

Abstract
Background
Human Papillomavirus (HPV) infection is considered a necessary step for the development of cervical cancer and >95% of all cervical cancers have detectable HPV sequences. We have recently demonstrated the efficacy of radioimmunotherapy (RIT) which targeted viral oncoprotein E6 in treatment of experimental cervical cancer We hypothesized that pre-treatment of tumor cells with various agents which cause cell death and/or elevation of E6 levels would increase the accumulation of radiolabeled antibodies to E6 in cervical tumors.
Methods
HPV-16 positive CasKi cells were treated in vitro with up to 6 Gy of external radiation, or proteasome inhibitor MG-132 or unlabeled anti-E6 antibody C1P5 and cell death was assessed. Biodistribution of 188Rhenium (188Re)-labeled C1P5 antibody was performed in both control and radiation MG-132 treated CasKi tumor-bearing nude mice.
Results
. 188Re-C1P5 antibody demonstrated tumor specificity and very low uptake and fast clearance from the major organs. The amount of tumor uptake was enhanced by MG-132 but was unaffected by pre-treatment with radiation. In addition, in vitro studies demonstrated an unanticipated effect of unlabeled antibody on the amount of cell death, a finding that was suggested by our previous in vivo studies in CasKi tumor model.
Conclusion
We demonstrated that pre-treatment of cervical tumors with proteasome inhibitor MG-132 and with unlabeled antibody to E6 can serve as a means to generate non-viable cancer cells and to elevate the levels of target oncoproteins in the cells for increasing the accumulation of targeted radiolabeled antibodies in tumors. These results favor further development of RIT of cervical cancers targeting viral antigens.
Keywords: Cervical cancer, viral antigens, radiolabeled antibodies, 188-Rhenium, chemotherapy
Introduction
More than 95% of all cervical cancers are associated with and caused by the Human papillomavirus (HPV) (1, 2), a discovery that led to Dr. zur Hausen receiving a Nobel Prize in 2008. Though HPV vaccination is now FDA approved, there are issues with implementation, access and changes in cervical cancer screening as a result of vaccination. Additionally, current vaccines are effective for preventing only HPV16 and 18 incident infection with little benefit in those women already infected with HPV16 or 18 or those infected with any other oncogenic HPV types. Increasing the potency of DNA vaccines is still among the most important challenges for DNA vaccine development (3). The impact of prophylactic vaccination on the incidence of the disease has yet to be determined while millions of women remain at risk for cervical carcinoma worldwide. HPV strains utilize viral oncoproteins E6 and E7 to immortalize epithelial cells in culture and increase cellular transformation in concert with other oncoproteins (4–6). The E6 and E7 oncoproteins are located intracellularly and bind to p53, promoting its rapid degradation via the ubiquitin-dependent pathway, while E7 oncoprotein binds to retinoblastoma (RB), thus causing ineffective cell growth regulation. By minimizing effects of tumor suppressor genes p53 and RB, more random mutations can occur, which can potentially lead to malignant transformation. Thus, E6 and E7 oncoproteins appear to be logical targets for targeted novel therapies for cervical cancer.
Radioimmunotherapy (RIT) is used experimentally for the treatment of various malignancies (7), and two radiolabeled antibodies have been approved for treatment of recurrent or refractory non-Hodgkin lymphoma (NHL). In a previous report, we demonstrated the feasibility of targeting E6 and E7 oncoproteins in experimental cervical cancer by using radiolabeled antibodies as selective mediators of tumor destruction (8). The distinctive features of this approach are: 1) the viral origin of target oncoproteins as opposed to “self” human antigens used in prior RIT approaches which obviates targeting host tissues, and 2) intracellular and, in fact, the intranuclear location of E6 and E7 oncoproteins. Targeting of intranuclear antigens is possible because degenerating and necrotic cells release their intranuclear contents and exhibit abnormal surface membrane permeability that allow reactivity of antibody with intracellular antigen -characteristics not found in normal cells. Thus, degenerating cells provide target material given that intracellular proteins dissipate from the damaged cell membrane and attracts the radiolabeled antibody, which further mediates destruction of viable tumor cells through long range beta emission of a radionuclide such as 188-Rhenium (188Re).
Clearly, the success this strategy will depend on the amount of target oncoproteins and their accessibility for binding antibody. Higher levels of target proteins and more non-viable cells releasing such protein would result in increased uptake of the radiolabeled antibody in the tumor. We investigated the use of external radiation, proteasome inhibitor MG-132 and pre-treatment with unlabeled antibody to E6 as distinct means to generate non-viable cancer cells and to elevate the levels of target oncoproteins in the cells for increasing the accumulation of radiolabeled antibodies in cervical cancer in nude mice.
