

Summary
Early noninfectious pulmonary complications represent a significant cause of mortality after hematopoietic cell transplantation (HCT). We tested the hypothesis that oral beclomethasone dipropionate (BDP) is effective for preventing early noninfectious pulmonary complications after allogeneic HCT. We retrospectively reviewed medical records of 120 patients, 60 in each treatment arm. to identify noninfectious and infectious pulmonary events and prospectively collected pulmonary function test results from all patients who participated in two randomized trials of oral BDP for treatment of acute gastrointestinal graft-versus-host disease. 17-beclomethasone monopropionate (17-BMP), the active metabolite of BDP, was evaluated in blood from the right atrium. Among placebo treated patients, 33 of 42 (79%) patients experienced a decrease of the DLCO from pretransplant to day 80 after transplant, compared to 27 of 49 (55%) BDP-treated patients (p=0.02). In the first 200 days after randomization, there were no cases of noninfectious pulmonary complications in BDP-treated patients, versus four cases among placebo-treated patients (p=0.04). Levels of 17-BMP were detected in atrial blood at steady state. Delivery of a potent glucocorticoid such as 17-BMP to the pulmonary artery after oral dosing of BDP may be useful in modulating pulmonary inflammation and preventing the development of noninfectious pulmonary complications after allogeneic HCT.
Keywords: idiopathic pneumonia syndrome, oral beclomethasone dipropionate, allogeneic hematopoietic cell transplantation
Pulmonary complications after allogeneic hematopoietic cell transplantation (HCT) remain a major cause of morbidity and mortality. Among the estimated 50,000 to 60,000 hematopoietic cell transplants performed each year (www.ibmtr.org), approximately 30% to 60% of the transplant recipients will experience a pulmonary complication.1,2 While significant progress has been made in the prevention, diagnosis, and treatment of opportunistic pulmonary infections, particularly infections caused by viral and fungal infections, no advances have been made in the prevention, diagnosis, and treatment of HCT-related early noninfectious pulmonary syndromes such as idiopathic pneumonia syndrome (IPS) and diffuse alveolar hemorrhage (DAH). These syndromes represent diffuse interstitial noninfectious pulmonary processes that are suspected to be caused by a spectrum of insults, including the toxic effects of conditioning regimens, immunologic cell-mediated injury, inflammatory cytokines, or occult pulmonary infections.3–14 When these syndromes occur, they are associated with a high mortality rate of 60–100%, despite aggressive support in a critical care setting and treatment with high-dose systemic corticosteroids.
Two multi-center randomized placebo-controlled, double-blind trials have been conducted to examine whether oral beclomethasone dipropionate (BDP) is an effective therapy for treatment of gastrointestinal graft-versus-host disease (GVHD).15,16 The results of these studies indicate that oral BDP is effective for treating gastrointestinal GVHD, allows rapid taper and less use of systemic corticosteroid therapy, and results in a 45% reduction in mortality risk at one year post-randomization. In addition to these benefits, the most recent multi-center trial revealed that BDP treatment was also associated with fewer cytomegalovirus reactivations (28% versus 39%), fungal infections, (7% versus 14%), and multiple bacteremic episodes (none versus 9%).16 Of interest to the pulmonary community was an observation in the phase II study, 12/67 (18%) patients on the placebo arm had developed noninfectious pulmonary infiltrative disorders, compared to 0/62 patients in the BDP treatment arm.17 Based upon this initial observation, we developed the hypothesis that oral BDP may be effective in preserving lung function and preventing the development of early noninfectious pulmonary complications after allogeneic HCT. However, recognizing the potential for misclassification among different centers, we decided to test this hypothesis by evaluating all patients who participated in both randomized trials conducted at our Center. The current analysis was restricted to patients treated at our Center because all patients at our Center were evaluated using standardized prospective pulmonary evaluations and a clinical evaluation algorithm in place during both randomized trials. In addition, our Center has near complete follow-up of all clinical events after discharge from our Center through the Long Term Follow-up Clinic.15
Methods
Patient selection
All Fred Hutchinson Cancer Research Center (FHCRC, also referred to as Center) patients enrolled in two randomized placebo-controlled trials of oral BDP for the treatment of acute gastrointestinal GVHD were included in the current analysis.15,16 The first study was a single-center, Phase 2 trial conducted between 1994 and 1996 that enrolled 60 patients; 29 received oral BDP 8 mg/day, 31 received placebo.15 The second study was a multi-center Phase 3 trial conducted between 2001 and 2005 that enrolled a total of 129 patients, including 60 patients at the FHCRC, of whom 31 patients received oral BDP and 29 patients received placebo.16 Patients enrolled at other institutions were not included in this analysis because of the lack of pulmonary function monitoring and potential differences in clinical definitions of pulmonary syndromes (see below). Both protocols and the current retrospective review were approved by our Institutional Review Organization.
Description of Randomized, Placebo-Controlled, Double-Blind Trials of Oral BDP
Details of the two randomized trials have been previously described.15,16 Briefly, patients with biopsy-proven acute gastrointestinal GVHD received an induction course of prednisone (1 mg/kg/day for 10 days) plus either oral BDP pills (8 mg/d in four divided doses, half as a gastric-release formulation and half as an enteric-coated formulation) or placebo. Patients whose symptoms were controlled at study day ten continued on study drug while the prednisone dose was rapidly tapered. Patients whose symptoms required additional prednisone were considered treatment failures; study drug was discontinued on the day of treatment failure. The duration of treatment with study drug was 30 days with a 10-day follow-up period in the first trial15 and 50 days with a 30-day follow-up period in the second trial.16 Based upon recommendations from the U.S. Food & Drug Administration, all patients in the Phase III study were followed for clinical events until day 200 after randomization. At our Center, detailed information about patient outcomes during the first year after transplant is gathered by our Long-Term Follow-Up office. Although the day 200 post-randomization endpoint had not been pre-specified in the protocol for the Phase II study of oral BDP vs. placebo, all patients in the Phase II study were also followed through our Long Term Follow-Up office using identical clinical protocols.
