

Abstract
Recessive mutations at the mouse pirouette (pi) locus result in hearing loss and vestibular dysfunction due to neuroepithelial defects in the inner ear. Using a positional cloning strategy, we have identified mutations in the gene Grxcr1 (glutaredoxin cysteine-rich 1) in five independent allelic strains of pirouette mice. We also provide sequence data of GRXCR1 from humans with profound hearing loss suggesting that pirouette is a model for studying the mechanism of nonsyndromic deafness DFNB25. Grxcr1 encodes a 290 amino acid protein that contains a region of similarity to glutaredoxin proteins and a cysteine-rich region at its C terminus. Grxcr1 is expressed in sensory epithelia of the inner ear, and its encoded protein is localized along the length of stereocilia, the actin-filament-rich mechanosensory structures at the apical surface of auditory and vestibular hair cells. The precise architecture of hair cell stereocilia is essential for normal hearing. Loss of function of Grxcr1 in homozygous pirouette mice results in abnormally thin and slightly shortened stereocilia. When overexpressed in transfected cells, GRXCR1 localizes along the length of actin-filament-rich structures at the dorsal-apical surface and induces structures with greater actin filament content and/or increased lengths in a subset of cells. Our results suggest that deafness in pirouette mutants is associated with loss of GRXCR1 function in modulating actin cytoskeletal architecture in the developing stereocilia of sensory hair cells.
Introduction
Studies of mouse genetic models have defined several genes that are required for normal maturation of stereocilia, the specialized actin-filament-rich microvilli of mechanosensory cells in the inner ear.1 In the normal inner ear, bundles containing 50 to 300 stereocilia are organized in a staircase arrangement at the apical surface of sensory cells, with multiple fine connections along their lengths that link neighboring stereocilia with one another.2,3 Deflection of bundles by auditory or vestibular stimuli and the associated gating of cation channels in the plasma membrane near the tip of individual stereocilium play key roles in mechanotransduction.4 The core of each stereocilium is composed of tightly packed bundles of actin filaments of the same relative orientation.5 During development, increases in the length and width of stereocilia require elongation of existing actin filaments, nucleation of additional filaments, and incorporation of these filaments into the core.6,7 Elongation occurs through addition of actin monomers at the barbed ends of filaments, which are positioned near the tips of stereocilia.8 Genes implicated in stereocilia maturation include those that are required for normal polarity of the bundle, for bundle organization and cohesion, and for appropriate growth of individual stereocilia.1
Mice homozygous for mutations at the pirouette (pi) locus exhibit vestibular defects and profound deafness.9–12 The stereocilia in early postnatal pirouette mutants have a distinctive thin appearance with some shortening, suggesting that the affected gene is necessary for processes that mainly increase the diameter of stereocilia and to some extent increase stereocilia growth during maturation.10–12 In the present study, we have used a positional cloning approach to identify mutations in the gene Grxcr1 (glutaredoxin cysteine-rich 1) that are responsible for the pirouette phenotype. This gene is expressed in neuroepithelial cells in the inner ear and encodes a protein with a domain that suggests a role in cellular processes influenced by reduction-oxidation (redox) regulation. Expression of GRXCR1 in cultured cell lines indicates localization to actin-filament-rich structures at the cell periphery. Expression of the protein in cochlear explant tissue reveals targeting to hair cell stereocilia or microvilli of nonsensory cells. Together with the stereocilia pathology in sensory hair cells of affected pirouette mice, this cellular localization suggests a role for GRXCR1 in regulation of actin filament architecture in hair cell stereocilia. We have also identified potential pathogenic variants of human GRXCR1 that are associated with congenital hearing loss, suggesting an evolutionarily conserved role for the gene in sensory function.
Material and Methods
Mutant Mice
The original pi mutation arose spontaneously on a C3H strain of mice9 and was maintained by repeated backcrossing to C57BL/6J mice. The resulting congenic strain (B6.C3-piJ; referred to hereafter as pi) was obtained from the cryopreservation laboratory at The Jackson Laboratory (Bar Harbor, ME, USA). The transgene insertion allele, pitg370, was generated by microinjection of a GABAA receptor β3 transgene into (C57BL/6J × C3H) F2 embryos.13 A second transgene insertion allele, pitde, was generated by microinjection of a human placental alkaline phosphatase genomic construct into (C57BL/6J × CBA/Ca) F2 embryos.14 Two previously uncharacterized pirouette mutations arose spontaneously at The Jackson Laboratory: nm2766 (pi2J) arose in a colony of DBA/2J mice; nm3325 (pi3J) arose in a colony of BKS.Cg-m+/+Leprdb/J mice. Each of the strains was maintained by homozygote-to-heterozygote matings. All mice were cared for in accordance with institutional animal-care standards.
Genetic Markers and Genotype Analysis in Mouse
F2 offspring (529 total; 133 [25.1%] affected) from (pi × CAST/EiJ) F1 intercrosses were genotyped for simple sequence length polymorphism (SSLP) markers from central chromosome 5 as previously described.15 Single-strand conformational polymorphisms (SSCP) were identified for Atp8a1 and Kctd8 as described previously,15 and genotypes were determined for F2 progeny with observed recombinations between D5Mit234 and D5Mit185. Sequences of primers used for SSCP analysis, as well as all other primers and hybridization probes mentioned below, are described in Table S1, available online. Genetic linkage analysis was performed as previously described.15 For localization of the nm2766 mutation, we performed a genome-wide linkage screen of 51 F2 progeny from an intercross of (DBA/2J-nm2766 × CAST/EiJ) F1 mice.
Gene Identification, cDNA Sequence Assembly, and Expression Studies
The exon content in the region was evaluated by Genscan analysis,16 by sequence similarity searches of public databases via the BLAST algorithm,17 and by scrutiny of public annotation of assembled mouse18 and human19 genomic sequence available from the ENSEMBL20 and UCSC Genome Browser21 projects. Predicted exons were verified by sequence analysis of available cDNA clones and by RT-PCR amplification with gene-specific primer sets. Templates for RT-PCR were prepared from brain and cochlear RNA obtained from normal and mutant mice, via methods described previously.15 In situ hybridization was performed on cochlear sections as described previously,22 with the use of α-[35S]UTP-labeled cRNA probes derived from sense and antisense templates of nucleotides 463 to 992 of the Grxcr1 cDNA.
Genomic DNA Analysis
pi2J and pi3J Alleles
Primer sets designed from genomic sequence within and upstream of Grxcr1 were used to amplify DNA from control mice and from mice homozygous for the pi2J and pi3J mutations.
pitg370 Allele
Southern blots prepared with genomic DNA from pitg370 homozygotes and control strains were hybridized sequentially with radiolabeled DNA probes, as previously described.15 Probes were derived by genomic PCR from intron 2 of Grxcr1; the second probe was a 1.2 kb SwaI-PvuII fragment of the pitg370 transgene construct.13
pitde Allele
Southern blots prepared with genomic DNA from pitde homozygotes and control strains were hybridized sequentially with probes derived by genomic PCR from intron 1 of Grxcr1, followed by a vector DNA probe (pGEM7zf; Promega, Madison, WI, USA), which contains sequences identical to a portion of the pitde transgene construct.
pi Allele
Southern blots prepared with genomic DNA from pi homozygotes and control strains were hybridized with radiolabeled DNA probes derived from intron 1 of Grxcr1 and from sequences more telomeric of the gene. Nested primers designed from exon 1 sequences were used for performing 3′ rapid amplification of cDNA ends (RACE).23 The positions of the estimated deletion (pi2J and pi3J) and inversion (pi) breakpoints in the mutants were determined from the July 2007 (mm9) genome data compiled at the Mouse Genome Browser, which used the Build 37 assembly (NCBI and the Mouse Genome Sequencing Consortium).