Materials and Methods
Cell line, antibodies and reagents
CasSki cell line was obtained from American Type Culture Collection (Manassas, VA). Cells were grown in RPMI-1640 medium containing 10% FBS (Sigma) and 1% Penicillin-streptomycin solution (Sigma, penicillin 10,000 U and streptomycin 10mg/ml) at 37°C in a 5% CO2 incubator. This cell line was derived from an HPV-16 positive human cervical cancer that expresses both E6 and E7 oncogenic proteins. A murine antibody C1P5 (IgG1) to HPV-16 E6 + HPV-18 E6 was procured from Abcam; human-mouse chimeric antibody ch-TNT3 (IgG1) directed against a universal nuclear antigen was a gift from Dr. Alan Epstein (University of Southern California School of Medicine, Los Angeles, CA). Proteasome inhibitor MG-132 was obtained from Calbiochem; BD Matrigel™ Basement Membrane Matrix – from BD Biosciences.
Tumor model
All animal studies were carried out in accordance with the guidelines of the Institute for Animal Studies at the Albert Einstein College of Medicine. Thirty six-week-old athymic Nu/Nu CD1 nude mice, purchased from Charles River Laboratories, were randomized into groups of 5 mice and 107 cells were injected subcutaneous into the right flank of each mouse. For experimental development of a tumor model the cells were also mixed before inoculation with combinations of RPMI medium, FBS, Matrigel and MG-132. Mice in Group 1 were inoculated only with 107 cells; for other groups mice were injected with 107 cells mixed with: 3μg/ml MG-132 - Group 2; 80% FBS - Group 3; 80% Matrigel–Group 4; 3μg/ml MG-132 and 80% FBS - Group 5; and 3μg/ml MG-132 and 80% Matrigel - Group 6. Tumor volume was calculated using length × width × height and divided by 2. The biodistribution experiments were carried out when the tumors volume reached an average of 1 cm3.
Antibodies radiolabeling
The beta-emitter 188Re with a half life of 16.9 hours was produced from beta decay of 188-Tungsten parent (half life 69 days) using a 188W/188Re generator (Oak Ridge National Laboratory, Oak Ridge, TN). After 188Re was eluted in the form of sodium perrhenate, the antibodies were labeled with 188Re directly through binding of reduced 188Re to the generated –SH groups on the antibodies as previously described (8).
Biodistribution of 188Re-labeled C1P5 and ch-TNT3 mAbs in non-tumor bearing mice
C1P5 mAb to E6 and control mAb ch-TNT3 were radiolabeled with 188Re as described above with the specific activity of 1 μCi/μg and 20 μCi 188Re-C1P5 or 188Re-ch-TNT3 were administered to non-tumor bearing nude mice via intraperitoneal injection (IP). At 6 and 24 hrs, 8 mice (4 mice per antibody group) were sacrificed, blood and major organs were collected at necropsy, weighed and counted in a LKB 1282 Compugamma Universal Gamma Counter. Injected dose per gram tissue expressed as percentage (% ID/g) was calculated according to the equation:
where cpm are counts per minute.
Determination of effects of radiation on CasKi cells
For in vitro study CaSki cells were cultivated in a 24-well plate to a total volume of 1×105 cells/ml per well with RPMI medium supplemented with 10% FBS and 1% Penicillin/Streptomycin and allowed to grow at 37°C in 5% CO2 incubator overnight. The medium in each well was removed, and the cells were washed with PBS three times. Five hundred μl fresh PBS was added and irradiation with the doses of 0, 2, 4 and 6 Gy was performed on Philips Orthovoltage MGC 40 Unit operated at 320 kVp, 5.0 mA at 1.8 focal setting. After irradiation, PBS was removed and fresh medium was added, and the cells were placed back in 5% CO2 incubator and grown at 37°C, collected at different time points and cell viability was determined by the Trypan Blue dye exclusion method by using a counting chamber (Neubauer hemocytomer) under the light microscope (Nikon Eclipse TS100).
For in vivo study, the tumor-bearing mice were immobilized using a cylindrical lead shield that allowed the tumors to be exposed sparing surrounding areas. Philips Orthovoltage MGC 40 Unit operated at the identical to in vitro experiments settings was used to administer 6 Gy of radiation. Three days after irradiation the mice were injected IP with 20 μCi 188Re-C1P5 mAb and the bio-distribution studies were performed 24 hrs later as described above.
