART if their CD4 cell count and percentage declined to <50% of baseline (mean of entry and week 3 values). The primary endpoint was the VL setpoint, defined as the average (on log10 scale) of the last two scheduled VL evaluations during weeks 10–13 of ATI (in one subject, only the week 10 VL value was used because he resumed ART after that point). The proportion of subjects with VL setpoint <5000 copies/mL was also a planned analysis. The study was designed to have 80% power to detect a difference in VL of 0.74 log10 c/mL between study arms (assuming 0.6 log10 c/mL variability estimate) [21].
2.2. ELISPOT assays
Interferon gamma (IFN-γ) ELISPOT assays were performed as described [22] with the following modifications. PBMCs were plated at 200,000 cells/well in the presence or absence of vaccinia viruses (Aventis Behring) encoding HIV-1 Gag, Pol, Env or Nef, or with parental (WR) vaccinia vector at a MOI of 2 viral particles/cell. For positive controls, Staphylococcus enterotoxin A (SEA, Sigma) or vaccinia encoding cytomegalovirus (CMV) pp65 antigen were used. Average values for duplicate wells were multiplied by 5 to calculate the number of spot-forming cells (SFC)/million PBMCs. One subject was excluded due to lack of virologic control during vaccinations; four subjects were excluded because the SEA control for baseline samples was below a threshold of 1000 SFC/million PBMC. We additionally excluded 34 specimens because of a low SEA control, and 7 specimens due to high background (>100 SFC/million PBMC for media or WR control). The final ELISPOT data set consisted of 196 specimens from 24 subjects. An ELISPOT response to an HIV-1 antigen was defined as >30 SFC/million PBMC and >3-fold over the maximum of the WR or media controls. For the baseline ELISPOT level, the geometric mean of the week 0 and 3 values was determined. A positive post-vaccination fold-change in the magnitude of the ELISPOT response was defined as having ≥30 SFC/million PBMC during or after vaccination (weeks 7, 11, and 16–19) along with a threefold increase in SFC/million PBMC compared to baseline. In addition to evaluating the response to individual HIV-1 antigens (Gag, Env, Nef, Pol), we also summed the responses to these four antigens (summed ELISPOT response) at different time points. The association between fold-change ELISPOT response after vaccination and VL setpoint was a planned analysis.
2.3. Lymphocyte proliferation assays (LPA)
LPA were performed on fresh blood specimens as previously described [23]. Stimulants included pokeweed mitogen, candida antigen, tetanus toxoid, KLH, and five pools of HIV-1-peptides (15-mers overlapping by 11) based on regions of sequence rich in T cell epitopes identified in the Los Alamos HIV-1 sequence database [24]. The pools contained from 61 to 89 different peptides. The stimulation index (SI), defined as the geometric mean counts per minute (CPM) in the stimulated replicates divided by the geometric mean CPMs for media alone, was used for analysis of the response.
2.4. Statistical methods
All tests were two-sided, 5% level, and not adjusted for multiple testing. Arms were compared using exact Wilcoxon Rank Sum tests for continuous endpoints or Fisher's exact tests for proportions. Rank-based correlations were used to assess correspondence between responses. Time-to-event endpoints were compared with log-rank tests.
3. Results
3.1. Baseline characteristics
Twenty-nine HIV-1-infected subjects were randomized to receive vaccinations with either DCs pulsed with ALVAC-HIV vCP1452 + KLH (arm A, n = 14) or ALVAC-HIV vCP1452 + KLH alone (arm B, n = 15). Twenty-eight subjects were male and one subject was female. Fifty-two percent were white non-Hispanics, 24% were black, 21% were Hispanic and 3% were more than 1 race. The median (Q1, Q3) age was 43 (38, 48) years. Median duration of ART was 7.3 years in arm A and 6.4 years in arm B. The baseline CD4 cell count did not differ significantly between arms A and B (median of 664 and 724 cells/mm3, respectively). The median nadir CD4 cell count was 344 and 522/mm3 in arms A and B, respectively (p = 0.10). The CD8 cell count was lower in subjects randomized to arm A compared with subjects in arm B (median 796/mm3 vs. 994/mm3, p = 0.004).
3.2. Safety of the vaccines and ATI
All 29 subjects completed vaccinations and underwent ATI. One subject in arm A withdrew during ATI because of development of grade 4 neutropenia. Two subjects in arm B did not complete the ATI: one restarted ART and one went off-study at his request. A total of 26 subjects completed at least 10 weeks of ATI, 13 in each treatment arm, and achieved a VL setpoint (Fig. 2).
Fig. 2.