Pulmonary function testing
All PFTs were performed at our Center according to American Thoracic Society guidelines.18–20 Pulmonary function assessments included forced vital capacity (FVC), one-second forced expiratory volume (FEV1), total lung capacity (TLC), and carbon monoxide diffusion capacity (DLCO), which was adjusted for hemoglobin level at the time PFTs were obtained. All PFT values were expressed as a percent of the predicted value calculated according to published equations.21,22 As part of our standard practice, pretransplantation and day-80 PFTs were obtained regardless of the presence or absence of symptoms. For purposes of the current study, we considered PFTs performed between days 60 and 100 as valid for the day-80 PFT. Change in lung function was assessed by comparing PFT parameters performed prior to start of conditioning to those obtained post-transplantation at day 80. Analysis of change in lung function was done on an intent-to-treat basis, by randomization assignment to oral BDP or placebo, with maintenance of the study blind.
Clinical follow-up and categorization of pulmonary disease after hematopoietic cell transplantation
All patients in both trials were monitored according to the Standard Practice Manual of the FHCRC. While at our Center, this included daily evaluation for pulmonary symptoms, the presence of which would prompt physiologic and radiographic evaluation, followed by pulmonary and infectious diseases subspecialty consultation when deemed appropriate. Microbiologic studies are pursued according to recommendations from these services. Upon discharge from our Center, the patients are followed by the Long Term Follow-Up office, which remains in close contact with the patient and their healthcare provider for the first year after transplant. Should pulmonary issues arise, recommendations are made according to our Standard Practice Manual. All recommendations and clinical events are documented in our electronic records, which were reviewed for case designation.
While blinded to randomization assignment, two investigators (JWC and MS) reviewed all patients’ pulmonary radiologic records accumulated from the time of randomization. The medical records of all patients with abnormal pulmonary radiological findings were reviewed to determine whether the pulmonary disease was a clinically significant noninfectious or infectious syndrome. Clinically significant noninfectious pulmonary syndromes included idiopathic pneumonia syndrome (IPS), defined as widespread alveolar injury in the absence of active lower respiratory tract infection after HCT23; diffuse alveolar hemorrhage (DAH), defined as IPS with bronchioalveolar lavage showing progressively bloodier return5; or biopsy-proven cryptogenic organizing pneumonia (COP) otherwise known as bronchiolitis obliterans organizing pneumonia.(BOOP) 24,25 All clinically significant pulmonary infections required microbiologic documentation of an infectious agent in the respiratory tract via bronchoscopy or resolution of pulmonary abnormalities after empirical antibiotic therapy with no addition of immunosuppressive agents.
Measurement of 17-beclomethasone monopropionate (17-BMP) in blood from the right atrium
Four FHCRC patients enrolled in the Phase 3 randomized study had participated in an analysis of BDP pharmacokinetics. At study day 50, blood was drawn from the right atrium via an indwelling Hickman catheter at frequent intervals after a morning dose of oral BDP 2 mg (1 mg each in gastric-release and enteric-coated tablets). Blood samples were collected over EDTA, plasma was collected after centrifugation at 4°C, and aliquots were frozen at −20°C until analysis for BDP and 17-BMP using high-pressure liquid chromatography and mass spectroscopy (MDS Pharma Services, Montreal, Canada). Pharmacokinetic parameters were calculated using WinNonlin v.2.1 (Pharsight Corp, Palo Alto CA). Plasma concentration-time data were plotted and non-compartmental parameters were calculated using the linear trapezoidal rule. Estimates for half-lives were obtained using regression analysis by specifying an appropriate range of time points for the most linear portions of the log concentration-linear time data.
Statistical analysis
All statistical analyses were performed using SAS (version 8.2; SAS Institute, Cary, NC), R (freeware; version 2.0.1), and S-Plus (version 6.2; Insightful, Seattle, WA) software. P-values ≤ 0.05 were considered statistically significant. Change in pulmonary function test parameters was analyzed by comparing the proportion of patients in the treatment groups who experienced a decrease of each PFT parameter from pretransplantation to day 80 using the chi-square test. Average change from baseline to day 80 was compared using the two-sample t-test. Cumulative incidence curves were used to summarize the probability of the time-to-event endpoints noninfectious and infectious pulmonary events, where death without the respective pulmonary event was considered a competing risk. For these endpoints, the hazard ratio (HR) for failure was estimated based on a Cox proportional hazards model that included a term for treatment group. No additional covariates were included in the model for noninfectious complications due to the low number of events in the study. Patients alive without failure for the pulmonary time-to-event endpoints at day 200 after randomization were censored at these times as appropriate.
Results
There were 120 randomized patients from our Center available for this analysis, 60 received oral BDP and 60 received placebo. The characteristics of these patients are summarized in Table 1. In the analysis of change in PFTs from baseline to day 80, eight patients who did not meet the minimum five-day criterion of treatment with study drug prior to their day-80 PFT were not included (five in placebo group, three in BDP group). The median time to randomization was 33.5 days after HCT (range, 19–105) for the Phase 2 study and 31 days (range, 16–89) for the Phase 3 study.