Protein Sequence Analysis
Sequence comparisons to protein databases were performed with the BLAST-PSI algorithm17 at the NCBI server. Orthologous proteins were identified from annotated sequence databases at NCBI and at the Ensembl Genome Server. Multiple sequence alignments were made with the use of CLUSTALV, with MegAlign software (Lasergene; Madison, WI, USA). Domain structure of the GRXCR1 protein sequence was evaluated with the use of the Conserved Domains Database.24 Comparisons to the SCOP protein domain database and prediction of secondary structure were performed with the 3D-PSSM algorithm25 via a server at the Imperial College of Science, Technology and Medicine, London, UK.
Grxcr1 Fusion Constructs
The entire 870 bp open reading frame of Grxcr1, plus partial 5′ and 3′ UTR sequences, were amplified from inner ear cDNA and cloned into the EcoRI and BamHI sites of pECFP-N1 and pEGFP-N1, upstream of the coding region for cyan (CFP) or green fluorescent protein (GFP) (BD Biosciences, Palo Alto, CA, USA).
Cell Culture and Expression Studies
COS-7 cells were transfected at subconfluency on glass coverslips with a Grxcr1-CFP construct, or a control pECFP-N1 vector, with the use of FuGene (Roche; Indianapolis, IN, USA). Cells were fixed, permeabilized, and incubated with an antibody specific for GFP or CFP (Chemicon; Temecula, CA, USA), secondary anti-rabbit IgG antibodies conjugated to Alexa 488 (Molecular Probes; Eugene, OR, USA), and rhodamine-phalloidin (Molecular Probes; Eugene, OR, USA). Differentiated LLC-PK1-CL4 (CL4) epithelial cells were transfected with a Grxcr1-GFP construct, fixed, permeabilized and incubated with Texas Red-phalloidin.26 Cells were analyzed with a Zeiss LSM 510 confocal microscope.
Cochlear Explant Studies
For explant studies, cochleae were dissected from C57BL/6J mice at postnatal day 4 (P4), cultured for 1 to 2 days in Dulbecco's modified Eagle's medium supplemented with 7% fetal bovine serum, transfected with a Helios gene gun (Biorad; Hercules, CA, USA) with the Grxcr1-GFP construct attached to 1.0 μm gold particles (Biorad), and processed for immunohistochemistry 24 hr later, as previously described.27 The images from explant transfections are representative of GRXCR1-GFP expression data from nine independent experiments (total number of expressing cell types: 9 inner hair cells [IHCs], 13 outer hair cells [OHCs], 11 vestibular hair cells, and more than 30 nonsensory epithelial cells).
GRXCR1 Antibodies and Tissue Immunocytochemistry
A peptide designed from the primary sequence of mouse GRXCR1 (N-NEQEKDQDNLLVLART-C) was covalently linked to KLH and injected into New Zealand rabbits (Proteintech Group; Chicago, IL, USA). Antiserum was purified with the use of an affinity column containing full-length recombinant GRXCR1, via previously described methods.28 Sensory epithelia from the inner ears of postnatal and adult mice were prepared by dissection of temporal bones, followed by removal of the otic capsule along with the lateral wall and tectorial membrane. With the use of a fine needle, organ of Corti spirals were isolated, fixed in 4% paraformaldehyde for 2 hr, washed in PBS, permeabilized for 20 min in 0.4% or 0.5% Triton X-100, and pretreated with 5% goat serum or 5% goat serum plus 2% BSA in PBS. Samples were then treated with anti-GRXCR1 antiserum or control preimmune serum, washed in PBS, and incubated with Alexa 568- or Alexa 488-conjugated anti-rabbit secondary antibodies (Molecular Probes) and with Alexa 488-phalloidin or rhodamine-phalloidin, respectively. After washing in PBS, samples were mounted in Prolong Gold (Molecular Probes) and analyzed with a Zeiss LSM 510 confocal microscope. Images were processed with Adobe Photoshop.
Mutation Screen in Human Pedigrees
Primers designed from sequence flanking each human GRXCR1 exon were used for amplifying genomic DNA from affected members of the original but unpublished DFNB25 family and from a panel of 400 probands of consanguineous Iranian parentage who were diagnosed with congenital severe-to-profound deafness presumed to be due to autosomal-recessive inheritance. Primer sequences are listed in Table S1. Sequences of the amplified products were compared to the public reference assembly NCBI Build 36.1 and to GRXCR1 sequences derived from 192 individuals from the Centre d'Étude du Polymorphisme Humain (CEPH) reference population and from 96 individuals with normal hearing from the Iranian population. All procedures were approved by human research institutional review boards at the Welfare Science and Rehabilitation University and the Iran University of Medical Sciences, Tehran, Iran, and the University of Iowa, Iowa City, Iowa, USA. All participants provided informed consent.
Results
Five Mutant Alleles of Pirouette
Previous mapping studies localized the original pi mutation to the central region of chromosome 5.11,29,30 Recently, we identified two additional mouse strains (nm2766 and nm3325) that exhibit similar vestibular and auditory deficits due to spontaneous recessive mutations. Analysis of progeny from an intercross of (DBA/2J-nm2766 × CAST/EiJ) F1 mice demonstrated linkage to microsatellite markers in an overlapping region of chromosome 5, with the following order: centromere – D5Mit79 – 8.1 ± 2.9 cM – D5Mit300 – 4.0 ± 2.0 cM – [D5Mit235, nm2766] – 3.0 ± 1.7 cM – D5Mit205 – telomere. A complementation test was carried out by crossing an affected male homozygote with a female heterozygous for the original pi allele. Of 18 total offspring, ten (56%) lacked a startle response to loud noise at weaning age and exhibited vestibular dysfunction. This failure to complement, along with the coincident linkage data, indicates that nm2766 is allelic to pi. Similarly, all 12 offspring from matings between nm3325 and nm2766 homozygotes exhibited vestibular and auditory defects, indicating noncomplementation of the mutations, consistent with a third spontaneous alteration at the pi locus. The nm2766 and nm3325 strains are therefore designated as pi2J and pi3J, respectively. Together with two transgene insertion mutants, pitg370 and pitde,11 five alleles of pirouette have now been described.
High-Resolution Genetic Mapping of the Pirouette Locus
Analysis of F2 offspring of a (pi × CAST/EiJ) F1 intercross defined a 0.2 cM candidate interval for the pi locus, between D5Mit111 and D5Mit185 (Figure S1). We identified two genes in this region of approximately 1.2 Mb (Figure 1A). One, Mm.332422, is located on the centromeric side of the region and is composed of four exons that encode a protein with sequence similarity to glutaredoxin proteins and a C-terminal cysteine-rich region (Grxcr1, glutaredoxin cysteine-rich 1). The other gene, Kctd8, located on the telomeric side, is composed of two exons separated by a large intron of 270 kb. This gene encodes a product with a protein-protein interaction domain also present in potassium channels.31
Figure 1.