Determination of effects of MG-132 proteasome inhibitor on CasKi cells
In our previous in vitro study we established using Western blot technique that treatment of CasKi cells with 2.5 – 10μg/mL MG-132 for 3–6 hrs significantly elevated the levels of target oncoprotein E6 in these cells (8). In this study we investigated whether the same treatment also made cells nonviable. For this purpose, the cells were grown in 24-well plates as described above, MG-132 solution was then added at 0, 1, 2, 5, 10 or 25 μg/ml. The cells were incubated under the same conditions for 3 or 6 hours and the cell viability was determined by the Trypan Blue dye exclusion method.
For in vivo investigation of the influence of MG-132 on the tumor uptake of 188Re-C1P5 mAb, a group of 4 mice carrying the CaSki tumors was injected IP with 20 μg MG-132 and another group of 4 tumor-bearing mice was used as control. Three hours later, all mice in both groups were injected IP with 20 μCi 188Re-C1P5 mAb and 24 hrs later the biodistribution was performed as above.
Determination of effects on unlabeled C1P5 antibody on CasKi cells
Previously we observed a pronounced effect of unlabeled C1P5 antibody on slowing down the growth CasKi tumors in nude mice (8). Here we performed in vitro experiments aimed at establishing whether unlabeled C1P5 causes cell death. CasKi cells were grown in 24-well plates as above and then treated with 0.013, 0.025 or 0.40μg/mL of C1P5 antibody or with the same concentration of the control mAb ch-TNT3. One 24-well plate was untreated and another one was treated with 5 μL of lysis buffer as per the lactate dehydrogenase (LDH) kit guidelines at each time point. Culture supernatant was collected at 1 – 30 hrs and frozen. The culture supernatant was then thawed and cells were removed by centrifugation, at 250 g for 10 min. The 100 μL of the provided reaction mixture was then added to the cell-free supernatant and incubated while being protected from light for approximately 40 min at room temperature. Controls were prepared as per the LDH Roche Cytotoxicity Detection Kit instructions. The absorbance at 492 nm was then measured using an ELISA reader (Roche Applied Science).
Statistical analysis
Non-parametric Wilcoxon Rank Sum test was used to compare organ and tumor uptake in the bio-distribution studies. The differences were considered statistically significant when p-values were < 0.05.
Results
Matrigel promoted the growth of CaSki tumors
We chose the HPV-16 positive CaSki cell line because it reliably expresses both E6 and E7 oncoproteins in vitro and in vivo (8). However, in our experience it took approximately 60 days from the inoculation of mice with CasKi to tumors reaching 3–5 mm in diameter (8). Consequently, we sought means to shorten this time by identifying experimental approaches that increased the tumorigenicity of the CasKi cells. The use of Matrigel, a solubilized tissue basement membrane matrix significantly enhances the tumorigenicity of a wide variety of cancer cell lines in vivo, namely breast, ovarian, endometrioid, lung, prostate, glioblastoma as well as reports of enhancing growth of primary breast material from patients (9), but its effect on cervical carcinoma lines has not been studied. Therefore we elected to test the effect of combinations of Matrigel, as well as RPMI medium, Fetal Bovine Serum (FBS) and MG-132, on the tumorigenicity of CaSki cells.
Matrigel promoted the formation of CaSki tumors (Fig 1a). Groups 4 and 6 each had 100% tumor growth with the effect of MG-132 demonstrating 100% growth at 14 days compared to day 21 in Matrigel and medium alone. However, the overall tumor size was larger in Group 4, with no MG-132 (Fig 1b). FBS alone (Group 5) seemed to inhibit tumor growth over time, as evidenced by almost total regression of tumor during the observation period. It is possible that in combination with MG-132, some inhibitory effects of FBS may be overcome, however, the overall tumor size (Fig. 1b) was not significantly enhanced.
Figure 1.

Influence of various tumor growth promoters on growth of CasKi tumors in nude mice. Matrigel promoted the growth of CasKi tumors more than other agents. a) percentage of mice with tumors; b) average tumor size, cm3 in various groups. Mice in Group 1 were inoculated only with 107 CasKi cells; for other groups mice were injected with 107 cells mixed with: 3μg/ml MG-132 - Group 2; 80% FBS - Group 3; 80% Matrigel – Group 4; 3μg/ml MG-132 and 80% FBS - Group 5; and 3μg/ml MG-132 and 80% Matrigel - Group 6.