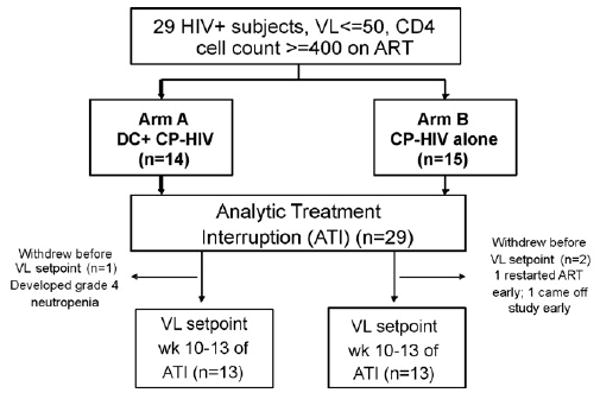
Subject disposition. VL, viral load; DC, dendritic cell; CP-HIV, canarypox-HIV; Wk, week; ART, antiretroviral therapy.
There were no grade 3 or higher adverse events during the course of study vaccinations. One subject (in arm A) experienced transient viremia during the vaccinations, which returned to <50 c/mL without a change in his regimen. During the ATI, two subjects developed a ≥grade 3 event: one subject in arm B with a history of immune thrombocytopenic purpura had a decline in platelet count to 44,000/mm3 at week 12 of ATI, and one subject in arm A developed an absolute neutrophil count of 264/mm3 at week 5 of ATI.
Three subjects reinitiated ART or withdrew from the study during ATI (see Fig. 2). Two subjects (both in arm B) met criteria for restarting ART (CD4 cell count and percentage >50% below baseline) at the end of 12 weeks of ATI; both restarted ART and achieved a VL<400c/mL. After completing 12 weeks of ATI, 11 additional subjects reinitiated ART and had sufficient follow-up during the study to assess VL response; of these, 10 achieved a VL <400c/mL and one subject had persistent viremia on ART. One subject reinitiated ART near the end of the study (27 weeks after starting ATI) but did not have sufficient follow-up to assess VL response. Twelve subjects did not reinitiate ART during the study.
3.3. Immunologic changes during vaccinations and ATI
During the vaccinations, the CD4 cell count declined slightly: the median change from baseline to pre-ATI CD4 cell count (last value before discontinuing ART) was −73/mm3 in arm A and −23/mm3 in arm B (p = 0.10). During the 12-week ATI, there was a large drop in CD4 cell count: the median change in CD4 cell count from pre-ATI to the end of ATI was −188/mm3 in arm A and −260/mm3 in arm B (p = 0.36).
We assessed the impact of vaccination on LPA response to tetanus, candida, pokeweed, KLH and five pools of HIV-1 peptides. Subjects who received KLH-pulsed DCs (arm A) had a significantly greater increase in KLH LPA response prior to ATI than subjects who received KLH without DCs (arm B) (median SI fold-change 5.7 vs. 1.8, p = 0.007) (Fig. 3). Following ATI, the LPA response to KLH declined in both treatment groups (data not shown). There was no apparent increase in LPA response to the HIV-1 peptides or the other antigens in either treatment arm.
Fig. 3.
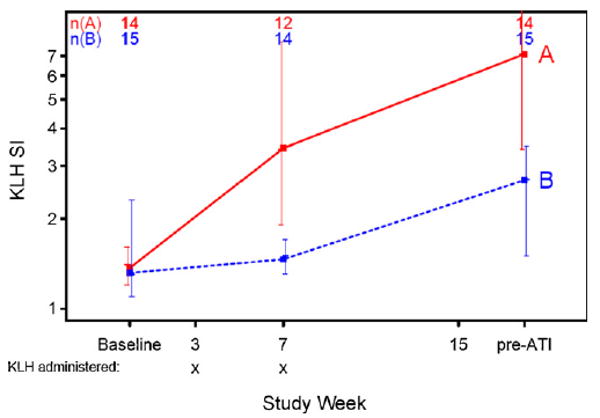
Lymphoproliferative responses to keyhole limpet hemocyanin (KLH) during vaccinations. Median values are plotted and bars represent the 25th–75th percentiles. SI, stimulation index; ATI, analytic treatment interruption.
To assess whether addition of DCs to CP-HIV improved virus-specific CD8 cell responses, we compared the ELISPOT responses to HIV-1 antigens in the two treatment arms. Prior to immunization, there was no difference in the number of SFC/million PBMC between subjects in arms A and B. Pooling arms, 58% (14/24) of subjects had a baseline ELISPOT response to HIV-1 Gag, Pol, Env or Nef (as defined in Section 2). Following vaccination, 64% (7/11) of subjects developed a new Pol response, 53% (9/17) developed a new Gag response, 29% (6/21) developed a new Env response and 22% (4/18) developed a new Nef response. Development of a new ELISPOT response to at least one HIV-1 antigen did not differ by treatment arm (six subjects in arm A and seven subjects in arm B); there was also no difference between the treatment arms in terms of the number of subjects developing a new response at the level of the specific HIV-1 antigen.