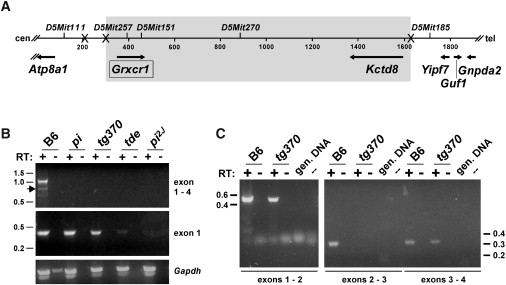
Grxcr1 Cochlear Expression Is Altered in the Pirouette Alleles
(A) Combined genetic and sequence-based map is shown, with Xs indicating the relative positions of recombination breakpoints detected in a (pi × CAST/EiJ) F1 intercross. Map length is indicated in 200 kb increments. The gray box represents the nonrecombinant region that is ≤ 0.2 cM in size (95% confidence interval). In addition to the two genes located within the nonrecombinant region (Grxcr1 and Kctd8), four genes are located in the immediate flanking regions: Atp8a1 (U75321),60Yipf7 (AF217188),61Guf1 (AK084627), and Gnpda2 (AK016785). The predicted transcriptional orientation of each gene is indicated by an arrow. Centromeric (cen) and telomeric (tel) orientation of the chromosome is indicated.
(B) RT-PCR products were amplified from cochlear RNA of C57BL/6J (B6) control mice and from homozygous pirouette mutants with the use of Grxcr1 primers complementary to sequences in exon1 and exon 4 (top panel) or within exon 1 (middle panel) and with the use of control Gapdh primers (bottom panel). Sequence analysis indicated that a minor product amplified from B6 RNA represents an alternatively spliced transcript lacking exon 2 (arrow). Positions of molecular-size standards are indicated at left in kilobases.
(C) RT-PCR products were amplified from cochlear RNA of C57BL/6J (B6) control mice and from homozygous pitg370 mutants with the use of Grxcr1 primers complementary to sequences in exons 1 and 2 (left), in exons 2 and 3 (middle), and in exons 3 and 4 (right). Control PCR reactions that lacked template (–) were also performed, along with reactions with genomic DNA used as template. RT indicates the presence or absence of reverse transcriptase in corresponding reactions.
Grxcr1 Mutations in the Five Pirouette Alleles
Given that chromosomal insertion of transgenes often alters the levels or sizes of endogenous transcripts, we evaluated expression of Kctd8 and Grxcr1 in brain and cochlear tissues from the transgenic pirouette alleles (pitg370 and pitde), as well as from two of the three spontaneous allelic strains (pi and pi2J). No alterations of Kctd8 transcripts were observed in either RNA blot or RT-PCR analyses in any of the four alleles (data not shown). The sequence of the Kctd8 transcript from the original pi allele also did not exhibit any coding region alterations (data not shown). None of the mutants, however, produced full-length Grxcr1 transcripts (exons 1–4) in the cochlea, although we could detect transcripts containing exon 1 sequences in pi, pitg370, and pitde (Figure 1B). We did not detect transcripts containing exons 2, 3, or 4 in the pi or pitde alleles (data not shown), suggesting the presence of mutations that block normal transcription or splicing of the gene in or near intron 1. We detected Grxcr1 transcripts from homozygous pitg370 mice that were correctly spliced between exons 1 and 2 and between exons 3 and 4, but we failed to detect transcripts with normal splicing between exons 2 and 3 (Figure 1C), consistent with a transgene insertion within intron 2.
To determine the basis for these alterations in Grxcr1 expression, we analyzed DNA for evidence of mutations in each of the pirouette strains. In pi2J homozygotes, genomic PCR demonstrated a deletion with a maximum size of approximately 200 kb that includes exon 1, nearly 125 kb of upstream sequence, and the majority of intron 1 (Figure 2A). The deletion breakpoint in intron 1 is within 12 kb of exon 2. Using a set of 33 primer pairs across this region, we identified a deletion in the pi3J strain with the same breakpoints as those present in pi2J (Figure S2 and Table S1). Analysis of normal genomic sequence in the region indicated tandem arrays of nearly full-length L1 repeat elements near the centromeric and telomeric deletion breakpoints, supporting nonhomologous recombination between these repeats as a probable deletion mechanism in both mutant strains. Using Southern blot analyses with probes derived from Grxcr1 and from the tde transgene, we detected a genomic rearrangement within intron 1 of pitde mice, consistent with insertion of a transgene array 32 kb upstream of exon 2 (Figure 2B). Similar Southern blot data indicated a genomic rearrangement within intron 2 of pitg370 mice, consistent with insertion of the tg370 transgene 25 kb downstream of exon 2 (Figure 2C). In the original pi allele, we identified hybrid transcripts in which exon 1 was spliced to a cryptic exon located over 600 kb telomeric of Grxcr1 (Figure 2D). The orientation of the cryptic exon and an associated splice site is consistent with a large inversion of genomic DNA in the region. We found evidence for genomic rearrangements in pi within intron 1 and telomeric of the cryptic exon, suggestive of inversion breakpoints in this allele (Figure 2E and Figure S3). The genetic distance of 0.3 cM (three recombinations per 1058 meioses) between the closest flanking recombinant markers D5Mit111 and D5Mit185, as determined in the pi mapping cross (Figure S1), and the physical distance of 1.6 Mb between these markers indicate a ratio of genetic distance to physical distance of 0.19 cM per Mb. This is relatively low as compared to the genome-wide average in mouse of approximately 0.5 cM per Mb and is consistent with a predicted suppressive effect on recombination of a large inversion in the mutant pi region.
Figure 2.
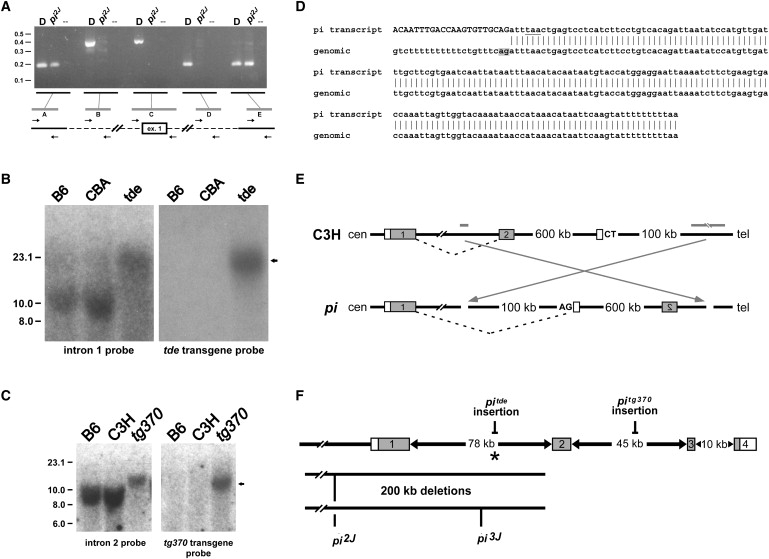
Genomic Alterations within Grxcr1 Are Present in Each of the Pirouette Alleles
(A) PCR products were amplified from genomic DNA of control DBA/2J (“D”) and homozygous pi2J mice, with the use of primer pairs (“A”–“E”) designed from sequences upstream of exon 1, immediately flanking exon 1, and within intron 1. The distance between pairs A and E in normal genomic DNA is approximately 200 kb and represents the maximum size of a deletion in the mutant. The distance between pairs B and D is approximately 175 kb and represents the minimal size of the deletion. The deletion breakpoint in intron 1 is located within the 9 kb region between pairs D (no amplification in pi2J DNA) and E (positive for amplification in both control and pi2J DNA), approximately 3 kb upstream of exon 2. Positions of molecular-size standards are indicated at left in kilobases.