Given that the Matrigel effects on the promotion of tumor growth were so pronounced - we performed a follow-up study of Matrigel inoculation alone into the flanks of nude mice to see if Matrigel was able to recruit murine epithelial cells contributing to tumorigenesis. However, no resultant tumor was noted (results not shown) strongly suggesting that Matrigel alone does not cause tumor formation. In fact it serves as a potent stimulator of tumor growth in the nude mouse cervical cancer model. Consequently, in all these studies we initiated CasKi tumors in nude mice using 107 CaSki cells mixed with Matrigel.
Antibodies to intracellular antigens cleared rapidly from tissues
Given our aim to investigate the in vitro effects of unlabeled mAb to E6 C1P5 on CasKi tumors cells, we needed to ascertain that ch-TNT3 mAb (TNT -Tumor Necrosis Treatment) mAb intended as a control for this and future RIT studies has similar C1P5 clearance patterns in normal organs. The mAbs were radiolabeled with 188Re and their clearance was studied at 6 and 24 hrs. Both mAbs demonstrated rapid clearance from the blood, liver, spleen, kidneys, stomach and muscle with 188Re-C1P5 clearing faster at 6 hrs especially from the kidneys and 188Re-ch-TNT3 – at 24 hrs (Fig. 2). We concluded that ch-TNT-3 had fast clearance due to low cross-reactivity with surface antigens and thus was a compatible control for intranuclear targeting.
Figure 2.
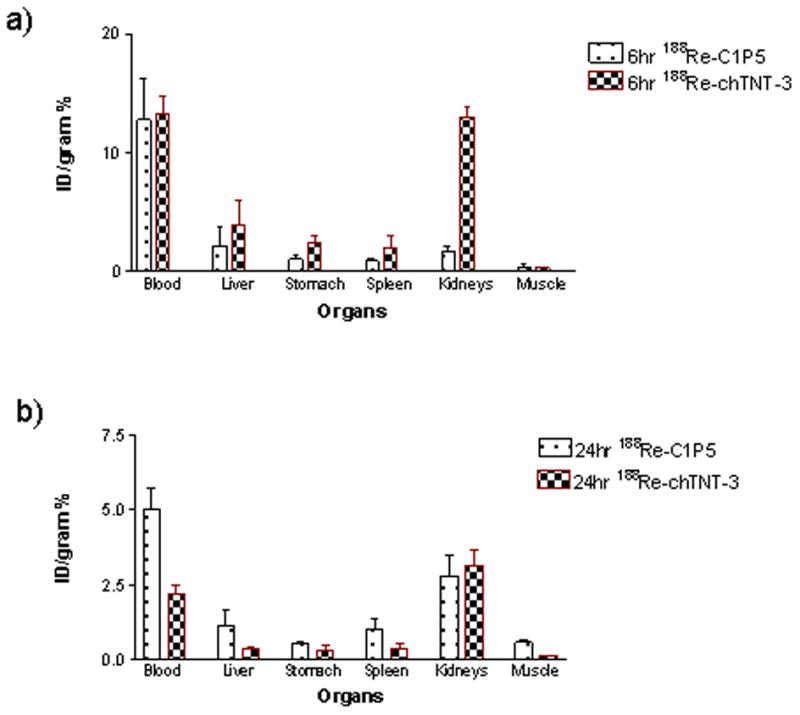
Biodistribution of anti-E6 188Re-C1P5 mAb and control 188Re-ch-TNT3 mAbs in non-tumor bearing nude mice: a) 6 hr post injection; b) 24 hr post injection. 188Re-C1P5 mAb cleared faster at 6 hrs than control 188Re-ch-TNT3 and displays low uptake in normal tissues.
External radiation did not increase the uptake of 188Re-C1P5 in CaSki tumors while MG-132 caused significant increase in tumor uptake
The results of in vitro study of cell viability after external radiation using doses of 0 – 6 Gy demonstrated that the highest number of cells with permeable membranes was produced by 6 Gy 72 hrs post-radiation (Fig. 3a). Therefore, we exposed CasKi tumors in mice to 6 Gy of radiation and administered 188Re-C1P5 3 days later. No increase in the tumor uptake post-radiation was found; in fact, there was a trend (p=0.055) towards a decrease in uptake (Fig. 3b).
Figure 3.