In terms of the magnitude of the change in ELISPOT response after vaccination, almost half of the subjects had a threefold or greater increase in summed ELISPOT response over baseline to HIV-1 antigens (Gag, Env, Nef and Pol), that was at least 30 SFC/million PBMC, at one or more time points: 5/12 (42%) in arm A vs. 5/12 (42%) in arm B (Fig. 4). There was no significant difference in the SFC/million PBMC at the pre-ATI time point between subjects in arm A and those in arm B. Although the sum of ELISPOT responses to HIV-1 antigens in subjects in arm A was higher than those in arm B at the pre-ATI time point (median 723 SFC/million PBMC vs. 255 SFC/million PBMC), this difference was not statistically significant (p = 0.27). There was also no significant difference in the fold-change from baseline to pre-ATI SFC between the two treatment arms.
Fig. 4.
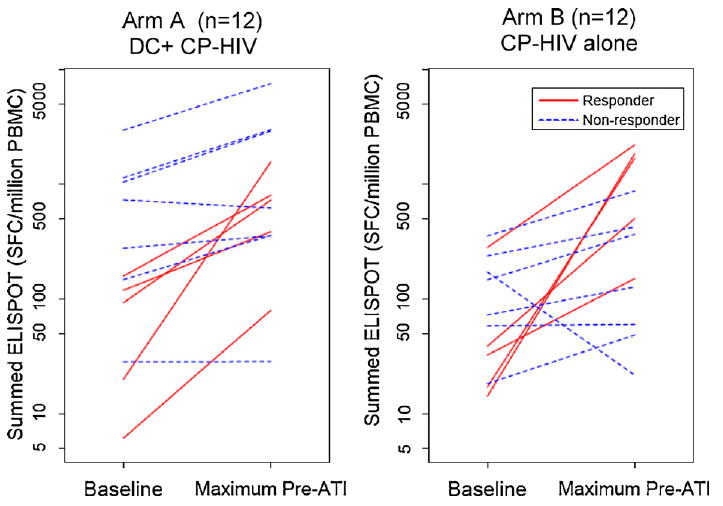
Summed ELISPOT responses to HIV-1 antigens in subjects who received DC + CP-HIV (group A) vs. subjects who received CP-HIV alone (group B). A responder is defined as a subject who has a ≥3-fold increase in summed ELISPOT response, that is also ≥30 spot-forming cells/106 peripheral blood mononuclear cells, to HIV-1 antigens from baseline to the maximum pre-ATI time point (i.e. maximum ELISPOT level at week 3, 7 or between weeks 16–19). ATI: analytic treatment interruption.
To evaluate the breadth of the immune response to different HIV-1 antigens, both before and after immunization, we examined whether the ELISPOT response to one HIV-1 vaccine antigen was associated with responses to other vaccine antigens. We found a strong correlation among the ELISPOT response (as measured by SFCs/million PBMCs) to Gag, Env, Pol and Nef at baseline (correlation coefficients 0.59–0.78, all p-values <0.005). The ELISPOT SFC responses to HIV-1 antigens after completion of vaccination were also highly correlated (correlation coefficients 0.62–0.82, all p-values <0.005); that is, subjects who responded to one HIV-1 antigen tended to also respond to other HIV-1 antigens. There was a strong correlation in the fold-change among ELISPOT responses to Gag, Env, Pol and Nef from baseline to the pre-ATI time point (correlation coefficients 0.58–0.80, all p-values <0.005). In contrast to the strong correlation among immune responses to HIV-1 antigens, there was not a significant association between the ELISPOT SFC responses to HIV-1 antigens and the ELISPOT SFC response to CMV at baseline or at the pre-ATI time point (correlation coefficients 0.12–0.41, all p-values >0.06).
3.4. Virologic control during ATI
Twenty-six subjects achieved a VL setpoint at the end of ATI (Fig. 2). The VL setpoint did not differ between subjects in arms A and B (medians 4.1 and 4.5 log10 c/mL, p = 0.20) (Table 1). In arm A, 4/13 subjects (31%) had a VL setpoint <5000 c/mL compared with 0/13 subjects in arm B (p = 0.096) (Fig. 5). If the last observed VL is carried forward for the three subjects who did not have an observed VL setpoint (because they either restarted therapy or went off-study), then 4/14 (29%) in arm A achieved VL setpoint <5000c/mL compared with 0/15 (0%) in arm B (p = 0.042) (Table 1). In the four subjects who achieved a VL setpoint <5000 c/mL at the end of ATI, the durability of viral control was limited: two of the four had a VL >5000c/mL on subsequent follow-up (7 weeks later).
Table 1.
