

Abstract
The reactions of a series of strained bicyclic and tricyclic one-carbon bridged lactams with organometallic reagents have been investigated. These amides permit isolation of a number of remarkably stable hemiaminals upon nucleophilic addition to the twisted amide bonds present in the lactam precursors. The factors that affect the stability of the resulting bridged hemiaminals are presented. In some cases, the hemiaminals were found to collapse to the open-form amino ketones in a manner expected for traditional carboxylic acid derivatives. Transannular N⋯C=O interactions were also observed in some 9-membered amino ketones. Additionally, tricyclic bridged lactams were found to react with some nucleophiles that typically react with ketones but not with planar amides. The effect of geometry on the reactivity of amide bonds and the amide bond distortion range that marks the boundary of amide-like and ketone-like carbonyl reactivity of lactams are also discussed.
INTRODUCTION
The tetrahedral intermediate formed in the addition of nucleophiles to carboxylic acid derivatives is typically a short-lived species that is sometimes detected but rarely isolated.1 Thus, although tetrahedral intermediates formed in the reaction of tertiary amides2 and N-methoxy-N-methylamides3 with organometallic reagents are relatively stable as their salts, and have been used extensively for synthesis of ketones, the tetrahedral intermediates themselves rapidly decompose upon protonation (Figure 1a). In addition, isolated tetrahedral intermediates would provide models for in vivo transacylation processes. Thus, the products of thiol-, alcohol- and amine-based nucleophiles with esters or amides are commonly encountered in enzymatic acylation reactions, but are difficult to investigate in the absence of enzyme stabilization.4
Figure 1.

a) Reaction of carboxylic acid derivatives with nucleophiles; b) examples of N⋯C=O interactions.
Transannular N⋯C=O interactions between tertiary amines and carbonyl groups5 were the primary tool in a fundamental study by Bürgi and Dunitz6 to designate the trajectory of the attack of nucleophiles on C=O bonds (Figure 1b). In this investigation, a set of conformationally-frozen amino-ketones kept the reactive groups in close proximity to limit the number of unproductive conformations. It is generally accepted that the Bürgi-Dunitz angle, so determined, is followed by nucleophiles in their attack on carboxylic acid derivatives, producing tetrahedral intermediates.7 Herein, we report that medium-bridged twisted amides8 can serve as model systems to monitor a transition from stable tetrahedral intermediates through intramolecular N⋯C=O interactions to unstable tetrahedral intermediates. As a result of this investigation, we also provide evidence that a rotation of an amide bond to ca. 50° (where 0° would correspond to planar amide and 90° to fully orthogonal amide bond)9 is close to the border from an amide-like to ketone-like switch of reactivity of amide carbonyl groups.
Recently, our laboratory described that one-carbon bridged amides undergo novel C-N bond cleavage reactions, which are a direct result of the distortion of amide linkage.10 We also found that despite significant twist values of the amide bonds, one-carbon bridged lactams are much less readily hydrolyzed by nucleophilic solvents when compared to fully twisted lactams.11 This unusual-to-twisted-amides property was ascribed to destabilization of the potentially formed carboxylic acid and amine functionalities by their placement on the opposite sides of the medium-sized ring, where they were additionally subjected to strong proximity effects. We hypothesized that such lactams might undergo facile nucleophilic addition reactions and additionally permit the isolation of stable tetrahedral intermediates. This supposition was supported by the very limited but important precedent obtained by Kirby8h and Coe8i groups in the study of highly constrained adamantane-like twisted amides, in which lactam transformations afforded stabilized hemiaminals.12
RESULTS AND DISCUSSION
Additions to bicyclic bridged amides
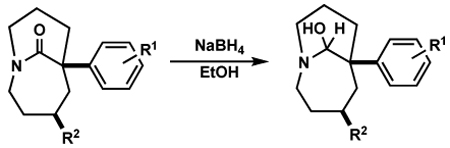
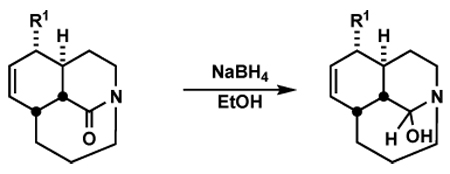
We began our study by investigating the behavior of one-carbon bridged amides in addition reactions of hydride, the smallest available nucleophile (Table 1). In agreement with our hypothesis, treatment of bridged bicyclic amide 1a with NaBH4 (1.0 equiv) in EtOH led to the formation of stable hemiaminal 2a in excellent yield (entry 1). Since planar amides are typically not reduced by NaBH4,13 this transformation occurs due to the increased reactivity of the bridged amide bond. The stability of 2a indicates that lone pairs of electrons at oxygen do not overlap with the σ*C–N bond in the bicyclic hemiaminal system. Furthermore, since the reduction stops at the tetrahedral intermediate stage, the bridged nitrogen atom is incapable of donation of its n electrons into the σ*C–O. We recently reported that a series of epoxy-hemiaminals are similarly stabilized.14 However, aside from Kirby and Coe precedents noted above, we are unaware of other conversions of non-adamantane twisted amides to a stable hemiaminal under mild, protic conditions.15
Table 1.