(B) Southern blots containing EcoRV-digested genomic DNA from control (C57BL/6J, B6; CBA/CaJ, CBA) and homozygous pitde mice were hybridized with a probe derived from genomic sequences 32 kb upstream of exon 2 (left panel), stripped, and rehybridized with a probe complementary to the tde transgene construct (right panel). The arrow indicates common-sized 20 kb fragments that hybridize with both probes only in pitde DNA, consistent with insertion of the tde transgene into this region of intron 1.
(C) Southern blots containing EcoRI-digested genomic DNA from control (C57BL/6J, B6; C3He/FeJ, C3H) and homozygous pitg370 mice were hybridized with a probe derived from genomic sequences 25 kb downstream of exon 2 (left panel), stripped, and rehybridized with a probe complementary to the tg370 transgene construct (right panel). The arrow indicates common-sized 13 kb fragments that hybridize with both probes only in pitg370 DNA, consistent with insertion of the tg370 transgene into this region of intron 2.
(D) The sequence of a hybrid Grxcr1 cochlear transcript detected in affected pi mice. The top sequence is from a 3′ RACE product amplified with the use of nested primers derived from exon 1 of Grxcr1. The upper-case nucleotides indicate identity to the 3′ end of exon 1. The underlined nucleotides indicate a stop codon that would result in a truncated GRXCR1 protein. Vertical bars represent identity to genomic sequence located 600 kb telomeric of Grxcr1 (bottom, in telomere-to-centromere orientation). The gray box denotes a cryptic splice acceptor signal; an adjacent polypyrimidine tract is also present.
(E) Structure of a predicted chromosomal inversion in pi. Gray bars represent the relative position in the WT background strain C3H of putative inversion breakpoints located in intron 1 of Grxcr1 and in a region approximately 700 kb telomeric (see Figure S3). Gray boxes indicate the first two exons of Grxcr1. The open boxes represent the cryptic exon detected by 3′ RACE. An inversion in the pi allele (arrows) would place the cryptic exon in the correct orientation for inclusion in the hybrid transcript with exon 1 of Grxcr1, with the use of the AG splice acceptor signal shown.
(F) Summary of the Grxcr1 mutations in each of the pirouette alleles. The asterisk indicates the position of the putative centromeric inversion breakpoint in the original pi allele.
Interestingly, we detected the centromeric rearrangement in pi with the same intron 1 hybridization probe used for detecting the pitde rearrangement, indicating that these alterations are within 10 kb of one another and, together with the presence of the telomeric deletion breakpoints in pi2J and pi3J approximately 20 kb downstream, suggesting instability of this chromosomal region. The genomic alterations in each of the pirouette alleles disrupt the Grxcr1 transcription unit, are consistent with the abnormal transcript profiles in the cochlea, and are likely to represent null alleles that produce no full-length GRXCR1 protein (see Figure 2F for summary). The five independent alterations identified in Grxcr1 therefore provide strong evidence that defects in this gene are the basis for auditory and vestibular dysfunction in the mutant pirouette mice.
Grxcr1 Encodes a Protein with Evolutionarily Conserved Domain Architecture
The Grxcr1 cDNA encodes a predicted protein of 290 amino acids that exhibits strong sequence identity with the human orthologous gene located on 4p12 (268/290; 92%) and with orthologs from other vertebrates, including chicken and zebrafish (Figure 3A). Additional sequence comparisons indicated a domain in the central portion of the protein with significant similarity to glutaredoxins (Figure 3B). Glutaredoxins are enzymes that reduce oxidized cysteines of cellular proteins, using glutathione as an electron donor.32,33 The region of similarity with GRXCR1 includes a putative catalytic site and other conserved residues required for glutaredoxin function34 (Figure 3B). The predicted secondary structure of this region in GRXCR1 is also similar to the “thioredoxin fold” demonstrated by direct structural studies of glutaredoxin.35 The C-terminal 55 amino acids of GRXCR1 contain two groups of four cysteines arranged in a putative zinc finger configuration36 (Figure 3A). Proteins similar in sequence to GRXCR1 were also identified in a wide range of more divergent metazoan species (Figure S4). Although different at their N termini, this group of proteins exhibits approximately 40% sequence identity across the region of glutaredoxin similarity and the cysteine-rich C terminus of GRXCR1, and it defines a conserved arrangement of these two domains.
Figure 3.

Grxcr1 Encodes a Potential “Redox” Protein
(A) The predicted amino acid sequence of mouse GRXCR1 is indicated with one-letter abbreviations and aligned with orthologs from human (NP_001073945), chicken (ENSGALT00000037467), and zebrafish (ENSDARP00000102510). Black or gray shading indicates positions at which residues from at least three species are identical or biochemically similar, respectively. The region of similarity with glutaredoxin proteins is indicated with a gray bar, and conserved cysteines in the C terminus are marked with asterisks.
(B) Alignment of the central region of mouse GRXCR1 with pig glutaredoxin (Protein Data Bank no. d1kte). The paired catalytic Cys residues found in many glutaredoxins are indicated with asterisks. Residues predicted to be required for contact with glutathione are indicated with arrows. Secondary structure information: c, random coil; h, alpha helix; e, beta sheet.
Expression of Grxcr1 in the Inner Ear
To determine the distribution of Grxcr1 transcripts in wild-type (WT) mice, we screened RNA derived from a panel of adult tissues by RT-PCR. Grxcr1 transcripts containing exons 1–4 were present in cochlear RNA but were absent from the other tissues that were evaluated (Figure S5A). We also detected Grxcr1 transcripts containing exons 2–4, but not exon 1, in brain tissue under conditions of increased template concentration or increased PCR cycle number (data not shown). In situ hybridization of an antisense Grxcr1 probe to inner ear sections from mice at P5 were consistent with selective Grxcr1 expression in sensory hair cells and/or their supporting cells (Figure S5B).
An antiserum raised against a GRXCR1 peptide was used to immunostain inner ear tissue from WT C57BL/6J mice. The major site of GRXCR1 immunoreactivity in the cochlea at P1 and P5 was sensory hair cells and their stereocilia bundles, with apparently higher levels of GRXCR1 in OHC stereocilia bundles at P1 (Figures 4A and 4B) and in IHC stereocilia at P5 (Figures 4D and 4E). In P1 mice, GRXCR1 immunoreactivity was observed in each row of stereocilia within bundles, including immature, shorter stereocilia (Figures 4A and 4B). GRXCR1 was also present in the shorter apical microvilli of hair cells of the early postnatal cochlea (Figures 4A–4C, arrows). In the adult, GRXCR1 localized throughout the length of stereocilia of both OHC and IHC, with apparently similar levels in both types of bundles (Figures 4G and 4H). This localization pattern was also observed in all vestibular hair cell stereocilia, including those in the utricular maculae (Figures 4J and 4K). Interestingly, GRXCR1 immunoreactivity was most prominent in immature stereocilia bundles (arrows) of vestibular hair cells, which are characterized by small bundle size and a relatively short kinocilium, a microtubule-based structure that lacks actin filaments. A small number of immature stereocilia bundles coexist with mature bundles in postnatal and adult mouse vestibular sensory epithelium. The specificity of the anti-GRXCR1 antiserum was supported by the absence of GRXCR1 immunoreactivity in stereocilia or microvilli of cochlear hair cells from pi3J/pi3J mice, which lack exon 1 that encodes the immunizing peptide (Figure S6). In addition, in organ of Corti explants transfected with Grxcr1-GFP, transfected hair cells exhibited stronger stereocilia immunoreactivity in comparison to the immunoreactivity of neighboring untransfected hair cells, consistent with the ability of the antibody to recognize both endogenous GRXCR1 and overexpressed GRXCR1-GFP fusion protein (Figure S6). The GRXCR1 immunoreactivity in kinocilia that we observed in cochlear hair cells of early postnatal mice and in vestibular hair cells (Figures 4B, 4E, and 4K; arrowheads) may be nonspecific, given that we also observed a weak signal with this antibody in kinocilia of mutant mice (data not shown).