Viral load (VL) setpoint at the end of analytic treatment interruption (ATI).
| Viral load setpoint | Arm | N | Median (Q1, Q3) (log10 copies/mL) | pa | Proportion of subjects with VL <5000 copies/mL | pb |
|---|---|---|---|---|---|---|
| Observed (as-treated) | A | 13 | 4.1 (3.5, 4.6) | 0.20 | 4/13 (31%) | 0.096 |
| B | 13 | 4.5 (4.1, 4.8) | 0/13 (0%) | |||
| Worst rankc (Intent-to-treat) | A | 14 | 4.3 (3.5, 4.9) | 0.17 | 4/14 (29%) | 0.042 |
| B | 15 | 4.6 (4.1, 5.2) | 0/15 (0%) | |||
| Carry forwardd (intent-to-treat) | A | 14 | 4.3 (3.5, 4.9) | 0.25 | 4/14 (29%) | 0.042 |
| B | 15 | 4.5 (4.1, 5.2) | 0/15 (0%) |
Wilcoxon Rank Sum test.
Fisher's exact test.
Assign the worst-rank of the primary endpoint to subjects who do not have observed VL endpoint.
Carry forward the last observed HIV-1 RNA value (at ATI weeks 4–6) during the ATI for three subjects who did not have an observed VL endpoint.
Fig. 5.
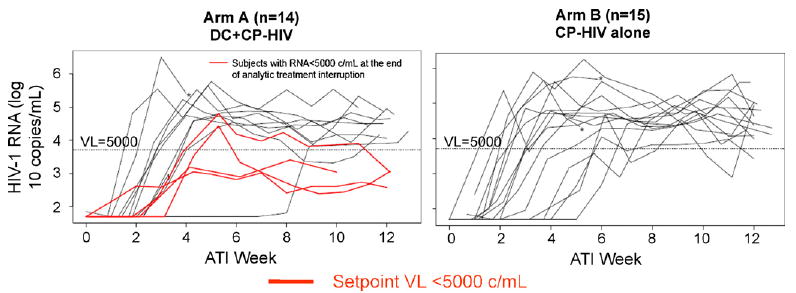
Viral load (VL) rebound during analytic treatment interruption. The horizontal dotted line represents a VL of 5000c/mL. The red lines (all in arm A) designate VL curves for subjects who had a setpoint VL<5000 c/mL. Three subjects restarted ART or went off-study prior to completion of the ATI (these subjects are designated with a “*” next to their VL curve). One subject resumed therapy after week 10 of the ATI.
There was no difference between subjects in arms A and B during ATI in the following measures of viral rebound: peak VL; lowest VL after peak VL; slope of initial VL rise; time to VL >50 c/mL; area under the curve log10 VL through week 12 of ATI. Subjects in arm A reached peak VL more quickly than subjects in arm B (median 36 days vs. 42 days, p = 0.027).
Of 10 subjects for whom there was data available on pre-ART VL, 7 had a lower VL setpoint at the end of ATI than their recorded pre-therapy VL. In these 10 subjects, the median (Q1, Q3) VL during ATI was 0.7 (0.2, 1.7) log10 c/mL lower than the pre-ART VL.
3.5. Immunologic response and VL setpoint
To assess whether there was evidence for immunologic control of viremia, we examined whether the immune response after vaccination was associated with VL setpoint at the end of ATI. Pooling arms, subjects with higher pre-ATI ELISPOT SFC response to HIV-1 Pol had a lower VL setpoint during ATI (r = −0.44, p = 0.046). Subjects with greater fold-increase in ELISPOT SFC responses to Gag and Nef after vaccination had a lower VL setpoint at the end of ATI (r = −0.50 and −0.54, p = 0.021 and 0.012, respectively). Subjects with a greater fold-change in summed ELISPOT SFC responses to HIV-1 antigens after vaccination had a lower VL setpoint at the end of ATI (r = −0.44, p = 0.047). Of note, there was no significant association between prevaccination ELISPOT SFC response to HIV-1 antigens and VL setpoint during ATI.
4. Discussion
In this randomized trial, we found that both CP-HIV and DC + CP-HIV vaccines are safe and immunogenic in HIV-1-infected patients. DCs improved LPA responses to the neoantigen KLH, suggesting that they are functioning as an adjuvant, but did not clearly enhance anti-viral CD8 ELISPOT responses when compared to CP-HIV alone. Although not reaching significance, there was a suggestion that subjects vaccinated with CP-HIV + DC had a higher rate of VL set-point <5000 c/mL during ATI than subjects immunized with CP-HIV alone; however, this control was transient. Subjects who had higher fold-increases in HIV-1-specific ELISPOT responses after vaccination had lower VL setpoints at the end of ATI. Enhancement of the response to one HIV-1 vaccine antigen after immunization was associated with boosting of responses to other vaccine antigens.