Table 1.
Characteristics of BDP and placebo treated patients
| Characteristics | Placebo (N=60) | BDP (N=60) |
|---|---|---|
| Median age (range) | 47 (12–66) | 40 (7–67) |
| Recipient: donor sex | ||
| M:M | 17 | 24 |
| M:F | 11 | 12 |
| F:F | 12 | 9 |
| F:M | 20 | 15 |
| Disease | ||
| CML | 13 | 15 |
| AML | 16 | 19 |
| MDS | 17 | 3 |
| ALL | 8 | 8 |
| NHL | 2 | 8 |
| Other | 4 | 7 |
| Donor type | ||
| Unrelated | 24 | 20 |
| Related matched | 31 | 32 |
| Related mismatched | 3 | 7 |
| Conditioning regimen | ||
| TBI-based myeloablative | 28 | 31 |
| Non-TBI-based myeloablative | 28 | 18 |
| Reduced intensity | 4 | 11 |
| Randomized study | ||
| Phase II | 29 | 31 |
| Phase III | 31 | 29 |
| Pulmonary function pretransplant * | ||
| FVC | 95.1 | 93.7 |
| FEV1 | 89.9 | 90.4 |
| TLC | 98.6 | 95.7 |
| DLCO | 90.9 | 83.6 |
| Pulmonary function at day 80 * | ||
| FVC | 93.0 | 93.8 |
| FEV1 | 89.2 | 90.3 |
| TLC | 96.7 | 96.9 |
| DLCO | 81.2 | 86.0 |
M=male; F=female; CML=chronic myeloid leukemia; AML=acute myeloid leukemia; MDS=myelodysplastic syndrome; ALL=acute lymphocytic leukemia; NHL=non-Hodgkins lymphoma; TBI=total body irradiation, FVC=forced vital capacity, FEV1=one second forced expiratory volume, TLC=total lung capacity, DLCO=diffusion capacity of carbon monoxide
Numbers represent the mean of the percent of predicted normal values;
Several patients were missing baseline and day-80, as indicated by the total number of patients contributing to the appropriate analyses summarized below and in Table 2. Overall, the majority of the patients had normal pulmonary function prior to transplantation, defined as a percent of the normal predicted value ≥ 80% [FVC 94/114 (82%); FEV1 88/114 (77%); TLC 105/113 (93%); DLCO 84/114 (74%)].
Table 2.
Change in pulmonary function from pretransplant to day 80
| PFT parameter | Placebo | BDP | p-value |
|---|---|---|---|
| FEV1 | |||
| N (%)* | 21/44 (48%) | 25/50 (50%) | 0.83 |
| Mean change (range) | −1.38 (−25, +19) | −0.05 (−22, +34) | 0.48 |
| FVC | |||
| N (%)* | 24/44 (55%) | 25/50 (50%) | 0.66 |
| Mean change (range) | −1.85 (−20, +16) | +0.34 (−30, +35) | 0.26 |
| TLC | |||
| N (%)* | 25/42 (60%) | 29/50 (58%) | 0.88 |
| Mean change (range) | −1.67 (−20, +23) | +1.41 (−25, +57) | 0.23 |
| DLCO | |||
| N (%)* | 33/42 (79%) | 27/49 (55%) | 0.02 |
| Mean change (range) | −7.95 (−40, +23) | +0.57 (−74, +115) | 0.08 |
FVC=forced vital capacity, FEV1=one second forced expiratory volume, TLC=total lung capacity, DLCO=diffusion capacity of carbon monoxide
N represents proportion of patients who experienced a decrease in that parameter
Changes in pulmonary function from pretransplantation to day 80 after transplantation are summarized in Table 2. The proportion of BDP- and placebo-treated patients who experienced a decrease (of any magnitude) of their pulmonary function from pretransplantation to day 80 after transplantation was similar for FVC, FEV1, and TLC, with no statistically significant differences. There was, however, a statistically significant difference in the proportion of patients who experienced a decrease of the DLCO (33 of 42 (79%) among placebo-treated patients, 27 of 49 (55%) in the BDP group, p = 0.02). Among placebo-treated patients the mean DLCO change was −7.95% (median, −7.17%; range − 40% to +24%), while the mean change from baseline to day 80 was +0.57% for BDP-treated patients (median, −0.61%; range −74% to +115%), the p-value did not exceed the 0.05 threshold (p = 0.08). These results were not qualitatively changed after adjusting for intensity of conditioning (reduced intensity vs. myeloablative) (p = 0.009 for proportion experiencing a decrease in DLCO decrease; p = 0.05 for the mean DLCO decrease) or after adjusting for age (p = 0.07 for proportion experiencing a decrease in DLCO decrease; p = 0.02 for the mean DLCO decrease).
Among 60 placebo-treated patients, four noninfectious complications occurred within the first 200 days after randomization (Figure 1). These cases were COP/BOOP (at day 39) and IPS (at days 69, 148, and 168). All of these cases occurred after a myeloablative conditioning regimen. Among 60 BDP-treated patients, there were no cases of noninfectious pulmonary complications during the first 200 days after randomization (Figure 1). When considered as a time-to-event endpoint, the risk of developing a noninfectious complication within the first 200 days after randomization was reduced among BDP-treated patients, (p = 0.04). With only 4 events, it was not possible to adjust for any factors such as intensity of conditioning or age.
Figure 1.