Figure 4.
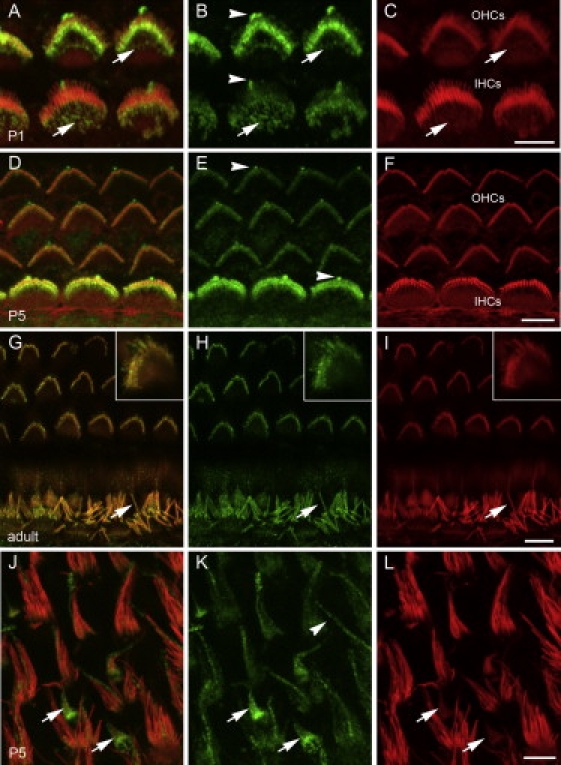
GRXCR1 Is Localized to Stereocilia on the Apical Surface of Sensory Hair Cells in the Inner Ear
Inner ear tissues from WT C57BL/6J mice at three different time points (P1, P5, and adult) were incubated with antiserum raised against GRXCR1 (green) and rhodamine-phalloidin (red). Panels at left are merged images.
(A–F) The major site of GRXCR1 immunoreactivity in the cochlea at P1 (A–C) and P5 (D–F) was sensory hair cells and their stereocilia bundles, with apparently higher levels of GRXCR1 in OHC stereocilia at P1 (A and B) and in IHC stereocilia at P5 (D and E). In P1 mice, GRXCR1 immunoreactivity was prominent in each row of stereocilia within bundles, including immature, shorter stereocilia that have lower relative levels of filamentous actin revealed by rhodamine-phalloidin staining (A–C). GRXCR1 immunoreactivity was also noted in apical microvilli of sensory cells at P1 (arrows, A–C) and in kinocilia at PI and P5 (arrowheads, B and E).
(G–I) In the adult, GRXCR1 was localized throughout the length of stereocilia of OHCs (inserts) and IHCs (arrows).
(J–L) GRXCR1 is localized along the length of vestibular hair cell stereocilia of the utricular macula. In vestibular epithelia, GRXCR1 staining was most prominent in immature stereocilia bundles (arrows). Immunoreactivity was also present in the kinocilia of vestibular sensory cells (arrowhead, K).
Scale bars represent 5 μm.
GRXCR1 Localization to Actin-Filament-Rich Structures in Transfected Cells
To begin characterizing the properties of GRXCR1, we transfected Grxcr1 fusion expression constructs into COS-7 fibroblasts, CL4 epithelial cells, and cultured explants of cochlear tissue derived from early postnatal mice. Treatment of the transfected fibroblast cells with rhodamine-phalloidin indicated that a significant fraction of GRXCR1-CFP protein colocalized with actin filaments in filopodia-like structures at the cortical and dorsal surfaces of the cells (Figure 5A). The number of dorsal projections was higher, and their actin filament content typically appeared more prominent, on cells expressing GRXCR1, suggesting a role for GRXCR1 in the induction and/or stabilization of these structures. In transfected CL4 cells, GRXCR1-GFP protein localized to the actin-filament-rich microvilli elaborated by these cells at their apical surface (Figure 5B). Grxcr1-GFP constructs delivered into IHCs of the cochlear explants produced fusion protein that was localized principally to stereocilia bundles (Figure 5C), consistent with immunolocalization of GRXCR1 in vivo. Similar GRXCR1-GFP localization was observed in transfected OHCs and vestibular hair cells (Figures S6E and S7B). In all instances, this fusion protein localized throughout the length of stereocilia in all rows of the transfected cells, including the immature shorter rows of stereocilia, which contain relatively lower levels of actin filaments (Figures 5C–5E, Figure S7A). This is consistent with the extensive GRXCR1 immunoreactivity detected in all stereocilia rows in early postnatal mice in vivo, including the immature rows (Figures 4A–4F, Figures S6B and S6C). GRXCR1-GFP fusion protein expressed in nonsensory epithelial cells in the transfected explants localized exclusively in apical microvilli. Of note, expression of the protein in nonsensory cells resulted in 2- to 5-fold increases in the length of microvilli relative to microvilli in untransfected cells. Figures 5F–5H depict a transfected nonsensory cell lateral to the sensory neuroepithelia of the organ of Corti. Similar microvillar elongation was also noted in supporting cells within the organ of Corti (Figures S7C and S7D) and in other types of nonsensory cells (Figure S7E). This is in contrast to GRXCR1-GFP expression in CL4 epithelial cells (Figure 5B) or sensory hair cells, in which such obvious changes in microvilli or stereocilia dimensions were not observed systematically. In transfected sensory hair cells, however, we occasionally noted an apparent moderate elongation of stereocilia (Figure S7A). Transfection of control GFP constructs resulted in expression of GFP throughout the entire cell and did not affect stereocilia or microvillar dimensions in sensory or nonsensory cells (data not shown).
Figure 5.
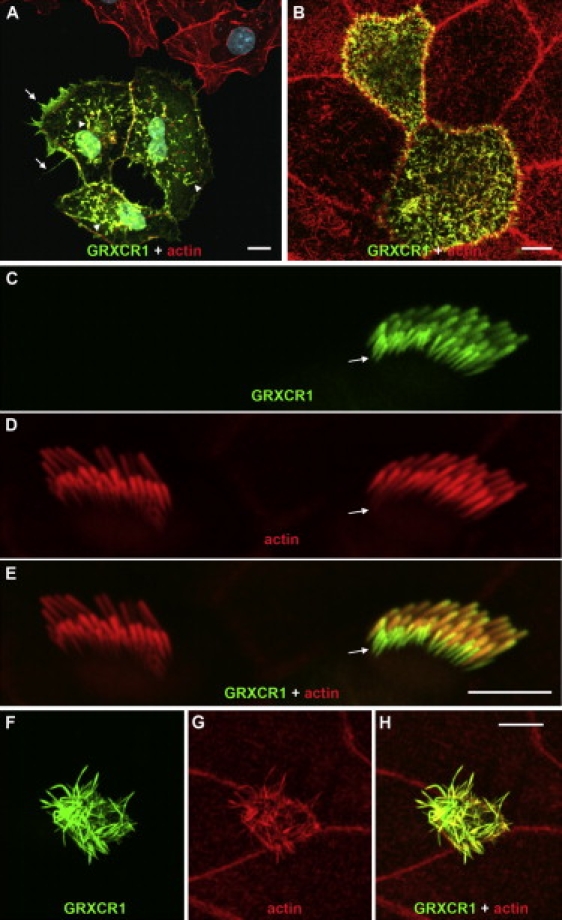
GRXCR1 Protein Localizes to Actin-Filament Bundles in Transfected Cells
(A) In COS-7 fibroblast cells transfected with Grxcr1-CFP, GRXCR1 (green) colocalizes with actin filaments (red) within filopodia-like structures at the cortical (arrows) and dorsal (arrowheads) surfaces of transfected cells. GRXCR1 did not colocalize significantly with actin stress fibers (data not shown). The number and actin filament content of dorsal projections in the GRXCR1-positive cells are increased relative to that of neighboring untransfected cells.