Although we could not show that adding DCs improved the immunogenicity of CP-HIV, we did find that greater fold-increase in HIV-1-specific ELISPOT responses after vaccination were associated with lower VL setpoint at the end of ATI. The ELISPOT response to HIV-1 antigens prior to vaccination was not associated with VL setpoint during ATI, suggesting that it is vaccine-induced immune responses that are important in lowering VL. However, because there was not a no-treatment control group in this study, we cannot be certain of the contribution of the vaccine interventions on control of viremia. Nevertheless, our findings suggest that inducing an immune response to HIV-1 through therapeutic vaccination may have a role in controlling viremia. Defining the association between functional immunologic measures and virologic control in preventive and therapeutic HIV-1 vaccine studies may lead to a better understanding of the mechanism by which the immune system contains HIV-1 replication.
Why did DCs not boost immunogenicity of CP-HIV in this trial? The fact that lymphoproliferative responses to KLH, a neoantigen, were increased in subjects randomized to group A indicates that the DCs were functioning effectively as a vaccine adjuvant. HIV-1-specific LPA responses were not increased after vaccination, perhaps because antigen was not presented effectively to CD4 cells by this vector or because subjects had clonal exhaustion or deletion of HIV-1-specific CD4 cells. The inability of DCs to improve HIV-1-specific ELISPOT responses, which predominantly measure virus-specific CD8 cells, may also be due to a defect in HIV-1-infected patients in CD4 cell help necessary for induction of CD8 cell responses [25] or may be due to inadequate antigen dose. These hypotheses could be tested by measuring immune responses to CP-HIV-pulsed DCs in healthy subjects and by testing different dosages of antigen-pulsed DCs.
What should we make of the slightly higher rate of transient control of viremia in the DC group? One possibility is that this finding is due to chance. Another possibility is that there are effects of DCs on immune function that are not measured by the standard ELISPOT assay, which only measures IFN-γ production [26]. We know that the function of CD8 cells - as measured by antigen-induced proliferation and production of other cytokines and mediators, for example – is more important than their absolute quantity in controlling HIV-1 [27]. Future studies will evaluate whether the immune responses elicited by DC combined with CP-HIV are of higher quality than those induced by CP-HIV alone.
This study had several limitations. First, because of the sample size, small effects of DCs on VL setpoint or ELISPOT response could not be excluded. Second, information on pre-ART VL was only available in a minority of subjects (10 of 29), in many cases because subjects had started ART prior to the era of VL measurement. Third, because the study was designed to evaluate whether addition of DC to a CP-HIV vaccine lowered VL setpoint during ATI, we did not include a no-treatment control arm. In a recent study, HIV-1-infected patients on ART who received CP-HIV had higher VL during ATI than subjects who received placebo [10]. In future trials designed to examine the ability of a therapeutic vaccine to control HIV-1 replication, a placebo arm is necessary.
Previous studies of DC vaccination in HIV-1-infected patients have used peptides, chemically or heat-inactivated HIV-1 as the vaccine immunogen [28–30]. Like the current study, these trials found DC vaccination to be safe and well tolerated. In a study of HIV-1-infected patients on ART, HIV-1 peptide-pulsed DCs led to an increased frequency of HIV-1-specific IFN-γ-positive cells; there was no ATI in this trial [28]. In a second study, 4 of 12 subjects who received DCs pulsed with heat-inactivated HIV-1 had VL setpoints during ATI that was >0.5 log10 c/mL lower than pre-ART VL, but the overall vaccine responses were weak [29]. Finally, in a provocative study, 8 of 18 HIV-1-infected subjects who were not taking ART had prolonged VL reduction after receiving DCs pulsed with chemically inactivated HIV-1 [30]. These studies demonstrate that DC vaccination is well tolerated, but the optimal approach in HIV-1-infected patients is not known.
How should the results of the current trial guide the development of future DC vaccine studies? Clearly, the immunogenicity of DC vaccines must be optimized. New antigen delivery methods – such as mRNA transfection of DCs [31,32] or linking antigens to antibody fragments that enhance delivery to DCs [33] – may result in more functional immune responses. Studies of new methods to improve antigen presentation – such as in situ activation of DCs or use of cytokine and Toll-like receptor agonists as adjuvants –are warranted.
It must be recognized that the correlates of control of viremia are imperfectly understood, as highlighted by the recent failure of a recombinant adenovirus type-5 HIV-1 vaccine to prevent acquisition of HIV-1 or lower VL in those who became infected, despite eliciting CD8 cell responses in HIV-1-negative subjects [34]. More precise understanding of how the body controls HIV-1, perhaps through in-depth evaluation of HIV-1 controllers or studies of genetic associations with virologic control, may define the types of immune responses we should aim to elicit through vaccination. The ability to induce those responses will depend on expanding our knowledge of how to manipulate the innate and adaptive immune system using DCs and other approaches. Studies of new interventions in pilot human studies and, eventually, randomized treatment interruption trials of the type reported here, will then be necessary to determine whether such approaches lead to immunologic control of HIV-1.