Hydride Addition to Bridged Amides
| entry | bridged amide | product | yield [%] |
|---|---|---|---|
 | |||
| 1 | (1a) R1 = H, R2 = t-Bu | (2a) 90 | |
| 2 | (1b) R1 = 4-(MeO), R2 = t-Bu | (2b) 91 | |
| 3 | (1c) R1 = 3,5-(MeO)2, R2 = t-Bu | (2c) 94 | |
| 4 | (1d) R1 = 3,4-(OCH2O), R2 = t-Bu | (2d) 95 | |
 | |||
| 5 | (1e) R1 = H | (2e) 91 | |
| 6 | (1f) R1 = 4-BrC6H4 | (2f) 88 | |
 | |||
| 7 | (1g) R1 = H, R2 = t-Bu | (2g) 24 | (3g) 52 |
| 8 | (1h) R1 = SMe, R2 = H | (2h) 40 | (3h) 48 |
| 9 | (1i) R1 = SPh, R2 = H | (2i) 62 | (3i) 34 |
To examine the effect of structure on the stability of hemiaminals, the reduction was extended to a number of bridged amides. Bridged lactam substrates were prepared via the intramolecular Schmidt reaction as previously reported,8l generally as the minor component of a mixture also containing the regioisomeric fused lactam; the additional tert-butyl group was incorporated to enhance the proportion of bridged product obtained. Thus, bicyclic amides with electron-rich aromatic rings in the α-position as well as tricyclic lactams smoothly underwent the reduction providing isolable hemiaminals in all cases studied (Table 1, entries 2–6). However, an α-unsubstituted bridged amide and amides with sulfides in the α position led to a mixture of hemiaminals and primary alcohols (entries 7–9). This outcome suggests that although these structure types display similarly enhanced reactivity to fully planar lactams and thus reaction with NaBH4, the products more readily collapse under the reaction conditions to afford aldehydes that undergo further in situ reduction.
Remarkably, reduction of bridged amides decorated with α-electron-withdrawing substituents afforded formamides (Scheme 1). In these examples, ready hemiaminal formation ultimately results in the formation of anions, stabilized by sulfonyl and 4-nitrophenyl groups (1j and 1k) or allylic olefin and amide bond (1l). The hemiaminal intermediate may well be in equilibrium with the corresponding amino aldehyde intermediate, but the observed products likely predominate because the anionic intermediate is readily protonated, rendering the overall conversion irreversible. Overall, the results from Table 1 and Scheme 1 demonstrate that the nature of the α-substituent plays a key role in determining the fate of tetrahedral intermediates generated from these bicyclic systems.
Scheme 1.

Reduction of Bridged Lactams with Cleavage of C–C Bond
Using amide 1a as a test substrate, we also briefly investigated the role of a hydride source and reaction conditions on the formation and fates of stable hemiaminals (see Supporting Information (SI) for full details).16 A number of reducing reagents (including LiBH4, LiAlH4, DIBAl-H and BH3) afforded isolable hemiaminal 2a. Interestingly, the reduction rate with NaBH4 was found to be qualitatively slower in methanol as a solvent than in ethanol, which contrasts with the reduction of typical carbonyl groups by NaBH4.17 In addition, the reduction of 1a was suppressed when CeCl3 was utilized as an additive.18 It is possible that the decreased reaction rates are due to the increased basicity of the non-planar amide bond nitrogen, leading to subsequent hydrogen bonding or metal coordination to the amide bond nitrogen. We have previously demonstrated that the facility of N-protonation, -alkylation, or -metallation occurred to a much greater degree in bridged lactams than in normal amides,10 however it is not clear why in the present case coordination to the nitrogen did not increase the carbonyl reactivity by inductive effect. Importantly, when a one-carbon higher homologue of 1a ([5.3.1] ring system) was treated with NaBH4, reduction was not observed, indicating that the [5.3.1] scaffold is not sufficiently distorted to permit hydride addition under these conditions.
Since hemiaminals are versatile synthetic intermediates,19 we examined the behavior of these bridged hemiaminals in a variety of settings (Scheme 2). Noteworthy transformations included the oxidation of 2a to the parent amide 1a and, conversely, full reduction to amine 5 (most likely proceeding via the intermediacy of a rarely encountered bridgehead iminium ion).20 This latter possibility was also consistent with the isolation of aminals 6 and 7 as single diastereoisomers from a mixture of hemiketal isomers in acidic methanol and by the determination that hemiaminal 2a readily epimerizes upon treatment with acid. Both reactions could occur either via the intermediate bridged iminium ion or through the acid-promoted opening to the aldehyde and re-closure to the more thermodynamically favored isomer. All of these reactions bode well for synthetic applications of bridged hemiaminals having this [4.3.1] ring system.
Scheme 2.