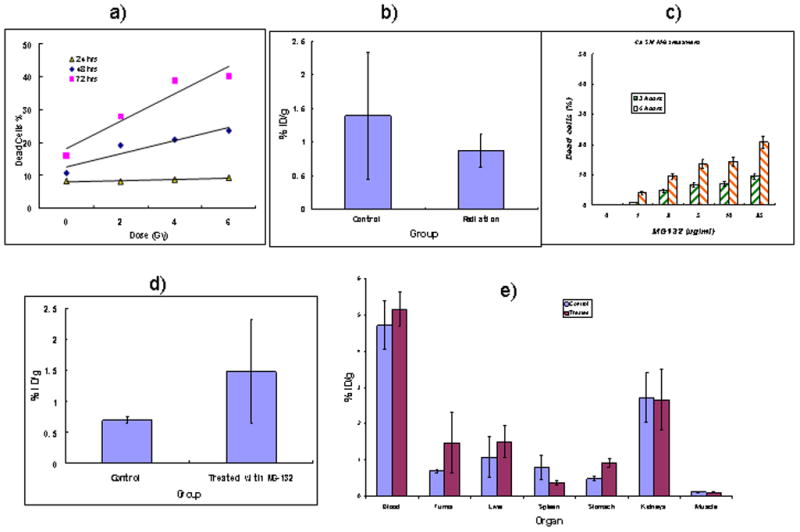
Influence of external radiation and proteasome inhibitor MG-132 on CasKi cell death in vitro and on tumor uptake and biodistribution of 188Re-C1P5 mAb in vivo: a) number of dead cells post radiation exposure; b) 24 hr uptake of 188Re-C1P5 mAb in CasKi tumors in mice exposed to 6 Gy external radiation and given 188Re-C1P5 mAb 3 days later; c) number of dead cells post MG-132 treatment; d) 24 hr uptake of 188Re-C1P5 mAb in CasKi tumors in mice treated with 20 μg MG-132 radiation and given 188Re-C1P5 mAb 3 hrs later; e) organ uptake of 188Re-C1P5 mAb in untreated and MG-132-treated CasKi tumor bearing mice. Pre-treatment of tumors with MG132 promotes the uptake of 188Re-C1P5 mAb while external radiation does not.
Treatment of CasKi cells in vitro with various concentrations of proteasome inhibitor MG-132 made approximately 8% of cells non-viable after 3 hrs (Fig. 3c). Though 6 hr treatment produced more dead cells (Fig. 3c), however, the elevation of E6 expression in pre-treated cells was shown to be the highest at 3 hrs with significant decline in expression at 6 hrs (8). Based on these data a 3 hr time point was chosen and CasKi tumor-bearing mice were injected IP with 20 μg MG-132, so the blood concentration would be 10 μg/ml, assuming a mouse blood volume of 2 mL and administered 188Re-C1P5 3 hrs later. Treatment with MG-132 resulted in more than a 2-fold increase in tumor uptake (P=0.03) (Fig. 3d). Importantly, no significant elevation in 188Re-C1P5 mAb uptake was observed in normal tissue (Fig. 3e).
Unlabeled C1P5 mAb was cytotoxic to CasKi cells in vitro
In our previous report (8), we noted inhibition of tumor growth in the nude mouse model when 30 μg (15 μg/ml blood) of unlabeled (“cold”) E6 antibody was administered. Here we investigated the cytotoxic range of C1P5 concentrations by measuring LDH released in the supernatant of treated cells as an indirect measure of cell death. The highest concentration of 0.4 μg/ml emulated approximately 2% ID/gram uptake of antibody in the tumor observed in our biodistribution experiments. Cytotoxicity was concentration dependent. There was no effect at 0.013 and 0.025 μg/ml C1P5 while concentration of 0.4 μg/ml had pronounced effects on cell death (Fig. 4a). When the same concentrations of control mAb ch-TNT-3 were applied – the significant lysis of cells was observed already at 0.025 μg/ml with 0.4 μg/ml causing an amount of cellular death comparable to that from the C1P5 treatment (Fig. 4b).
Figure 4.