Acknowledgments
We would like to acknowledge the contributions of all the members of the ACTG A5130 team, including Jennifer Nowak, Ken Braun, Jessica Hass, Marla Dondero, Karen Regardie. We would like to acknowledge the outstanding work of the study staff at Massachusetts General Hospital, particularly Amy Sbrolla, Kathy Habeeb, Nicole Burgett, Gil Roy, Daniel Kavanagh, Doug Kwon and Bruce Walker, and at Beth Israel Hospital, New York, especially Gwen Constantini, Sondra Middleton, Donald Garmon, and Scott Barnett. We would like to acknowledge Crystal M. Cruz, Angelica Angiulli and Francesca Angiulli at the New York University Cancer Institute Vaccine Center for expert technical assistance with the ELISPOT assays. We also gratefully acknowledge Aurora Kiviat and Betsy Wonderly for assistance in preparing this manuscript. Finally we would like to thank the 11 study subjects in Boston and 18 subjects in New York who participated in this trial.
Footnotes
Part of the information in this manuscript was previously presented at the 17th International AIDS Conference, Mexico City, Mexico, 3–8 August 2008.
This study was supported by the National Institutes of Health (AIO69472, A146370, AI68636, AI68634, AI38858, AI38855, AI066992-04). This study was also supported by grants from the Burroughs Welcome Foundation (NB), Elizabeth Glaser Pediatric AIDS Foundation (NB), and the Doris Duke Distinguished Clinical Scientist Award (NB). The trial is registered with ClinicalTrials.gov (http://clinicaltrials.gov), identifier: NCT00026624.
Publisher's Disclaimer: This article appeared in a journal published by Elsevier. The attached copy is furnished to the author for internal non-commercial research and education use, including for instruction at the authors institution and sharing with colleagues.
Other uses, including reproduction and distribution, or selling or licensing copies, or posting to personal, institutional or third party websites are prohibited.
In most cases authors are permitted to post their version of the article (e.g. in Word or Tex form) to their personal website or institutional repository. Authors requiring further information regarding Elsevier's archiving and manuscript policies are encouraged to visit: http://www.elsevier.com/copyright
References
- 1.Walsh SRBN, Gandhi RT. Dendritic cells and the promise of therapeutic vaccines for human immunodeficiency virus (HIV)-1. Current HIV Reports. 2003:205–16. doi: 10.2174/1570162033485285. [DOI] [PubMed] [Google Scholar]
- 2.O'Neill DW, Bhardwaj N. Exploiting dendritic cells for active immunotherapy of cancer and chronic infections. Mol Biotechnol. 2007;36:131–41. doi: 10.1007/s12033-007-0020-6. [DOI] [PubMed] [Google Scholar]
- 3.Russell ND, Graham BS, Keefer MC, McElrath MJ, Self SG, Weinhold KJ, et al. Phase 2 study of an HIV-1 canarypox vaccine (vCP1452) alone and in combination with rgp120: negative results fail to trigger a phase 3 correlates trial. J Acquir Immune Defic Syndr. 2007;44:203–12. doi: 10.1097/01.qai.0000248356.48501.ff. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fleury B, Janvier G, Pialoux G, Buseyne F, Robertson MN, Tartaglia J, et al. Memory cytotoxic T lymphocyte responses in human immunodeficiency virus type 1 (HIV-1)-negative volunteers immunized with a recombinant canarypox expressing gp 160 of HIV-1 and boosted with a recombinant gp160. J Infect Dis. 1996;174:734–8. doi: 10.1093/infdis/174.4.734. [DOI] [PubMed] [Google Scholar]
- 5.Clements-Mann ML, Weinhold K, Matthews TJ, Graham BS, Gorse GJ, Keefer MC, et al. Immune responses to human immunodeficiency virus (HIV) type 1 induced by canarypox expressing HIV-1MN gp120, HIV-1SF2 recombinant gp120, or both vaccines in seronegative adults. NIAID AIDS Vaccine Evaluation Group. J Infect Dis. 1998;177:1230–46. doi: 10.1086/515288. [DOI] [PubMed] [Google Scholar]
- 6.Evans TG, Keefer MC, Weinhold KJ, Wolff M, Montefiori D, Gorse GJ, et al. A canarypox vaccine expressing multiple human immunodeficiency virus type 1 genes given alone or with rgp120 elicits broad and durable CD8+ cytotoxic T lymphocyte responses in seronegative volunteers. J Infect Dis. 1999;180:290–8. doi: 10.1086/314895. [DOI] [PubMed] [Google Scholar]