Synthetic Transformations of Bridged Hemiaminals
We next evaluated the stability of hemiaminals formed from the addition of more sterically demanding organometallic reagents. Thus, treatment of bicyclic amide 1a with MeLi afforded amino ketone 8a (Table 2, entry 1). Not surprisingly, an increase in steric hindrance at the hemiaminal carbon shifted the equilibrium from the hemiaminal toward the aminoketone species. Similarly, the reaction of 1a with secondary and tertiary organolithiums furnished the corresponding amino ketones (entries 4 and 5). Interestingly, in all cases the addition stopped at the ketone stage despite the presence of a large excess of nucleophile. Re-subjection of the aminoketone 8a to the reaction conditions did not result in the formation of the tertiary alcohol, suggesting that the steric hindrance around the quaternary carbon prohibits further addition of the organometallic reagent. However, it is also possible that the initially formed addition product persists in the reaction mixture prior to workup. While it is known that planar tertiary amides react with organometallic reagents,1a the high yields obtained in the transformations of such sterically-congested bridged lactams are noteworthy and likely reflect the increased electrophilicity of the distorted amide bonds.
Table 2.
Organometallic Addition to Bicyclic Amides
 | ||||
|---|---|---|---|---|
| entry | amino-ketone | R | reagent | yield [%] |
| 1 | 8a | Me | MeLi | 89 |
| 2 | 8a | Me | MeMgI | 73 |
| 3 | 8b | n-Bu | n-BuLi | 83 |
| 4 | 8c | sec-Bu | sec-BuLi | 93 |
| 5 | 8d | tert-Bu | tert-BuLi | 80 |
Additions to tricyclic bridged amides
The behavior of the bicyclic hemiaminals corresponding to amino-ketones 8a–d prompted us to examine the behavior of their tricyclic analogues. We reasoned that the additional scaffolding afforded by the six-membered ring attached to the bridged system could further stabilize the hemiaminal form of the product.11 Indeed, exposure of 1e to MeLi afforded hemiaminal 9a in good yield (Table 3, entry 1). Increased bulk close to the reactive amide bond did not influence the reaction and 1m furnished the corresponding hemiaminal product (entry 2). Remarkably, addition of a secondary organolithium also provided stable hemiaminals (entries 3 and 4). Ultimately, addition of tert-butyllithium finally tipped the balance in favor of the ketone-containing product (Table 3, entries 5 and 6).
Table 3.
Organometallic Addition to Tricyclic Amides
 | |||||
|---|---|---|---|---|---|
| entry | amide | hemiaminal/ amino-ketone |
R2 | reagent | yield [%] |
| 1 | 1e | 9a | Me | MeLi | 95 |
| 2 | 1m | 10a | Me | MeLi | 92 |
| 3 | 1e | 9b | sec-Bu | sec-BuLi | 80 |
| 4 | 1m | 10b | sec-Bu | sec-BuLi | 88 |
| 5 | 1e | 9c | tert-Bu | tert-BuLi | 90 |
| 6 | 1m | 10c | tert-Bu | tert-BuLi | 90 |
Hemiaminals 9a–10b are some of the most sterically hindered tetrahedral intermediates isolated to date.1a Although, it is possible that these structures exist in equilibrium with the corresponding amino ketones, the predominance of the hemiaminal form is secured by NMR21 and IR22 spectra. The major species observed by NMR is characterized by a hemiaminal carbon resonance at about 86–88 ppm in the 13C NMR. The ketone peak is not detected in the 13C NMR and only in two instances were very weak peaks corresponding to the CO group observed in the IR (see below and SI for additional discussion regarding IR spectra). Although the analogous equilibrium is also possible in the transannular amino tert-butylketones 9c and 10c, in these cases the open-form amino ketones seem to be predominant as evidenced by 13C NMR and IR spectroscopy. Overall, these results indicate that the steric contribution can override the inherent stability of tetrahedral intermediates provided by the scaffolding effects.
We wished to directly compare the reactivity of distorted and planar amides in reactions with organometallic reagents. Thus, a couple of fused amides (obtained as complementary products to the bridged lactams in the intramolecular Schmidt reactions)8l were subjected to the same conditions utilized for transformations of bridged lactams (Scheme 3). The reaction of fused bicyclic amide 11 with MeLi afforded enamine 12.23 However, the dehydration was not general and addition of n-BuLi to 11 resulted in a complex mixture of products including the starting amide, ketone, enamine, and alcohol. Similarly, the reaction of a tricyclic fused amide 13 with MeLi led to an inseparable 3:1:1 mixture of enamine, ketone and alcohol, with the subsequent reduction affording amine 14. The very different results obtained from fused vs. bridged lactams emphasize the effect of the amide bond geometry on the outcome of nucleophilic addition reactions to amide bonds.
Scheme 3.