(B) GRXCR1-GFP colocalizes with actin filaments in the apical microvilli of transfected CL4 epithelial cells but does not appreciably alter microvilli dimensions.
(C–E) In transfected cochlear explants from WT mice, GRXCR1-GFP localized along the entire length of stereocilia of an inner hair cell, including the immature shorter row (arrows), which have lower actin filament content than that of longer rows. Stereocilia dimensions in GRXCR1-positive stereocilia bundles appeared similar to those of a neighboring untransfected hair cell (D, left).
(F–H) GRXCR1-GFP also colocalizes with actin filaments in the apical microvilli of nonsensory epithelial cells in transfected explant cultures. The GRXCR1-positive microvilli were substantially longer than those of neighboring untransfected cells.
Scale bars represent 10 μm (A) and 5 μm (B, E, and H).
Missense Variants in Human GRXCR1 Are Associated with Nonsyndromic Hearing Loss
The human GRXCR1 gene is located within a region on chromosome 4p15.3-q12 that was defined as the candidate interval for the nonsyndromic hearing loss locus DFNB25 (Hereditary Hearing Loss Homepage). Although we did not find coding sequence mutations in the original DFNB25 family, in our screen of 400 probands of consanguineous Iranian parentage diagnosed with congenital severe-to-profound deafness, we identified five missense substitutions of GRXCR1. Of these changes, two result in the amino acid substitutions p.G51E (c.152G>A) and p.G91V (c.272G>T) and are likely to be neutral, given that these variants were found in control populations and are in residues that are not well conserved through evolution. The other three variants were identified in two pedigrees. The missense variants p.G64S (c.190G>A) and p.F153V (c.457T>G) were identified in the homozygous state in affected individual IV-1 in family A (Figure 6A). These residues are located in the N-terminal region and the glutaredoxin-related domain of GRXCR1, respectively. Both sites are well conserved through evolution. The third variant, p.P38L (c.113C>T), also located in the N-terminal domain, was found in the homozygous state in affected individual IV-1 in family B (Figure 6B). This residue is less conserved, but like p.G64S and p.F153V, p.P38L variants were not found in a total of 96 ethnically matched individuals with normal hearing or in another control population of 192 individuals. In both families, parents of the probands were heterozygous for the corresponding variant alleles. We were unable to assess allele segregation in siblings because DNA samples were unavailable.
Figure 6.
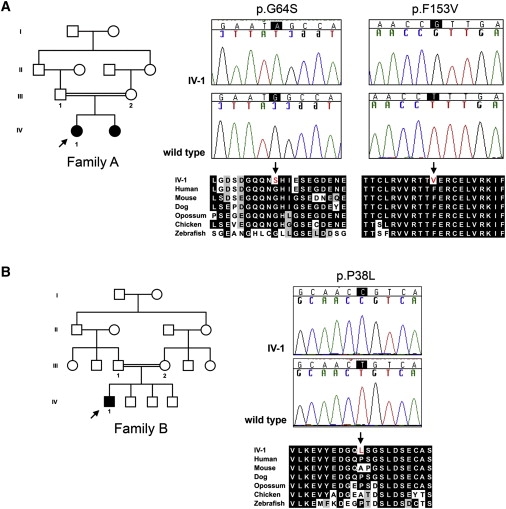
Missense Variants in Human GRXCR1 Are Associated with Congenital Nonsyndromic Hearing Loss
(A and B) Consanguineous pedigrees, sequence chromatograms of probands (arrows), and partial alignments of GRXCR1 encoded by genomic sequences from probands and controls and from other vertebrate species are shown for the double homozygous variants p.G64S (c.190G>A) and p.F153V (c.457T>G) (A) and for the single variant p.P38L (c.113C>T) (B).
Discussion
We have identified mutations in five independent alleles of pirouette that prevent synthesis of full-length Grxcr1 transcripts and protein and are likely therefore to represent null alleles at the locus. This hypothesis is supported by the identical pathologies observed in the inner ears of the mutant mice (data not shown and refs.11,12). Grxcr1 transcripts are expressed in sensory neuroepithelia in the inner ear, and GRXCR1 protein is localized to the stereocilia of sensory hair cells, consistent with the specific pathologies identified in affected pirouette mice. Ectopic expression of GRXCR1 in cultured cells indicates selective targeting to actin-filament-rich structures and suggests a possible role in regulating the turnover or stability of these dynamic structures that could account for the defects in actin architecture observed in sensory cells of pirouette mice. The localization of GRXCR1 protein to filopodia-like projections, microvilli, and stereocilia suggests that GRXCR1 has a preference for structures that contain parallel actin bundles.37 Consistent with such a preference, we did not observe significant association of GRXCR1 with actin stress fibers (data not shown), which feature a qualitatively different type of actin bundle. The additional pathology noted in a subset of pirouette sensory cells was the actin-filament-rich cytocaud,10,11 also supporting a role for GRXCR1 in regulating actin filament architecture.
The similarity of the central region of GRXCR1 with glutaredoxin proteins suggests a potential functional relationship with this family of proteins. Glutaredoxins are found in both prokaryotes and eukaryotes and catalyze reduction of disulfide bonds in cellular proteins through exchange reactions between cysteine thiol groups of the enzymes and their disulfide substrates.32,33 Genetic and biochemical studies have implicated glutaredoxins and related proteins in several physiological processes, including the activation of metabolic enzymes by reduction of specific cystine pairs, in cellular stress responses to reactive oxygen species through a more general reducing effect on cysteine thiols of oxidized proteins,38 and in control of signal transduction pathways by regulation of the redox state of key target proteins.39–41 Glutaredoxin proteins typically require dual cysteines (CXXC; X = any amino acid) in their active sites.33 Although thiol transferase activity has been demonstrated for a number of redox proteins with single cysteines in their active sites (e.g.,34,42), the comparable region in GRXCR1 and a conserved group of metazoan proteins (F/YXXC) (Figure S4) may lack enzymatic activity. The putative thioredoxin fold in GRXCR1 could instead act as a protein-protein interface, as predicted for a similar domain in the PICOT protein.43
Concurrent increases in length and diameter of stereocilia occur during late embryogenesis and early postnatal life in mammals.44,45 GRXCR1 localization in stereocilia bundles in sensory cells of early postnatal mice is consistent with a local role for this protein in the process of stereocilia growth. The relatively higher levels of GRXCR1 immunoreactivity in bundles of OHCs versus IHCs at P1 in comparison to the higher levels in IHCs versus OHCs at P5 may reflect differences in the developmental stage of stereocilia thickening and growth in the two types of hair cells. The presence of GRXCR1 in adult stereocilia suggests that the protein may have a role in maintenance of the adult bundle.