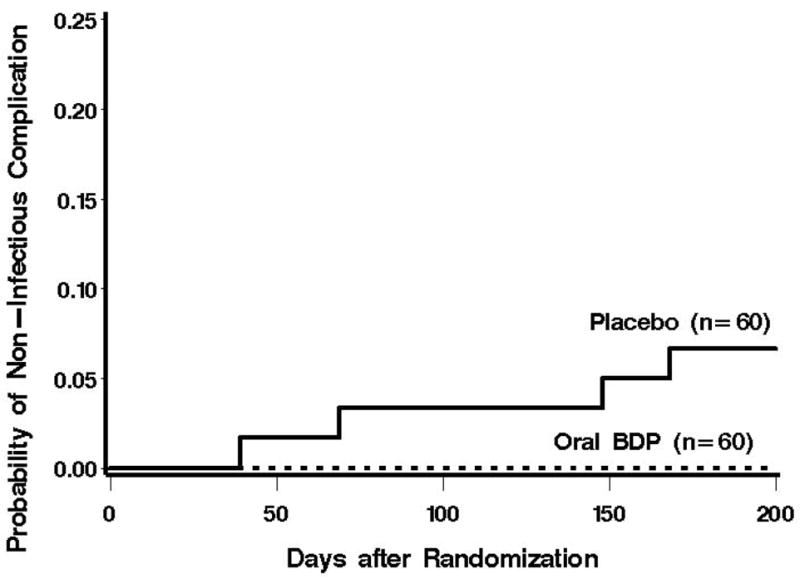
Cumulative incidence of non-infectious pulmonary complications after treatment randomization. Prior to day 200 after treatment randomization, no cases of non-infectious pulmonary complications occurred among the BDP-treated patients.
The causes of pulmonary infections are summarized in Table 3. Among 60 placebo-treated patients, 5 cases of pulmonary infection occurred within the first 200 days after randomization (Figure 2). Among 60 BDP-treated patients, 6 cases of pulmonary infection occurred during the first 200 days after randomization (Figure 2). When considered as a time-to-event endpoint, the risk of developing a pulmonary infection within 200 days following randomization was not statistically significantly different between BDP and placebo groups (HR = 1.21 (0.37–3.96, p = 0.75)).
Table 3.
Causes of pulmonary infection according to treatment group
| Placebo | Oral BDP | ||
|---|---|---|---|
| Cause of infection | Day post-randomization | Cause of infection | Day post-randomization |
| Fungal, nonspecific | 41 | Legionella spp. | 27 |
| Aspergillus fumigatus | 59 | Polymicrobial bacteria | 31 |
| Unknown cause | 125 | Pseudomonas aeruginosa | 48 |
| Aspergillus spp, | 142 | Candida glabrata | 83 |
| Unknown cause | 143 | Unknown cause | 85 |
| Unknown cause | 104 | ||
Figure 2.
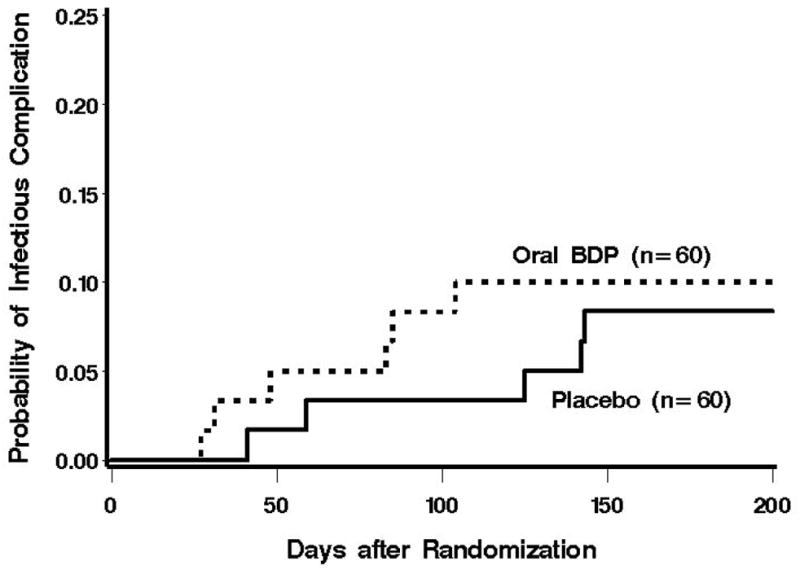
Cumulative incidence of pulmonary infections after treatment randomization. There was not a statistically significant difference in the risk of pulmonary infections between the two treatment groups.
In four patients, peak concentrations of 17-BMP in right atrial blood were achieved at a median of 1.5 hours after oral dosing of BDP 2 mg on the morning of study day 50. The median Cmax was 1738 pg/mL (range 632–3701). Median steady state exposure to 17-BMP was 5347 pg.hr/mL (range 3273–5201) as estimated by AUC0–4 hr. The median half-life of 17-BMP was 6.3 hours (range 3.1–6.8). No BDP was detected in right atrial blood.
Discussion
The current results suggest that in the context of the original randomized, placebo-controlled trials of oral BDP, patients randomized to receive oral BDP were more likely to experience a preservation of pulmonary diffusing capacity and fewer episodes of noninfectious pulmonary complications compared to placebo-treated patients. These results are consistent with what is known about the pathophysiology of lung function loss and idiopathic lung injury in the aftermath of allogeneic HCT and the pharmacology of oral BDP and its potent metabolite, 17-BMP.