Organometallic Addition to Planar Analogues of Bridged Amides
Observation of N⋯C=O interactions
We hypothesized that the conformation of the 9-membered ring in 8a (Table 2, entry 1) should favor the placement of the reactive nitrogen and ketone groupings on the same side of the ring, and thus permit observation of transannular interactions between the amine and carbonyl groups under appropriate reaction conditions.5 It is generally accepted that N⋯C=O interactions between amines and electrophilic ketones or aldehydes result in formation of a pseudo-tetrahedral hemiaminal-type carbon adopting a hybridization state between sp2 and sp3 with a partial bonding being formed between the reactive moieties.5b Although this type of transannular interaction has been utilized extensively in conformational analysis,21b mechanistic physical-organic chemistry,5a, 5b total synthesis projects24 and medicinal chemistry,5f, 5h–i, 5l–n the majority of nitrogen-carbonyl transannular interactions reported so far involve electrophiles placed directly on a ring or otherwise conformationally restricted tropane-type structures. In contrast, the currently studied one-carbon bridged amides would provide reasonably flexible systems with the carbonyl moved one-carbon away from the ring. The transannular hypothesis was also supported by our previous finding that amino acids resulting from hydrolysis of some of the one-carbon bridged amides are subjected to strong proximity effects induced by the placement of the reactive carboxylic acid and amine moieties on the opposite sides of the medium-size rings.11
We were pleased to find that upon treatment of 8a with MeOD-d4 a transannular N⋯C=O interaction takes place (Scheme 4). Thus, the carbon NMR spectrum of 8a in chloroform exhibits a single set of signals, with a peak at 211 ppm corresponding to the methyl ketone. These observations are consistent with a simple ketone-containing structure as drawn. In methanol, the carbonyl peak is no longer present in the 13C NMR, and other peaks are significantly broadened. We ascribe these characteristics to an N⋯C=O interaction affording a pseudo-tetrahedral hemiaminal-type carbon (8aa). Addition of DCl to the methanol solution terminated the equilibrium, affording a 9:1 mixture of protonated amino ketone 8ab and hemiaminal 8ac. Upon dissolving 8a in DMSO-d6, a ca. 3:1 mixture of amino ketone and hemiaminal was formed, further illustrating that the transannular closure is favored by increased solvent polarity.5m
Scheme 4.

Transannular Interaction in Bicyclic System
The transannular interaction between nitrogen and carbonyl group in 8a could be independently detected by UV spectroscopy (Figure 2).25, 5e, 5m–n When the UV spectrum of 8a was measured in chloroform, a weak absorption with the maximum at 241 nm was observed (ε = 793 L mol−1 cm−1), corresponding to the π → π*C=O transition. However, the spectrum of 8a in MeOH exhibited a new and significantly more intense absorption band appearing at a shorter wavelength (λmax = 211 nm, ε = 5688 L mol−1 cm−1), which could be ascribed to an nN → π*C=O absorption. The change in absorption of 8a in methanol is similar to that observed in earlier examples of N⋯C=O interactions. 25, 5e, 5m−n Furthermore, a detailed examination of the IR spectra22 of a series of bicyclic and tricyclic amino ketones and hemiaminals 8a–8d and 9a–10c also suggest the existence of the transannular interaction in the 9-membered ring systems (see SI for additional discussion).
Figure 2.

UV spectra in CHCl3 (solid line) and MeOH (dotted line) of 8a.
Although the above data are clearly consistent the presence of N⋯C=O interaction, we cannot completely rule out a dynamic bond breaking and forming event taking place between the amino ketone and hemiaminal, with the latter species being stabilized in polar solvents. This type of weak covalent character of N⋯C=O interaction, as contrasted to the traditionally accepted nN → π*C=O homoconjugation,5b has also been recently suggested in the literature.5l
It is noteworthy that this single bridged amide can be utilized to monitor a continuum of change: from isolable hemiaminal 2a (stable tetrahedral intermediate) to amino ketone 8a (resulting from the collapse of said tetrahedral intermediate) through N⋯C=O interaction (8aa, MeOH). This picture also includes a progressive change from hemiaminals 9a–10b (stable tetrahedral intermediates) to amino ketones 9c and 10c (collapsed tetrahedral intermediates).
As noted above, we hypothesized that the difference in stability between the collapsed amino ketone 8a (Table 2, entry 1) and the stable hemiaminal 9a (Table 3, entry 1) could be due to the increased scaffolding effect gained by the presence of the additional cyclohexene ring in 9a. However, we also considered that it could arise from a difference in distortion parameters of the two parent amides. As previously established by X-ray crystallographic analysis, the tricyclic lactam 1e is characterized by Winkler-Dunitz distortion parameter,9 twist angle, τ = 51.5°,11 while a representative bicyclic lactam 1b is slightly less distorted (τ = 43.2°).26
Reactions with additional nucleophiles
To investigate this point we subjected bicyclic and tricyclic bridged amides to reactions with some oxygen and nitrogen nucleophiles, which typically do not react with planar amides (Scheme 5). Thus, reactions of tricyclic lactams with 1,3-propanediol and benzylamine afforded bridged ketal 15 and bridged imine 16, respectively, whereas fused bicyclic lactams were unreactive under these conditions as expected. In addition, the tricyclic 1e furnished bridged hydrazone 17. This particular reaction was also performed to allow comparison of one-carbon bridged lactams with an adamantane-type bridged amide reported by Coe and coworkers (Figure 3).8i Interestingly, although in the case of Coe’s amide a subsequent full reduction to the corresponding amine was carried out, we were unable to reduce 17 under Wolff-Kishner conditions.