Through analysis of mouse mutant strains, several genes have been implicated, similar to Grxcr1, in the control of stereocilia dimensions.46,47 The localization of the GRXCR1 protein along the entire length of stereocilia is similar to that of espin, an actin-bundling protein that localizes to the filament cores of stereocilia in sensory cells of the inner ear.48 Mutation of espin (Espn) in the jerker mouse strain is associated with shortened, thin stereocilia as well as profound deafness and balance defects.48,49 In transfected epithelial cells, espin cross-links increase the steady-state length of apical microvilli through effects on the treadmilling actin bundle at their core.26 Specific isoforms of espin may also impact actin filament elongation directly at barbed ends.50 We have previously demonstrated that espin and plastin 1 (fimbrin), a second actin-bundling protein normally present in stereocilia bundles,51,52 are also expressed in stereocilia of affected pirouette mice, indicating that gross alterations in the expression levels or localization of these two proteins do not appear to be involved in the pirouette pathology.11
Although the influence of GRXCR1 on actin-filament-rich structures could be due to a direct interaction with actin, similar to that of espin, the primary amino acid sequence of GRXCR1 does not contain any known actin-binding motifs.53 Further analysis of purified GRXCR1 protein will be required to uncover domains that affect actin filaments. The thin stereocilia with somewhat shortened length in pirouette mutant mice carrying loss-of-function Grxcr1 mutations indicate a critical role for GRXCR1 in development of normal diameter and growth of stereocilia. However, the extent of the shortening of pirouette stereocilia is much less than that of stereocilia in shaker 2 mice, in which deafness is associated with a myosin 15a motor mutation.54 In addition, the phenotypes of single and double mutants of pi and shaker 2 mice suggest at least some independence in differential regulation of stereocilia dimensions in these mouse models.11,12,55 Increases both in length and in diameter of actin-filament-rich processes require net actin monomer polymerization. For overall increases in structure length, the barbed ends of all actin filaments in the core must undergo a net increase in monomer addition, whereas a specific increase in diameter requires addition to newly nucleated filaments. Our demonstration that transfected GRXCR1 can induce elongation of microvilli in nonsensory cells in the cochlea and more robust dorsal actin-filament-rich structures in COS7 cells suggests that, in particular cellular contexts, GRXCR1 may increase actin polymerization. The lack of obvious increases in stereocilia diameter in WT transfected hair cells expressing GRXCR1-GFP is similar to the situation with GFP-myosin 15a expression. Myosin 15a is necessary for establishing normal stereocilia length in vivo but does not cause additional lengthening of stereocilia when it is overexpressed in WT hair cells.56 Endogenous myosin 15a and GRXCR1 are probably subject to developmental regulation by factors that could also control activity of overexpressed versions of both proteins in sensory cells. Similarly, the lack of significant increases in actin content upon GRXCR1 transfection of CL4 epithelial cells indicates that these cells may contain factors that modulate an effect of GRXCR1 on actin polymerization.
Mutations in genes that are critical for stereocilia development, such as MYO15A and ESPN, also underlie hearing loss in humans.57,58 A screen for GRXCR1 variants in individuals with congenital hearing loss identified at least two missense variants that may be pathogenic. The variants p.G64S and p.F153V were identified in the homozygous state in a single pedigree but were absent from a large number of control chromosomes. Although this pedigree is too small to provide supporting linkage evidence, these variants are plausible candidates for causative mutations. Both residues are highly conserved across a large collection of vertebrates, with the p.F153 site conserved in Grxcr1 homologs even in some plant species (Figure S4). The p.F153V variant is located in the central domain of GRXCR1 that is similar to glutaredoxin proteins. Sequence alignments suggest that this position corresponds to the N-terminal cysteine required for redox activity in dithiol glutaredoxins. The effect of this alteration on putative redox function of GRXCR1 is unclear at present. The p.G64S variant is located in the N-terminal region of GRXCR1, which overall is well conserved across vertebrates but diverges in nonvertebrate species. A third missense variant, p.P38L, is also located in the N-terminal divergent domain, but it is less well conserved. Although this variant was not found in a large collection of control chromosomes, it may represent a rare neutral polymorphism. The absence of mutations in the coding sequence of GRXCR1 in the original DNFB25 family is consistent with the possibility of mutations in other regions of this gene or with a second deafness-associated gene in this interval. Recently, Schraders et al. have found GRXCR1 mutations to underlie recessive, nonsyndromic hearing loss in additional families.59
Collectively, our data suggest that GRXCR1 plays a conserved, critical role in the mammalian ear. Evaluation of the potential effects of the p.G64S, p.F153V, and p.P38L variants on GRXCR1 function will be required to determine their potential role in hereditary hearing loss. Additional study of the biochemical and cellular function of GRXCR1 will provide insight into pathways that are critical for stereocilia development and for inner ear function.
Supplemental Data
Supplemental Data include seven figures and one table and can be found with this article online at http://www.ajhg.org.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
3D-PSSM Server, http://www.sbg.bio.ic.ac.uk/servers/3dpssm/
Conserved Domains Database, http://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml
Ensemble Genome Server, http://www.ensembl.org/index.html
Hereditary Hearing Loss Homepage, http://webhost.ua.ac.be/hhh/
Mouse Genome Browser, http://genome.ucsc.edu/cgi-bin/hgGateway
NCBI Server, http://www.ncbi.nlm.nih.gov/
Accession Numbers
The GenBank accession numbers for candidate gene sequences reported in this paper are AY616753 (mouse Grxcr1 [Mm.332422]) and AY615967 (mouse Kctd8).
Acknowledgments
We thank Margaret Lomax and Sally Camper for critical reading of the manuscript, and we thank Ursula Jakob, Jochen Schacht, Suhua Sha, and John Moran for helpful discussions. We acknowledge Alyce LaDeau and Kate Donnell for identification of the pi2J and pi3J mutants, Sandra Gray for help with mouse husbandry at The Jackson Laboratory, Tzy-Wen Gong for tissue panel RNA and inner ear dissection expertise, Robert J. Morell for in situ hybridization advice, and Bruce Donohoe and Chris Edwards for assistance with confocal microscopy analysis. We thank Sarah Davis, Joe Schacht, Junghan Kim, and Sue Lee for excellent technical assistance. We also appreciate the genomic sequencing of several BAC clones by the NIH Mouse Genome Sequencing Network, including the group headed by Bruce Birren and Ken Dewar at MIT. This work was supported by a grant from the Deafness Research Foundation (D.C.K.), by NIH-NIDCD grants R29-DC03049 (D.C.K.), R01-DC003049 (D.C.K.), R01-DC004314 (J.R.B.), R01-DC002842 (R.J.H.S.), T32-DC000011 (H.O.), and P30-DC05188 (D.C.K. and Y.R.), by NIH-NIDCD contract DC62108 (K.R.J.), and by NIH-NIDCD intramural funds Z01-DC000048 (T.B.F.).