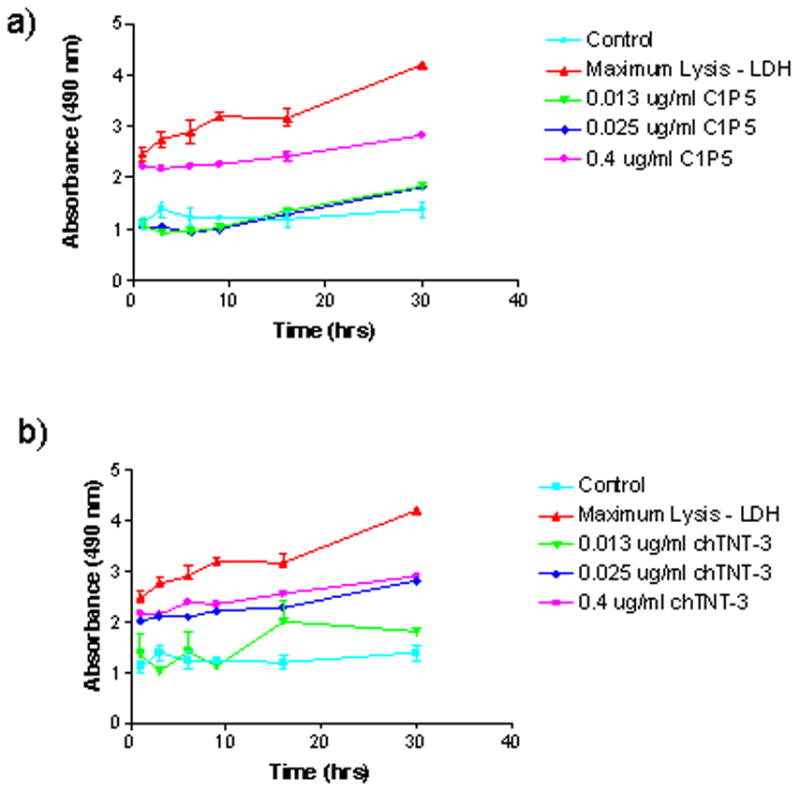
Results of LDH assay of CasKi cells treated with various concentrations of: a) anti-E6 C1P5 mAb; b) control ch-TNT3 mAb. Control–no antibody was added to the cells; max lysis the cells were treated with lysis buffer. Treatment with unlabeled anti-E6 C1P5 mAb causes cell death in CasKi cells.
Discussion
Despite the major advances in early detection via the cytological screening with aggressive management of pre-cancerous lesions and now a preventative type-specific HPV vaccine, cervical cancer remains the second leading cause of cancer death among women worldwide. For those women who have limited access to and poor medical care, cervical cancer is a significant cause of morbidity and mortality. Furthermore, the impact of surgery and the efficacy of radiation and chemotherapy for locally advanced cervical carcinoma is limited and novel, well-tolerated, therapeutic options are urgently needed.
Monoclonal antibodies have clinical efficacy in the treatment of some advanced cancers (7). Progress has been made in the application of monoclonal antibodies in treatment of gynecologic malignancies, such as ovarian cancer, however the success of these therapies has been limited (10). Currently, there are no clinical trials of monoclonal antibody-directed treatment of patients with cervical cancer.
We recently reported that RIT targeting viral antigens could be used in the treatment of a broad range of virus-associated tumors, such as cervical cancer (11, 12). Many virus-associated cancer cells express viral antigens either on their surfaces or intracellularly. Intracellular viral antigens are also potential targets for RIT, since tumor cell turnover is likely to result in the release of these proteins into tumor interstitial spaces. It is important to emphasize that when treating virally-associated cancers by targeting viral antigen - not every cell in the tumor needs to express viral antigens for a therapeutic effect. Long range emitters such as 188Re (emission range in tissue 10 mm) emit radiation in a 360° sphere and consequently can kill viable tumor cells in the vicinity of the antigen location via the so called “cross-fire” effect. As a therapeutic radionuclide 188Re has other attractive features such as a lack of accumulation in the bone marrow and rapid clearance through the kidneys (13, 14).
Our study has demonstrated that antibodies targeting viral antigens, which are also intracellular, have very low cross-reactivity with the normal tissues. The ability of such antibodies to bind to their respective targets retained by the non-viable cells would result in the increased residence times of the radiolabeled antibodies in the tumor. The clearance of 188Re-C1P5 from organs in nude mice was more pronounced at 6 hrs than that of control 188Re-ch-TNT3. The latter binds to intracellular universal nuclear antigen (15) and clears significantly faster from the tissues when compared to 188Re-labeled IgGs to membrane-associated antigens (16). Such low cross-reactivity with normal tissues of mAbs to viral antigens can potentially translate into low toxicity with RIT against viral targets.