Scheme 5.

Transformations of Bridged Amides
Figure 3.

Transformations of adamantane-type bridged amides.
Furthermore, although Kirby demonstrated that perfectly perpendicular 1-aza-2-adamantanone (τ = 90.5°) undergoes Wittig olefination (Figure 3),8h similar treatment of the currently investigated amides did not result in enamines (only starting amides were re-isolated). However, the corresponding bridged exocyclic enamine 18 could be conveniently prepared from 1a utilizing Petasis reagent (Scheme 6).27 Although some planar amides undergo olefination under Petasis conditions, to the best of our knowledge this is the first example of a direct olefination of a bridged bicyclic lactam with a metalloorganic reagent. It is noteworthy that the oxatitanacyclobutane intermediate did not collapse with the opening to the nine-membered ring system (in a fashion similar to 8a), and that the resulting enamine was stable to silica gel chromatography.
Scheme 6.

Petasis Olefination of Lactam 1a
Overall, these results clearly demonstrate that lactams having a twist angle of ca. 50° display reactivity patterns much closer to those expected for twisted rather than planar amides. It is interesting to note that the bridged structures resulting from nucleophilic addition reactions to these distorted lactams (Scheme 5) are also stabilized through proximity effects.
CONCLUSION
In summary, we have shown that tetrahedral adducts formed from the addition of nucleophiles to one-carbon medium-bridged twisted amides exhibit remarkable proximity-induced stability. These amides can serve as models to delineate the transition from stable tetrahedral intermediates to N⋯C=O interactions to unstable tetrahedral intermediates. In addition, this work provides the first experimental evidence regarding degrees of the amide bond distortion that mark the border between amide-like and ketone-like carbonyl reactivity of lactams. Importantly, lactams characterized by the midway rotated amide bonds undergo certain reactions typically associated with ketones but not planar amides. Our laboratory is currently investigating the chemistry of one-carbon bridged amides and their derivatives, including heteroatom-substituted tetrahedral intermediates.
Supplementary Material
Acknowledgements
This work was supported by the National Institute of General Medical Sciences (GM-49093). The authors thank Ruzhang Liu and Dr. Kevin Frankowski (University of Kansas) for helpful discussions, Dr. Justin Douglas (University of Kansas) for NMR assistance, Dr. Reza Esfandiary and Dr. Sangeeta Joshi (University of Kansas, Laboratory for Macromolecular and Vaccine Stabilization) for help with UV measurements.
Footnotes
Supporting Information Available: Experimental details and characterization data for new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For reviews, see: Adler M, Adler S, Boche G. J. Phys. Org. Chem. 2005;18:193–209. Deslongchamps P. Sterneoelectronic Effects in Organic Chemistry. Pergamon Press; 1983. For examples of isolated tetrahedral intermediates, see: Evans DA, Borg G, Scheidt KA. Angew. Chem., Int. Ed. 2002;41:3188–3191. doi: 10.1002/1521-3773(20020902)41:17<3188::AID-ANIE3188>3.0.CO;2-H. Kirby AJ, Komarov IV, Feeder N. J. Am. Chem. Soc. 1998;120:7101–7102. Adler M, Marsch M, Nudelman NS, Boche G. Angew. Chem., Int. Ed. 1999;38:1261–1263. doi: 10.1002/(SICI)1521-3773(19990503)38:9<1261::AID-ANIE1261>3.0.CO;2-E.