- 7.Jin X, Ramanathan M, Jr, Barsoum S, Deschenes GR, Ba L, Binley J, et al. Safety and immunogenicity of ALVAC vCP1452 and recombinant gp160 in newly human immunodeficiency virus type 1-infected patients treated with prolonged highly active antiretroviral therapy. J Virol. 2002;76:2206–16. doi: 10.1128/jvi.76.5.2206-2216.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jacobson JM, Pat Bucy R, Spritzler J, Saag MS, Eron JJ, Jr, Coombs RW, et al. Evidence that intermittent structured treatment interruption, but not immunization with ALVAC-HIV vCP1452, promotes host control of HIV replication: the results of AIDS Clinical Trials Group 5068. J Infect Dis. 2006;194:623–32. doi: 10.1086/506364. [DOI] [PubMed] [Google Scholar]
- 9.Kinloch-de Loes S, Hoen B, Smith DE, Autran B, Lampe FC, Phillips AN, et al. Impact of therapeutic immunization on HIV-1 viremia after discontinuation of antiretroviral therapy initiated during acute infection. J Infect Dis. 2005;192:607–17. doi: 10.1086/432002. [DOI] [PubMed] [Google Scholar]
- 10.Autran B, Murphy RL, Costagliola D, Tubiana R, Clotet B, Gatell J, et al. Greater viral rebound and reduced time to resume antiretroviral therapy after therapeutic immunization with the ALVAC-HIV vaccine (vCP1452) Aids. 2008;22:1313–22. doi: 10.1097/QAD.0b013e3282fdce94. [DOI] [PubMed] [Google Scholar]
- 11.Gandhi RT, Altfeld M. The quest for an HIV-1 therapeutic vaccine. J Infect Dis. 2005;192:556–9. doi: 10.1086/432014. [DOI] [PubMed] [Google Scholar]
- 12.Kilby JM, Bucy RP, Mildvan D, Fischl M, Santana-Bagur J, Lennox J, et al. A randomized, partially blinded phase 2 trial of antiretroviral therapy, HIV-specific immunizations, and interleukin-2 cycles to promote efficient control of viral replication (ACTG A5024) J Infect Dis. 2006;194:1672–6. doi: 10.1086/509508. [DOI] [PubMed] [Google Scholar]
- 13.Engelmayer J, Larsson M, Lee A, Lee M, Cox WI, Steinman RM, et al. Mature dendritic cells infected with canarypox virus elicit strong anti-human immunodeficiency virus CD8+ and CD4+ T-cell responses from chronically infected individuals. J Virol. 2001;75:2142–53. doi: 10.1128/JVI.75.5.2142-2153.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deeks SG, Walker BD. Human immunodeficiency virus controllers: mechanisms of durable virus control in the absence of antiretroviral therapy. Immunity. 2007;27:406–16. doi: 10.1016/j.immuni.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 15.Tartaglia J, Benson J, Cornet B, Cox W, El Habib R, Excler J, et al. Potential improvements for poxvirus-based immunization vehicles. In: Girard M, Dodet B, editors. Onzieme Colloque des Cent Grades. Paris, France: Pasteur Merieux, Marnes-La-Coquette; 1997. pp. 187–97. [Google Scholar]
- 16.Marovich MA, Mascola JR, Eller MA, Louder MK, Caudrelier PA, El-Habib R, et al. Preparation of clinical-grade recombinant canarypox-human immunodeficiency virus vaccine-loaded human dendritic cells. J Infect Dis. 2002;186:1242–52. doi: 10.1086/344302. [DOI] [PubMed] [Google Scholar]
- 17.Dhodapkar MV, Steinman RM, Sapp M, Desai H, Fossella C, Krasovsky J, et al. Rapid generation of broad T-cell immunity in humans after a single injection of mature dendritic cells. J Clin Invest. 1999;104:173–80. doi: 10.1172/JCI6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dhodapkar MV, Krasovsky J, Steinman RM, Bhardwaj N. Mature dendritic cells boost functionally superior CD8(+) T-cell in humans without foreign helper epitopes. J Clin Invest. 2000;105:R9–14. doi: 10.1172/JCI9051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Franchini G, Gurunathan S, Baglyos L, Plotkin S, Tartaglia J. Poxvirus-based vaccine candidates for HIV: two decades of experience with special emphasis on canarypox vectors. Expert Rev Vac. 2004;3:S75–88. doi: 10.1586/14760584.3.4.s75. [DOI] [PubMed] [Google Scholar]
- 20.Goepfert PA, Horton H, McElrath MJ, Gurunathan S, Ferrari G, Tomaras GD, et al. High-dose recombinant Canarypox vaccine expressing HIV-1 protein, in seronegative human subjects. J Infect Dis. 2005;192:1249–59. doi: 10.1086/432915. [DOI] [PubMed] [Google Scholar]