To increase the amount and accessibility of target oncoproteins in the tumors we investigated the pre-treatment of tumors with external radiation, proteasome inhibitor MG-132 and unlabeled mAb C1P5. Exposure to external radiation did not increase the uptake of 188Re-C1P5 in tumors, possibly because of timing issues, vascular damage which would prevent good circulation of antibodies in the tumor or inability of external radiation to damage nuclear membrane to release intracellular E6 oncoprotein. In contrast, pre-treatment with proteasome inhibitor MG-132 increased the tumor uptake of 188Re-C1P5 > 2-fold. In this setting, the proteasome inhibitor MG-132 reduces the degradation of ubiquitin-conjugated proteins in mammalian cells without affecting ATPase or isopeptidase activities. MG-132 has been reported to result in increased levels of E6 and E7 proteins in cervical cancer cells (17, 18). It is possible that pre-treatment of tumor-bearing mice with a “classical” chemotherapeutic agent such as cisplatin would also result in killing some of the tumor cells and lead to increased accessibility of target E6 protein. We have recently shown that pre-treatment of experimental melanoma tumors with dacarbazine (DTIC) rendered some tumor cells non-viable which made intracellular melanin pigment more accessible for the radiolabeled melanin-binding antibody (19). In our previous study (8) we have demonstrated in vitro that MG132 treatment also increased expression of E7 oncoprotein in other human cervical cancer cell lines such as HeLa and SiHa and we intend to perform experiments in mice bearing HeLa and SiHa tumors in the future.
Interestingly, exposure of CasKi cells to unlabeled C1P5 mAb at concentrations of 0.4 μg/mL which would be achieved in the tumor at approximately 2% ID/gram uptake (30 μg injected per mouse) caused significant death of the cells according to the results of the LDH assay and may explain the pronounced effect of unlabeled C1P5 on CasKi tumors in mice in our previous study (8). It is also possible that the cytotoxic effect of C1P5 on the cells contributed to the overall effect of RIT on CasKi tumors in mice (8) by increasing the number of non-viable cells in the tumor. The mechanism of this cytotoxicity is unknown and remains in need of further exploration.
In summary, our data demonstrated that pre-treatment of cervical cancers with the proteasome inhibitor MG-132 and with unlabeled antibody to E6 generated increased cell death of non-viable cancer cells. This resulted in elevated levels of target oncoproteins in the cells and increasing the accumulation of radiolabeled antibodies in these cervical cancers in mice. These data will contribute to further development of RIT of cervical cancers by targeting viral antigens and add a potential therapeutic option for patients with this disease.
Acknowledgments
Sources of support: E. Dadachova is Sylvia and Robert S. Olnick Scholar in Cancer Research and this research was supported by the Mary Kay Ash Foundation grant, Einstein NIH-designated Cancer Center and NIH grant AI60507. A. Casadevall is supported by the NIH grants AI33774 and AI33142.
Footnotes
Financial disclosure: PCT International Patent Application PCT/US2007/025491: E. Dadachova, A. Casadevall, H. Strickler and R.D. Burk Radioimmunotherapy and imaging of tumor cells that express viral antigens
References
- 1.Walboomers JM, Jacobs MV, Manos MM, Bosch FX, Kummer JA, Shah KV, Snijders PJ, Peto J, Meijer CJ, Munoz N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol. 1999;189:12–19. doi: 10.1002/(SICI)1096-9896(199909)189:1<12::AID-PATH431>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 2.Bosch FX, Manos MM, Munoz N, Sherman M, Jansen AM, Peto J, Schiffman MH, Moreno V, Kurman R, Shah KV. Prevalence of human papillomavirus in cervical cancer: a worldwide perspective. International biological study on cervical cancer (IBSCC) Study Group. J Natl Cancer Inst. 1995;7(87):796–802. doi: 10.1093/jnci/87.11.796. [DOI] [PubMed] [Google Scholar]