- 2.(a) Braude EA, Evans EA. J. Chem. Soc. 1955:3334–3337. [Google Scholar]; (b) Evans EA. J. Chem. Soc. 1956:4691–4692. [Google Scholar]; (c) Izzo PT, Safir SR. J. Org. Chem. 1959;24:701–703. [Google Scholar]; (d) Hwang YC, Chu M, Fowler FW. J. Org. Chem. 1985;50:3885–3890. [Google Scholar]; (e) Myers AG, Yang BH, Chen H, McKinstry L, Kopecky DJ, Gleason JL. J. Am. Chem. Soc. 1997;119:6496–6511. [Google Scholar]
- 3.(a) Nahm S, Weinreb SM. Tetrahedron Lett. 1981;22:3815–3818. [Google Scholar]; (b) Balasubramaniam S, Aidhen IS. Synthesis. 2008:3707–3738. [Google Scholar]
- 4.(a) Bruice TC, Benkovic SJ. Bioorganic Mechanisms. New York: Benjamin; 1966. [Google Scholar]; (b) Bender ML. Mechanisms of Homogenous Catalysis from Protons to Proteins. New York: Wiley; 1971. [Google Scholar]; (c) Fersht A. Structure and Mechanism in Protein Science. New York: 1995. [Google Scholar]; (d) Bender ML. Chem. Rev. 1960;60:53–113. [Google Scholar]; (e) Jencks WP. Chem. Rev. 1972;72:705–718. [Google Scholar]; (f) Williams A, Douglas KT. Chem. Rev. 1975;75:627–649. [Google Scholar]; (g) Blow DM. Acc. Chem. Res. 1976;9:145–152. [Google Scholar]; (h) Capon B, Ghosh AK, Grieve DMA. Acc. Chem. Res. 1981;14:306–312. [Google Scholar]; (i) McClelland RA, Santry LJ. Acc. Chem. Res. 1983;16:394–399. [Google Scholar]; (j) Brown RS, Bennet AJ, Slebockatilk H. Acc. Chem. Res. 1992;25:481–488. [Google Scholar]
- 5.For reviews, see: Leonard NJ. Acc. Chem. Res. 1979;12:423–429. Rademacher P. Chem. Soc. Rev. 1995;24:143–150. For examples, see: Leonard NJ, Fox RC, Oki M, Chiavarelli S. J. Am. Chem. Soc. 1954;76:630–631. and the following papers in the series. Leonard NJ, Morrow DF, Rogers MT. J. Am. Chem. Soc. 1957;79:5476–5479. Bell MR, Archer S. J. Am. Chem. Soc. 1960;82:151–155. Daum SJ, Martini CM, Kullnig RK, Clarke RL. J. Med. Chem. 1975;18:496–501. doi: 10.1021/jm00239a012. McCrindle R, McAlees AJ. J. Chem. Soc., Chem. Commun. 1983:61–62. Carroll JD, Jones PR, Ball RG. J. Org. Chem. 1991;56:4208–4213. Griffith R, Bremner JB, Titmuss SJ. J. Comput. Chem. 1997;18:1211–1221. Kirby AJ, Komarov IV, Bilenko VA, Davies JE, Rawson JM. Chem. Commun. 2002:2106–2107. doi: 10.1039/b206639d. Gautier A, Pitrat D, Hasserodt J. Bioorg. Med. Chem. 2006;14:3835–3847. doi: 10.1016/j.bmc.2006.01.031. Pilme J, Berthoumieux H, Robert V, Fleurat-Lessard P. Chem.--Eur. J. 2007;13:5388–5393. doi: 10.1002/chem.200601250. Waibel M, Hasserodt J. J. Org. Chem. 2008;73:6119–6126. doi: 10.1021/jo800719j. Waibel M, Pitrat D, Hasserodt J. Bioorg. Med. Chem. 2009;17:3671–3679. doi: 10.1016/j.bmc.2009.03.059.
- 6.(a) Bürgi HB, Dunitz JD, Shefter E. J. Am. Chem. Soc. 1973;95:5065–5067. [Google Scholar]; (b) Bürgi HB, Dunitz JD, Lehn MJ, Wipff G. Tetrahedron. 1974;30:1563–1572. [Google Scholar]; (c) Bürgi HB, Dunitz JD. Acc. Chem. Res. 1983;16:153–161. [Google Scholar]
- 7.For important study with 1-azaadamantane derivative, see: Davies JE, Kirby AJ, Komarov IV. Helv. Chim. Acta. 2003;86:1222–1233.
- 8.For reviews, see: Hall HK, Elshekeil A. Chem. Rev. 1983;83:549–555. Lease TG, Shea KJ. Advances in Theoretically Interesting Molecules. Vol. 2. JAI Press Inc.; 1992. pp. 79–112. Greenberg A, Breneman CM, Liebman JF, editors. Amide Linkage: Selected Structural Aspects in Chemistry, Biochemistry,and Materials Science. New York: Wiley; 2000. Clayden J, Moran WJ. Angew. Chem., Int. Ed. 2006;45:7118–7120. doi: 10.1002/anie.200603016.For representative examples, see: Grigg R, Sridharan V, Stevenson P, Worakun T. J. Chem. Soc., Chem. Commun. 1986:1697–1699. Williams RM, Lee BH, Miller MM, Anderson OP. J. Am. Chem. Soc. 1989;111:1073–1081. Lease TG, Shea KJ. J. Am. Chem. Soc. 1993;115:2248–2260. Kirby AJ, Komarov IV, Feeder N. J. Chem. Soc., Perkin Trans. 2. 2001:522–529. Bashore CG, Samardjiev IJ, Bordner J, Coe JW. J. Am. Chem. Soc. 2003;125:3268–3272. doi: 10.1021/ja028152c. Tani K, Stoltz BM. Nature. 2006;441:731–734. doi: 10.1038/nature04842. Ribelin TP, Judd AS, Akritopoulou-Zanze I, Henry RF, Cross JL, Whittern DN, Djuric SW. Org. Lett. 2007;9:5119–5122. doi: 10.1021/ol7023373. Yao L, Aubé J. J. Am. Chem. Soc. 2007;129:2766–2767. doi: 10.1021/ja068919r. Szostak M, Aubé J. Org. Lett. 2009;11:3878–3881. doi: 10.1021/ol901449y. Szostak M, Yao L, Aubé J. Org. Lett. 2009;11:4386–4389. doi: 10.1021/ol901771b.For ab initio studies, see: Greenberg A, Venanzi CA. J. Am. Chem. Soc. 1993;115:6951–6957. Greenberg A, Moore DT, DuBois TD. J. Am. Chem. Soc. 1996;118:8658–8668.