Our finding that DLCO decrease appears to be less pronounced among the BDP-treated patients is interesting. The DLCO measures the patient’s ability to absorb alveolar gases into the capillary blood flow, reflecting alveolar membrane thickness, hematocrit level, cardiac output and heterogeneity in the distribution of the diffusion capacity to regional ventilation and perfusion (in patients with pulmonary disease).26,27 It is well known that systemic inflammation, such as that which can occur in the early post-transplant period, can result in pulmonary inflammation, which ultimately can cause thickening of the pulmonary interstitium and decreased DLCO. In such a setting, direct delivery of BDP to the pulmonary interstitium via the pulmonary artery may serve to reduce inflammation within the lungs, thereby minimizing the DLCO reduction commonly observed after allogeneic HCT.
According to a National Institutes of Health workshop, IPS is loosely defined as “evidence of widespread alveolar injury in the absence of active lower respiratory tract infection” 3. In the modern era of hematopoietic cell transplantation, early HCT-related noninfectious pulmonary complications such as IPS generally occur within the first six months after transplantation,24 with a reported incidence of 3 to 15%.28–30 There is an accumulating body of evidence from murine models of lung injury after allogeneic HCT that strongly suggests that inflammation plays a significant role in the pathogenesis of IPS. Some of these murine studies have established a causal role for TNF-α in the development of IPS, where administration of a TNF-α binding protein (rhTNFR:Fc) reduces the progression of lung injury during the four-to-six-week period after HCT.12,31–33 Other studies have also suggested that lipopolysaccharide (LPS), which often gains access to systemic circulation early in the post-transplantation period by translocating across gut mucosa damaged by conditioning regimens and acute GVHD,34–36 may result in a significant inflammatory cytokine milieu in the lung that results in lung injury.9,35 Collectively, these data provide strong evidence that overwhelming inflammation within the lungs likely plays a significant role in the pathogenesis of early noninfectious pulmonary complications after HCT. Direct delivery of a potent corticosteroid such as oral BDP to the lungs via the pulmonary circulation may attenuate the inflammation and prevent the development of IPS. An alternative biologic explanation for the positive effects of oral BDP on these pulmonary endpoints is that less gut GVHD reduces gut translocation of bacteria and endotoxin, and thus, reduces pulmonary exposure to translocated LPS and inflammatory cytokines. However, we feel that this mechanism is less likely than that of pulmonary delivery of the potent metabolite 17-BMP because the primary site of 17-BMP activity in patients enrolled in these protocols was the upper gut, not the colon, which is where Gram-negative bacteria primarily reside and LPS translocation occurs.
Oral formulations of BDP were developed for treatment of inflammatory bowel disease, where they proved to be effective.37–39 However, while inhaled BDP has been used as inhaled formulations for the treatment of asthma and chronic inflammatory airway disease for over 35 years,40 oral BDP does not seem to have been evaluated for the treatment of pulmonary diseases, perhaps because of the assumed adverse effects associated with systemic corticosteroid therapy. The formulations of oral BDP used in the randomized trials described here included a gastric-release pill for distribution of BDP to the stomach and upper small intestine, and an enteric-coated pill for distribution to the distal small intestine and colon. Presence of 17-BMP in the blood from the right heart, as demonstrated by our data, suggests that 17-BMP, a product of gastrointestinal mucosal hydrolysis of BDP, was likely absorbed in the gastrointestinal tract and entered the right heart via portal venous circulation. In the HCT situation, we speculate that steady-state delivery of 17-BMP to the pulmonary circulation and ultimately the interstitial space prior to the onset of clinically detectable disease was responsible for reducing pulmonary inflammation, reflected by the preservation of pulmonary diffusing capacity and by the absence of noninfectious pulmonary infiltrates within the first 200 days after randomization
Oral BDP also has several advantages over other corticosteroid options from a pharmacodynamics perspective. First, although BDP itself is a relatively weak immunosuppressive glucocorticoid, its active metabolite 17-BMP, is a highly potent glucocorticoid;17-BMP is 3.6-times more potent than triamcinolone-16,17-acetonide, the active ingredient in Azmacort inhalation aerosol (Kos Pharmaceuticals) and 450-times more potent than dexamethasone.41 Second, because 17-BMP’s glucocorticoid receptor-α activity is 13-times more potent than dexamethasone,42 it can be administered at significantly lower doses. Third, due to the pulmonary “first pass” effect, higher 17-BMP concentration can be achieved in the lung with relatively small doses. Small doses of 17-BMP can be delivered to the pulmonary artery in milligram quantities per day after ingestion of 8 mg oral BDP, which is four- to eight-fold higher than can be delivered via inhaled BDP. Finally, the relative bioavailability of BDP and its metabolites is much lower in comparison to more commonly used systemic corticosteroids. In a bioavailability study of oral BDP, no BDP was detectable in the plasma following oral administration. Although the total oral bioavailability of the active metabolite 17-BMP was 21–41%,43 systemic exposure to 17-BMP is limited by its protein binding and clearance, such that oral BDP 2 mg would give systemic exposure equivalent to a dosing schedule of oral prednisone of 2.5 mg or less than 1 mg of intravenous dexamethasone (DOR BioPharma, Inc., unpublished data). These factors, in conjunction with the fact that distribution of 17-BMP into the pulmonary interstitial space is likely considerably greater with pulmonary artery delivery than the inhaled route, makes oral BDP worthy of further investigation for pulmonary diseases rooted in any inflammatory pathophysiology.