- 3.Yan J, Reichenbach DK, Corbitt N, Hokey DA, Ramanathan MP, McKinney KA, Weiner DB, Sewell D. Induction of antitumor immunity in vivo following delivery of a novel HPV-16 DNA vaccine encoding an E6/E7 fusion antigen. Vaccine. 2009;14:431–440. doi: 10.1016/j.vaccine.2008.10.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaur P, McDougall JK, Cone R. Immortalization of primary human epithelial cells coned cervical carcinoma DNA containing human papillomavirus type E6/E7 open reading frames. J Gen Virol. 1989;70:1261–1266. doi: 10.1099/0022-1317-70-5-1261. [DOI] [PubMed] [Google Scholar]
- 5.Kaur P, McDougall JK. HPV-18 immortalization of human keratinocytes. Virology. 1989;173:302–310. doi: 10.1016/0042-6822(89)90247-x. [DOI] [PubMed] [Google Scholar]
- 6.Percoraro G, Morgan D, Defendi V. Differential effects of human papillomavirus type 6,16, and 18 DNAs on immortalization and transformation of human cervical epithelial cells. Proc Natl Acad Sci USA. 1989;86:563–567. doi: 10.1073/pnas.86.2.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharkey RM, Goldenberg DM. Targeted therapy of cancer: new prospects for antibodies and immunoconjugates. CA Cancer J Clin. 2006;56:226–243. doi: 10.3322/canjclin.56.4.226. [DOI] [PubMed] [Google Scholar]
- 8.Wang XG, Revskaya E, Bryan RA, Strickler HD, Burk RD, Casadevall A, Dadachova E. Treating cancer as an infectious disease–viral antigens as novel targets for treatment and potential prevention of tumors of viral etiology. PLoS ONE. 2007;2:e114. doi: 10.1371/journal.pone.0001114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.The use of Matrigel to Facilitate the Establishment of Human Cancer Cell Lines as Xenografts. S.P. Langdon Human Press Inc; Totawa, NJ: Methods in Molecular Medicine, vol. 88: Cancer Cell Culture: Methods and Protocols- Ch 27. [Google Scholar]
- 10.Balkwill F, Schlom J, Berek J, et al. Discussion: immunological therapeutics in ovarian cancer. Gynecol Oncol. 2003;88:S110–S113. doi: 10.1006/gyno.2002.6695. [DOI] [PubMed] [Google Scholar]
- 11.Casadevall A, Goldstein H, Dadachova E. Targeting host cells harbouring viruses with radiolabeled antibodies. Expert Opin Biol Ther. 2007;7:595–597. doi: 10.1517/14712598.7.5.595. [DOI] [PubMed] [Google Scholar]
- 12.Dadachova E, Wang XG, Casadevall A. Targeting the virus with radioimmunotherapy in virus-associated cancer. Cancer Biother Radiopharm. 2007;22:303–308. doi: 10.1089/cbr.2007.344. [DOI] [PubMed] [Google Scholar]
- 13.Hayes RL, Rafter J. Research Report. Medical Division, Oak Ridge Associated Universities; Tennessee. ORAU 101: 1965. Rhenium-188 as a possible diagnostic agent; pp. 74–77. [Google Scholar]
- 14.Iznaga-Escobar N. 188 Re-Direct labeling of monoclonal antibodies for radioimmuno-therapy of solid tumors: biodistribution, normal organ dosimetry, and toxicology. Nucl Med Biol. 1998;25:441–447. doi: 10.1016/s0969-8051(98)00008-0. [DOI] [PubMed] [Google Scholar]
- 15.Epstein AL, Khawli LA, Chen F-M, Hu P, Glasky MS, Taylor CR. Tumor necrosis imaging and treatment of solid tumors. In: Torchilin VP, editor. Handbook of Targeted Delivery of Imaging Agents. Vol. 4. Boca Raton, FL: CRC Press; 1995. p. 151. [Google Scholar]
- 16.Sharkey RM, Blumenthal RD, Behr TM, Wong GY, Haywood L, et al. Selection of radioimmunoconjugates for the therapy of well-established or micrometastatic colon carcinoma. Int J Cancer. 1997;72:477–485. doi: 10.1002/(sici)1097-0215(19970729)72:3<477::aid-ijc16>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 17.Kehmeier E, Ruhl H, Voland B, Stoppler MC, Androphy E, Stoppler H. Cellular steady-state levels of “high risk” but not “low risk” human papillomavirus (HPV) E6 proteins are increased by inhibition of proteasome dependent degradation independent of their p53- and E6AP-binding capabilities. Virology. 2002;299:72–87. doi: 10.1006/viro.2002.1502. [DOI] [PubMed] [Google Scholar]
- 18.Oh KJ, Kalinina A, Wang J, Nakayama K, Nakayama KI, Bagchi S. The papillomavirus E7 oncoprotein is ubiquitinated by UbcH7 and Cullin 1- and Skp2-containing E3 ligase. J Virol. 2004;78:5338–5346. doi: 10.1128/JVI.78.10.5338-5346.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Revskaya E, Jongco AM, Sellers RS, Howell RC, Koba W, Guimaraes AJ, Nosanchuk JD, Casadevall A, Dadachova E. Radioimmunotherapy of experimental human metastatic melanoma with melanin-binding antibodies and in combination with dacarbazine. Clin Cancer Res. 2009;15:2373–2379. doi: 10.1158/1078-0432.CCR-08-2376. [DOI] [PubMed] [Google Scholar]