- 9.Winkler FK, Dunitz JD. J. Mol. Biol. 1971;59:169–182. doi: 10.1016/0022-2836(71)90419-0. [DOI] [PubMed] [Google Scholar]
- 10.Lei Y, Wrobleski AD, Golden JE, Powell DR, Aubé J. J. Am. Chem. Soc. 2005;127:4552–4553. doi: 10.1021/ja050214m. [DOI] [PubMed] [Google Scholar]
- 11.Szostak M, Yao L, Aubé J. J. Org. Chem. 2009;74:1869–1875. doi: 10.1021/jo802192v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hemiaminals are observed or isolated only in special cases. For examples, see: Forlani L, Marianucci E, Todesco PE. J. Chem. Res., Synop. 1984:126–127. Chudek JA, Foster R, Young D. J. Chem. Soc., Perkin Trans. 2. 1985:1285–1289. El Mail R, Garralda MA, Hernandez R, Ibarlucea L, Pinilla E, Torres MR. Organometallics. 2000;19:5310–5317. Heine A, DeSantis G, Luz JG, Mitchell M, Wong CH, Wilson IA. Science. 2001;294:369–374. doi: 10.1126/science.1063601. Billard T, Langlois BR, Blond G. Eur. J. Org. Chem. 2001:1467–1471. doi: 10.1021/jo015587u. Suni V, Kurup MRP, Nethaji M. J. Mol. Struct. 2005;749:177–182. Dolensky B, Kvicala J, Paleta O. J. Fluorine Chem. 2005;126:745–751. Iwasawa T, Hooley RJ, Rebek J. Science. 2007;317:493–496. doi: 10.1126/science.1143272. Kawamichi T, Haneda T, Kawano M, Fujita M. Nature. 2009;461:633–635. doi: 10.1038/nature08326.For examples of hemiaminals stabilized due to electronic means, see: Chang YA, Sun TH, Chiang MY, Lu PJ, Huang YT, Liang LC, Ong CW. Tetrahedron. 2007;63:8781–8787. Troast DM, Porco JA. Org. Lett. 2002;4:991–994. doi: 10.1021/ol025558l. Boger DL, Keim H, Oberhauser B, Schreiner EP, Foster CA. J. Am. Chem. Soc. 1999;121:6197–6205. Lanman BA, Overman LE. Heterocycles. 2006;70:557–570. doi: 10.3987/com-06-s(w)16. Baldwin SW, Debenham JS. Org. Lett. 2000;2:99–102. doi: 10.1021/ol9911472. Norcross NR, Melbardis JP, Solera MF, Sephton MA, Kilner C, Zakharov LN, Astles PC, Warriner SL, Blakemore PR. J. Org. Chem. 2008;73:7939–7951. doi: 10.1021/jo8013512. Sakaguchi H, Tokuyama H, Fukuyama T. Org. Lett. 2007;9:1635–1638. doi: 10.1021/ol0631197. Kim J, Ashenhurst JA, Movassaghi M. Science. 2009;324:238–241. doi: 10.1126/science.1170777.
- 13.Brown HC, Rao BCS. J. Am. Chem. Soc. 1955;77:3164–3164. [Google Scholar]
- 14.Szostak M, Aubé J. J. Am. Chem. Soc. 2009;131:13246–13247. doi: 10.1021/ja906471q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.For examples of bridged amides that form unstable hemiaminals upon reduction, see: Denzer M, Ott H. J. Org. Chem. 1969;34:183–187. Levkoeva EI, Nikitskaya ES, Yakhontov LN. Dokl. Akad. Nauk. 1970;192:342–345. Levkoeva EI, Nikitskaya ES, Yakhontov LN. Khim. Geterotsikl. Soedin. 1971:378–384. For preparation of relevant 2-quinuclidinol derivatives, see: Bochow H, Schneider W. Ber. 1975;108:3475–3482. For reduction of a bridged lactam derived from 8-azabicyclo[3.2.1]octane scaffold, see: Baylis AM, Davies MPH, Thomas EJ. Org. Biomol. Chem. 2007;5:3139–3155. doi: 10.1039/b708910d. For additional examples of reduction of more relaxed ring systems of bridged lactams to hemiaminals, see: Ban Y, Yoshida K, Goto J, Oishi T. J. Am. Chem. Soc. 1981;103:6990–6992. Arata Y, Kobayashi T. Chem. Pharm. Bull. 1972;20:325–329. Büchi G, Coffen DL, Kocsis K, Sonnet PE, Ziegler FE. J. Am. Chem. Soc. 1965;87:2073–2075. For examples of reduction of more relaxed ring systems of bridged lactams to amines, see: Kozak JA, Dake GR. Angew. Chem., Int. Ed. 2008;47:4221–4223. doi: 10.1002/anie.200800522. Enders D, Lenzen A, Backes M, Janeck C, Catlin K, Lannou MI, Runsink J, Raabe G. J. Org. Chem. 2005;70:10538–10551. doi: 10.1021/jo0518093. Magnus P, Giles M, Bonnert R, Johnson G, McQuire L, Deluca M, Merritt A, Kim CS, Vicker N. J. Am. Chem. Soc. 1993;115:8116–8129. Amat M, Coll MD, Bosch J, Espinosa E, Molins E. Tetrahedron: Asymmetry. 1997;8:935–948.For examples of bridged hemiaminals used in total synthesis projects but prepared by methods other than reduction of amide bonds, see: Kutney JP, Balsevich J, Bokelman GH, Hibino T, Honda T, Itoh I, Ratcliffe AH, Worth BR. Can. J. Chem. 1978;56:62–70. Magnus P, Gazzard L, Hobson L, Payne AH, Rainey TJ, Westlund N, Lynch V. Tetrahedron. 2002;58:3423–3443.