Several limitations should be recognized when interpreting the results of this study. First, since the original trials were neither powered nor designed to address pulmonary issues, they are not optimal for examining the true potential of oral BDP as a prophylaxis therapy for noninfectious pulmonary complications. Such a study would require the intervention to be initiated at the time of transplant and probably with a longer duration of treatment, not to mention a substantially larger number of patients. Second, the size of the current study is limiting, not only with respect to statistical power, but also because we were not able to adjust for important factors that may influence the risk for developing noninfectious complications. For instance, despite the randomized design, more patients in the oral BDP arm of the phase III study received a reduced-intensity conditioning regimen, which has been previously found to be associated with a lower incidence of IPS.24 However, even based upon the conservative estimates among reduced intensity regimen recipients, it would be expected that IPS should have been observed in at least 2.2% of the placebo treated group (if not 8%).24 Finally, while the DLCO results are interesting, the DLCO data need to be interpreted with caution. The clinical implication of small percentage changes in DLCO, such as that we have detected, are not readily obvious, especially in a transplant setting, where there are many clinical variables that can influence the DLCO.
In summary, the current data suggest that oral BDP treatment was associated with a lower incidence of noninfectious pulmonary complications and that delivery of a potent glucocorticoid (17-BMP) to the pulmonary artery may be useful in modulating pulmonary inflammation after allogeneic HCT. We emphasize that we consider these data to be hypothesis generating due to the limitations cited above. As such, these findings are not sufficient to justify the use of oral BDP to prevent noninfectious pulmonary complications, but should guide future studies designed to definitively address the potential of BDP to lower the risk of noninfectious pulmonary complications and assess the potential adverse events associated with prolonged oral BDP use.
Acknowledgments
The randomized placebo-controlled trials on which these data are based were supported by Orphan Products Development Grants from the U.S. Food and Drug Administration (FD-R 000827 and FD-R 02599) and by Enteron Pharmaceuticals, Inc., a wholly owned subsidiary of DOR BioPharma, Ewing, NJ. This research was supported by grants from the National Institutes of Health, National Cancer Institute (CA18029 and CA15704).
Footnotes
Authors’ Responsibilities and Contributions: Jason Chien analyzed pulmonary function tests, reviewed clinical pulmonary events, and participated in the analysis of data and preparation of the manuscript. Miwa Sakai reviewed patient medical records for pulmonary events and participated in data analysis and preparation of the manuscript. Ted Gooley performed statistical analyses and participated in preparation of the manuscript. H. Gary Schoch provided clinical data from a computerized patient database. George McDonald initiated the research and participated in study design, analysis, and preparation of the manuscript.
Disclosure: George B. McDonald is a consultant to DOR BioPharma Inc. and has an equity position in the company.
Reference List
- 1.Cordonnier C, Bernaudin J-F, Bierling P, Huet Y, Vernant J-P. Pulmonary complications occurring after allogeneic bone marrow transplantation. A study of 130 consecutive transplanted patients. Cancer. 1986;58:1047–1054. doi: 10.1002/1097-0142(19860901)58:5<1047::aid-cncr2820580512>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 2.Jules-Elysee K, Stover DE, Yahalom J, White DA, Gulati SC. Pulmonary complications in lymphoma patients treated with high-dose therapy autologous bone marrow transplantation. Am Rev Respir Dis. 1992;146:485–491. doi: 10.1164/ajrccm/146.2.485. [DOI] [PubMed] [Google Scholar]
- 3.Clark JG, Hansen JA, Hertz MI, Parkman R, Jensen L, Peavy HH. NHLBI workshop summary: idiopathic pneumonia syndrome after bone marrow transplantation [Review] Am Rev Respir Dis. 1993;147:1601–1606. doi: 10.1164/ajrccm/147.6_Pt_1.1601. [DOI] [PubMed] [Google Scholar]
- 4.Shankar G, Cohen DA. Idiopathic pneumonia syndrome after bone marrow transplantation: the role of pre-transplant radiation conditioning and local cytokine dysregulation in promoting lung inflammation and fibrosis (Review) Int J Exp Pathol. 2001;82:101–113. doi: 10.1111/j.1365-2613.2001.iep0082-0101-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Afessa B, Tefferi A, Litzow MR, Krowka MJ, Wylam ME, Peters SG. Diffuse alveolar hemorrhage in hematopoietic stem cell transplant recipients (Review) Am J Respir Crit Care Med. 2002;166:641–645. doi: 10.1164/rccm.200112-141cc. [DOI] [PubMed] [Google Scholar]
- 6.Afessa B, Tefferi A, Litzow MR, Peters SG. Outcome of diffuse alveolar hemorrhage in hematopoietic stem cell transplant recipients. Am J Respir Crit Care Med. 2002;166:1364–1368. doi: 10.1164/rccm.200208-792OC. [DOI] [PubMed] [Google Scholar]