- 21.Coombs RW, Speck CE, Hughes JP, Lee W, Sampoleo R, Ross SO, et al. Association between culturable human immunodeficiency virus type 1 (HIV-1) in semen and HIV-1 RNA levels in semen and blood: evidence for compartmentalization of HIV-1 between semen and blood. J Infect Dis. 1998;177:320–30. doi: 10.1086/514213. [DOI] [PubMed] [Google Scholar]
- 22.Adams S, O'Neill DW, Nonaka D, Hardin E, Chiriboga L, Siu K, et al. Immunization of malignant melanoma patients with full-length NY-ESO-1 protein using TLR7 agonist imiquimod as vaccine adjuvant. J Immunol. 2008;181:776–84. doi: 10.4049/jimmunol.181.1.776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lederman MM, Connick E, Landay A, Kuritzkes DR, Spritzler J, St Clair M, et al. Immunologic responses associated with 12 weeks of combination antiretroviral therapy consisting of zidovudine, lamivudine, and ritonavir: results of AIDS Clinical Trials Group Protocol 315. J Infect Dis. 1998;178:70–9. doi: 10.1086/515591. [DOI] [PubMed] [Google Scholar]
- 24.Yusim K, Kesmir C, Gaschen B, Addo MM, Altfeld M, Brunak S, et al. Clustering patterns of cytotoxic T-lymphocyte epitopes in human immunodeficiency virus type 1 (HIV-1) proteins reveal imprints of immune evasion on HIV-1 global variation. J Virol. 2002;76:8757–68. doi: 10.1128/JVI.76.17.8757-8768.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lichterfeld M, Kaufmann DE, Yu XG, Mui SK, Addo MM, Johnston MN, et al. Loss of HIV-1-specific CD8+ T cell proliferation after acute HIV-1 infection and restoration by vaccine-induced HIV-1-specific CD4+ T cells. J Exp Med. 2004;200:701–12. doi: 10.1084/jem.20041270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Precopio ML, Betts MR, Parrino J, Price DA, Gostick E, Ambrozak DR, et al. Immunization with vaccinia virus induces polyfunctional and phenotypically distinctive CD8(+) T cell responses. J Exp Med. 2007;204:1405–16. doi: 10.1084/jem.20062363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Betts MR, Nason MC, West SM, De Rosa SC, Migueles SA, Abraham J, et al. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood. 2006;107:4781–9. doi: 10.1182/blood-2005-12-4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Connolly NC, Whiteside TL, Wilson C, Kondragunta V, Rinaldo CR, Riddler SA. Therapeutic immunization with human immunodeficiency virus type1 (HIV-1) peptide-loaded dendritic cells is safe and induces immunogenicity in HIV-1-infected individuals. Clin Vaccine Immunol. 2008;15:284–92. doi: 10.1128/CVI.00221-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garcia F, Lejeune M, Climent N, Gil C, Alcami J, Morente V, et al. Therapeutic immunization with dendritic cells loaded with heat-inactivated autologous HIV-1 in patients with chronic HIV-1 infection. J Infect Dis. 2005;191:1680–5. doi: 10.1086/429340. [DOI] [PubMed] [Google Scholar]
- 30.Lu W, Arraes LC, Ferreira WT, Andrieu JM. Therapeutic dendritic-cell vaccine for chronic HIV-1 infection. Nat Med. 2004;10:1359–65. doi: 10.1038/nm1147. [DOI] [PubMed] [Google Scholar]
- 31.Kavanagh DG, Kaufmann DE, Sunderji S, Frahm N, Le Gall S, Boczkowski D, et al. Expansion of HIV-specific CD4+ and CD8+ T cells by dendritic cells transfected with mRNA encoding cytoplasm- or lysosome-targeted Nef. Blood. 2006;107:1963–9. doi: 10.1182/blood-2005-04-1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Melhem NM, Liu XD, Boczkowski D, Gilboa E, Barratt-Boyes SM. Robust CD4+ and CD8+ T cell responses to SIV using mRNA-transfected DC expressing autologous viral Ag. Eur J Immunol. 2007;37:2164–73. doi: 10.1002/eji.200636782. [DOI] [PubMed] [Google Scholar]
- 33.Nchinda G, Kuroiwa J, Oks M, Trumpfheller C, Park CG, Huang Y, et al. The efficacy of DNA vaccination is enhanced in mice by targeting the encoded protein to dendritic cells. J Clin Invest. 2008;118:1427–36. doi: 10.1172/JCI34224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Watkins DI, Burton DR, Kallas EG, Moore JP, Koff WC. Nonhuman primate models and the failure of the Merck HIV-1 vaccine in humans. Nat Med. 2008;14:617–21. doi: 10.1038/nm.f.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]