- 16.Hudlicky M. Reductions in Organic Chemistry. American Chemical Society; 1996. [Google Scholar]
- 17.Brown HC, Mead EJ, Rao BCS. J. Am. Chem. Soc. 1955;77:6209–6213. [Google Scholar]
- 18.Gemal AL, Luche JL. J. Am. Chem. Soc. 1981;103:5454–5459. [Google Scholar]
- 19.For examples from recent literature, see: Scott MS, Luckhurst CA, Dixon DJ. Org. Lett. 2005;7:5813–5816. doi: 10.1021/ol052333c. Lauzon C, Charette AB. Org. Lett. 2006;8:2743–2745. doi: 10.1021/ol0607847. Nourry A, Legoupy S, Huet F. Tetrahedron Lett. 2007;48:6014–6018. Nordstrom LU, Vogt H, Madsen R. J. Am. Chem. Soc. 2008;130:17672–17673. doi: 10.1021/ja808129p. Beshore DC, Smith AB. J. Am. Chem. Soc. 2008;130:13778–13789. doi: 10.1021/ja804939r. Hsu HC, Hou DR. Tetrahedron Lett. 2009;50:7169–7171. Enck S, Kopp F, Marahiel MA, Geyer A. Org. Biomol. Chem. 2010 doi: 10.1039/b917549k. DOI: 10.1039/b917549k.
- 20.For recent examples, see: Yamazaki N, Suzuki H, Kibayashi C. J. Org. Chem. 1997;62:8280–8281. doi: 10.1021/jo9715579. Brummond KM, Lu JL. Org. Lett. 2001;3:1347–1349. doi: 10.1021/ol010029n. Hoffmann HMR, Frackenpohl J. Eur. J. Org. Chem. 2004:4293–4312. Yotphan S, Bergman RG, Ellman JA. J. Am. Chem. Soc. 2008;130:2452–2453. doi: 10.1021/ja710981b.
- 21.(a) Nakashimi TT, Maciel GE. Org. Magn. Reson. 1972;4:321–326. [Google Scholar]; (b) Hanisch P, Jones AJ, Casey AF, Coates JE. J. Chem. Soc., Perkin Trans. 2. 1977:1202–1208. [Google Scholar]
- 22.Leonard NJ, Oki M, Brader J, Boaz H. J. Am. Chem. Soc. 1955;77:6237–6239. [Google Scholar]
- 23.Quast U, Schneider W. Liebigs Ann. Chem. 1982:1501–1508. [Google Scholar]
- 24.(a) Leonard NJ, Sato T. J. Org. Chem. 1969;34:1066–1070. [Google Scholar]; (b) Heathcock CH, Blumenkopf TA, Smith KA. J. Org. Chem. 1989;54:1548–1562. [Google Scholar]; (c) Kim G, Chu-Moyer MY, Danishefsky SJ, Schulte GK. J. Am. Chem. Soc. 1993;115:30–39. [Google Scholar]; (d) Fleming JJ, McReynolds MD, Du Bois J. J. Am. Chem. Soc. 2007;129:9964–9975. doi: 10.1021/ja071501o. [DOI] [PubMed] [Google Scholar]; (e) Fürstner A, Korte A. Chem.-Asian J. 2008;3:310–318. doi: 10.1002/asia.200700288. [DOI] [PubMed] [Google Scholar]; (f) Becker MH, Chua P, Downham R, Douglas CJ, Garg NK, Hiebert S, Jaroch S, Matsuoka RT, Middleton JA, Ng FW, Overman LE. J. Am. Chem. Soc. 2007;129:11987–12002. doi: 10.1021/ja074300t. [DOI] [PubMed] [Google Scholar]
- 25.Leonard NJ, Oki M. J. Am. Chem. Soc. 1955 77;:6239–6240. [Google Scholar]
- 26.Szostak M. Ph. D. Dissertation. University of Kansas; 2009. [Google Scholar]
- 27.Petasis NA, Bzowej EI. J. Am. Chem. Soc. 1990;112:6392–6394. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.