- 7.Della Volpe A, Ferreri AJ, Annaloro C, Mangili P, Rosso A, Calandrino R, et al. Lethal pulmonary complications significantly correlate with individually assessed mean lung dose in patients with hematologic malignancies treated with total body irradiation. Int J Radiat Oncol Biol Phys. 2002;52:483–488. doi: 10.1016/s0360-3016(01)02589-5. [DOI] [PubMed] [Google Scholar]
- 8.Clark JG, Madtes DK, Hackman RC, Chen W, Cheever MA, Martin PJ. Lung injury induced by alloreactive Th1 cells is characterized by host-derived mononuclear cell inflammation and activation of alveolar macrophages. J Immunol. 1998;161:1913–1920. [PubMed] [Google Scholar]
- 9.Cooke KR, Kobzik L, Martin TR, Brewer J, Delmonte JJ, Crawford JM, et al. An experimental model of idiopathic pneumonia syndrome after bone marrow transplantation: I. The roles of minor H antigens and endotoxin. Blood. 1996;88:3230–3239. [PubMed] [Google Scholar]
- 10.Panoskaltsis-Mortari A, Taylor PA, Yaeger TM, Wangensteen OD, Bitterman PB, Ingbar DH, et al. The critical early proinflammatory events associated with idiopathic pneumonia syndrome in irradiated murine allogeneic recipients are due to donor T cell infusion and potentiated by cyclophosphamide. J Clin Invest. 1997;100:1015–1027. doi: 10.1172/JCI119612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clark JG, Madtes DK, Martin TR, Hackman RC, Farrand AL, Crawford SW. Idiopathic pneumonia after bone marrow transplantation: cytokine activation and lipopolysaccharide amplification in the bronchoalveolar compartment. Crit Care Med. 1999;27:1800–1806. doi: 10.1097/00003246-199909000-00016. [DOI] [PubMed] [Google Scholar]
- 12.Cooke KR, Hill GR, Gerbitz A, Kobzik L, Martin TR, Crawford JM, et al. Tumor necrosis factor-alpha neutralization reduces lung injury after experimental allogeneic bone marrow transplantation. Transplantation. 2000;70:272–279. doi: 10.1097/00007890-200007270-00006. [DOI] [PubMed] [Google Scholar]
- 13.Englund JA, Boeckh M, Kuypers J, Nichols WG, Hackman RC, Morrow RA, et al. Fatal human metapneumovirus infection in stem cell transplant recipients. Ann Intern Med. 2006;144:344–349. doi: 10.7326/0003-4819-144-5-200603070-00010. [DOI] [PubMed] [Google Scholar]
- 14.Broliden K. Parvovirus B19 infection in pediatric solid-organ and bone marrow transplantation (Review) Pediatr Transplant. 2001;5:320–330. doi: 10.1034/j.1399-3046.2001.00035.x. [DOI] [PubMed] [Google Scholar]
- 15.McDonald GB, Bouvier M, Hockenbery DM, Stern JM, Gooley T, Farrand A, et al. Oral beclomethasone dipropionate for treatment of intestinal graft-versus-host disease: a randomized, controlled trial. Gastroenterology. 1998;115:28–35. doi: 10.1016/s0016-5085(98)70361-0. [DOI] [PubMed] [Google Scholar]
- 16.Hockenbery DM, Cruickshank S, Rodell TC, Gooley T, Schuening F, Rowley S, et al. A randomized, placebo-controlled trial of oral beclomethasone dipropionate as a prednisone-sparing therapy for gastrointestinal graft-versus-host disease. Blood. 2007;109:4557–4563. doi: 10.1182/blood-2006-05-021139. [DOI] [PubMed] [Google Scholar]
- 17.McDonald GB. Oral beclomethasone dipropionate: a topically active corticosteroid for the treatment of gastrointestinal graft-versus-host disease following allogeneic hematopoietic cell transplantation (Review) Expert Opin Investig Drugs. 2007;16:1709–1724. doi: 10.1517/13543784.16.10.1709. [DOI] [PubMed] [Google Scholar]
- 18.Standardization of spirometry, 1994 update: American Thoracic Society. Am J Respir Crit Care Med. 1995;152:1107–1136. doi: 10.1164/ajrccm.152.3.7663792. [DOI] [PubMed] [Google Scholar]
- 19.Lung function testing: selection of reference values and interpretative strategies. American Thoracic Society. Am Rev Respir Dis. 1991;144:1202–1218. doi: 10.1164/ajrccm/144.5.1202. [DOI] [PubMed] [Google Scholar]
- 20.American Thoracic Society. Single-breath carbon monoxide diffusing capacity (transfer factor). Recommendations for a standard technique - 1995 update. Am J Respir Crit Care Med. 1995;152:2185–2198. doi: 10.1164/ajrccm.152.6.8520796. [DOI] [PubMed] [Google Scholar]
- 21.Crapo RO, Morris AH, Gardner RM. Reference spirometric values using techniques and equipment that meet ATS recommendations. Am Rev Respir Dis. 1981;123:659–664. doi: 10.1164/arrd.1981.123.6.659. [DOI] [PubMed] [Google Scholar]
- 22.Crapo RO, Morris AH. Standardized single breath normal values for carbon monoxide diffusing capacity. Am Rev Respir Dis. 1981;123:185–189. doi: 10.1164/arrd.1981.123.2.185. [DOI] [PubMed] [Google Scholar]
- 23.Freudenberger TD, Madtes DK, Curtis JR, Cummings P, Storer BE, Hackman RC. Association between acute and chronic graft-versus-host disease and bronchiolitis obliterans organizing pneumonia in recipients of hematopoietic stem cell transplants. Blood. 2003;102:3822–3828. doi: 10.1182/blood-2002-06-1813. [DOI] [PubMed] [Google Scholar]
- 24.Fukuda T, Hackman RC, Guthrie KA, Sandmaier BM, Boeckh M, Maris MB, et al. Risks and outcomes of idiopathic pneumonia syndrome after nonmyeloablative and conventional conditioning regimens for allogeneic hematopoietic stem cell transplantation. Blood. 2003;102:2777–2785. doi: 10.1182/blood-2003-05-1597. [DOI] [PubMed] [Google Scholar]
- 25.Epler GR, Colby TV, McLoud TC, Carrington CB, Gaensler EA. Bronchiolitis obliterans organizing pneumonia. N Engl J Med. 1985;312:152–158. doi: 10.1056/NEJM198501173120304. [DOI] [PubMed] [Google Scholar]
- 26.Crapo RO, Forster RE. Carbon monoxide diffusing capacity (Review) Clin Chest Med. 1989;10:187–198. [PubMed] [Google Scholar]
- 27.Yamaguchi K, Mori M, Kawai A, Takasugi T, Oyamada Y, Koda E. Inhomogeneities of ventilation and the diffusing capacity to perfusion in various chronic lung diseases. Am J Respir Crit Care Med. 1997;156:86–93. doi: 10.1164/ajrccm.156.1.9607090. [DOI] [PubMed] [Google Scholar]
- 28.Weiner RS, Bortin MM, Gale RP, Gluckman E, Kay HEM, Kolb H-J, et al. Interstitial pneumonitis after bone marrow transplantation. Ann Intern Med. 1986;104:168–175. doi: 10.7326/0003-4819-104-2-168. [DOI] [PubMed] [Google Scholar]
- 29.Crawford SW, Longton G, Storb R. Acute graft-versus-host disease and the risks for idiopathic pneumonia after marrow transplantation for severe aplastic anemia. Bone Marrow Transplant. 1993;12:225–231. [PubMed] [Google Scholar]
- 30.Kantrow SP, Hackman RC, Boeckh M, Myerson D, Crawford SW. Idiopathic pneumonia syndrome: changing spectrum of lung injury after marrow transplantation. Transplantation. 1997;63:1079–1086. doi: 10.1097/00007890-199704270-00006. [DOI] [PubMed] [Google Scholar]
- 31.Piguet P-F, Grau GE, Allet B, Vassalli P. Tumor necrosis factor/cachectin is an effector of skin and gut lesions of the acute phase of graft-vs-host disease. J Exp Med. 1987;166:1280–1289. doi: 10.1084/jem.166.5.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hildebrandt GC, Olkiewicz KM, Corrion LA, Chang Y, Clouthier SG, Liu C, et al. Donor-derived TNF-alpha regulates pulmonary chemokine expression and the development of idiopathic pneumonia syndrome after allogeneic bone marrow transplantation. Blood. 2004;104:586–593. doi: 10.1182/blood-2003-12-4259. [DOI] [PubMed] [Google Scholar]
- 33.Hildebrandt GC, Olkiewicz KM, Corrion L, Clouthier SG, Pierce EM, Liu C, et al. The role for TNF receptor type II in leukocyte infiltration into the lung during experimental idiopathic pneumonia syndrome. Biol Blood Marrow Transplant. 2008;14:385–396. doi: 10.1016/j.bbmt.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hill GR, Crawford JM, Cooke KR, Brinson YS, Pan L, Ferrara JL. Total body irradiation and acute graft-versus-host disease: the role of gastrointestinal damage and inflammatory cytokines. Blood. 1997;90:3204–3213. [PubMed] [Google Scholar]
- 35.Cooke KR, Hill GR, Crawford JM, Bungard D, Brinson YS, Delmonte J, Jr, et al. Tumor necrosis factor- alpha production to lipopolysaccharide stimulation by donor cells predicts the severity of experimental acute graft-versus-host disease. J Clin Invest. 1998;102:1882–1891. doi: 10.1172/JCI4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nestel FP, Price KS, Seemayer TA, Lapp WS. Macrophage priming and lipopolysaccharide-triggered release of tumor necrosis factor during graft -versus-host disease. J Exp Med. 1992;175:405–413. doi: 10.1084/jem.175.2.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Levine DS. Immune modulating therapies for idiopathic inflammatory bowel diseases. Adv Pharmacol. 1994;25:171–234. doi: 10.1016/s1054-3589(08)60432-9. [DOI] [PubMed] [Google Scholar]
- 38.Raj RM, Hendrix TR, Moskaluk C, Levine DS, Pasricha PJ. Treatment of idiopathic lymphocytic enterocolitis with oral beclamethasone dipropionate. Am J Gastroenterol. 1997;92:147–149. [PubMed] [Google Scholar]
- 39.Rizzello F, Gionchetti P, D’Arienzo A, Manguso F, Di Matteo G, Annese V, et al. Oral beclometasone dipropionate in the treatment of active ulcerative colitis: a double-blind placebo-controlled study. Aliment Pharmacol Ther. 2002;16:1109–1116. doi: 10.1046/j.1365-2036.2002.01298.x. [DOI] [PubMed] [Google Scholar]
- 40.Brogden RN, Pinder RM, Sawyer PR, Speight TM, Avery GS. Beclomethasone dipropionate inhaler: a review of its pharmacology, therapeutic value and adverse effects. I: Asthma. Drugs. 1975;10:166–210. doi: 10.2165/00003495-197510030-00002. [DOI] [PubMed] [Google Scholar]
- 41.Harris DM. Properties and therapeutic uses of some corticosteroids with enhanced topical potency. J Steroid Biochem. 1975;6:711–716. doi: 10.1016/0022-4731(75)90057-6. [DOI] [PubMed] [Google Scholar]
- 42.Wurthwein G, Rohdewald P. Activation of beclomethasone dipropionate by hydrolysis to beclomethasone-17-monopropionate. Biopharm Drug Dispos. 1990;11:381–394. doi: 10.1002/bdd.2510110503. [DOI] [PubMed] [Google Scholar]
- 43.Daley-Yates PT, Price AC, Sisson JR, Pereira A, Dallow N. Beclomethasone dipropionate: absolute bioavailability, pharmacokinetics and metabolism following intravenous, oral, intranasal and inhaled administration in man. Br J Clin Pharmacol. 2001;51:400–409. doi: 10.1046/j.0306-5251.2001.01374.x. [DOI] [PMC free article] [PubMed] [Google Scholar]