References
- 1.Petit C., Richardson G.P. Linking genes underlying deafness to hair-bundle development and function. Nat. Neurosci. 2009;12:703–710. doi: 10.1038/nn.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lim D.J., Anniko M. Developmental morphology of the mouse inner ear. A scanning electron microscopic observation. Acta Otolaryngol. 1985;422(Supplement):1–69. [PubMed] [Google Scholar]
- 3.Goodyear R.J., Marcotti W., Kros C.J., Richardson G.P. Development and properties of stereociliary link types in hair cells of the mouse cochlea. J. Comp. Neurol. 2005;485:75–85. doi: 10.1002/cne.20513. [DOI] [PubMed] [Google Scholar]
- 4.Gillespie P.G., Müller U. Mechanotransduction by hair cells: models, molecules, and mechanisms. Cell. 2009;139:33–44. doi: 10.1016/j.cell.2009.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flock A., Cheung H.C. Actin filaments in sensory hairs of inner ear receptor cells. J. Cell Biol. 1977;75:339–343. doi: 10.1083/jcb.75.2.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tilney L.G., Tilney M.S., Saunders J.S., DeRosier D.J. Actin filaments, stereocilia, and hair cells of the bird cochlea. III. The development and differentiation of hair cells and stereocilia. Dev. Biol. 1986;116:100–118. doi: 10.1016/0012-1606(86)90047-3. [DOI] [PubMed] [Google Scholar]
- 7.Tilney L.G., DeRosier D.J. Actin filaments, stereocilia, and hair cells of the bird cochlea. IV. How the actin filaments become organized in developing stereocilia and in the cuticular plate. Dev. Biol. 1986;116:119–129. doi: 10.1016/0012-1606(86)90048-5. [DOI] [PubMed] [Google Scholar]
- 8.Schneider M.E., Belyantseva I.A., Azevedo R.B., Kachar B. Rapid renewal of auditory hair bundles. Nature. 2002;418:837–838. doi: 10.1038/418837a. [DOI] [PubMed] [Google Scholar]
- 9.Woolley G.W., Dickie M.M. Pirouetting mice. J. Hered. 1945;36:281–284. [Google Scholar]
- 10.Beyer L.A., Odeh H., Probst F.J., Lambert E.H., Dolan D.F., Camper S.A., Kohrman D.C., Raphael Y. Hair cells in the inner ear of the pirouette and shaker 2 mutant mice. J. Neurocytol. 2000;29:227–240. doi: 10.1023/a:1026515619443. [DOI] [PubMed] [Google Scholar]
- 11.Odeh H., Hagiwara N., Skynner M.J., Mitchem K., Beyer L., Allen N., Brilliant M., Lebart M.C., Dolan D.F., Raphael Y. Characterization of two transgene insertional mutations at pirouette, a mouse deafness locus. Audiol. Neurootol. 2004;9:303–314. doi: 10.1159/000080701. [DOI] [PubMed] [Google Scholar]
- 12.Erven A., Skynner M.J., Okumura K., Takebayashi S.I., Brown S.D., Steel K.P., Allen N.D. A novel stereocilia defect in sensory hair cells of the deaf mouse mutant Tasmanian devil. Eur. J. Neurosci. 2002;16:1433–1441. doi: 10.1046/j.1460-9568.2002.02213.x. [DOI] [PubMed] [Google Scholar]
- 13.Hagiwara N., Katarova Z., Siracusa L.D., Brilliant M.H. Nonneuronal expression of the GABA(A) beta3 subunit gene is required for normal palate development in mice. Dev. Biol. 2003;254:93–101. doi: 10.1016/s0012-1606(02)00030-1. [DOI] [PubMed] [Google Scholar]
- 14.Skynner M.J., Drage D.J., Dean W.L., Turner S., Watt D.J., Allen N.D. Transgenic mice ubiquitously expressing human placental alkaline phosphatase (PLAP): an additional reporter gene for use in tandem with beta-galactosidase (lacZ) Int. J. Dev. Biol. 1999;43:85–90. [PubMed] [Google Scholar]
- 15.Mitchem K.L., Hibbard E., Beyer L.A., Bosom K., Dootz G.A., Dolan D.F., Johnson K.R., Raphael Y., Kohrman D.C. Mutation of the novel gene Tmie results in sensory cell defects in the inner ear of spinner, a mouse model of human hearing loss DFNB6. Hum. Mol. Genet. 2002;11:1887–1898. doi: 10.1093/hmg/11.16.1887. [DOI] [PubMed] [Google Scholar]
- 16.Burge C., Karlin S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997;268:78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- 17.Altschul S.F., Madden T.L., Schäffer A.A., Zhang J., Zhang Z., Miller W., Lipman D.J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Waterston R.H., Lindblad-Toh K., Birney E., Rogers J., Abril J.F., Agarwal P., Agarwala R., Ainscough R., Alexandersson M., An P., Mouse Genome Sequencing Consortium Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 19.Lander E.S., Linton L.M., Birren B., Nusbaum C., Zody M.C., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W., International Human Genome Sequencing Consortium Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 20.Hubbard T., Barker D., Birney E., Cameron G., Chen Y., Clark L., Cox T., Cuff J., Curwen V., Down T. The Ensembl genome database project. Nucleic Acids Res. 2002;30:38–41. doi: 10.1093/nar/30.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kurima K., Peters L.M., Yang Y., Riazuddin S., Ahmed Z.M., Naz S., Arnaud D., Drury S., Mo J., Makishima T. Dominant and recessive deafness caused by mutations of a novel gene, TMC1, required for cochlear hair-cell function. Nat. Genet. 2002;30:277–284. doi: 10.1038/ng842. [DOI] [PubMed] [Google Scholar]
- 23.Frohman M.A., Dush M.K., Martin G.R. Rapid production of full-length cDNAs from rare transcripts: amplification using a single gene-specific oligonucleotide primer. Proc. Natl. Acad. Sci. USA. 1988;85:8998–9002. doi: 10.1073/pnas.85.23.8998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marchler-Bauer A., Anderson J.B., DeWeese-Scott C., Fedorova N.D., Geer L.Y., He S., Hurwitz D.I., Jackson J.D., Jacobs A.R., Lanczycki C.J. CDD: a curated Entrez database of conserved domain alignments. Nucleic Acids Res. 2003;31:383–387. doi: 10.1093/nar/gkg087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kelley L.A., MacCallum R.M., Sternberg M.J. Enhanced genome annotation using structural profiles in the program 3D-PSSM. J. Mol. Biol. 2000;299:499–520. doi: 10.1006/jmbi.2000.3741. [DOI] [PubMed] [Google Scholar]
- 26.Loomis P.A., Zheng L., Sekerková G., Changyaleket B., Mugnaini E., Bartles J.R. Espin cross-links cause the elongation of microvillus-type parallel actin bundles in vivo. J. Cell Biol. 2003;163:1045–1055. doi: 10.1083/jcb.200309093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Belyantseva I.A., Boger E.T., Friedman T.B. Myosin XVa localizes to the tips of inner ear sensory cell stereocilia and is essential for staircase formation of the hair bundle. Proc. Natl. Acad. Sci. USA. 2003;100:13958–13963. doi: 10.1073/pnas.2334417100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bartles J.R., Wierda A., Zheng L. Identification and characterization of espin, an actin-binding protein localized to the F-actin-rich junctional plaques of Sertoli cell ectoplasmic specializations. J. Cell Sci. 1996;109:1229–1239. doi: 10.1242/jcs.109.6.1229. [DOI] [PubMed] [Google Scholar]
- 29.Dickie M.M., Woolley G.W. Linkage studies with the pirouette gene. J. Hered. 1946;37:335–337. doi: 10.1093/oxfordjournals.jhered.a105550. [DOI] [PubMed] [Google Scholar]
- 30.Lane P.W. Linkage groups 3 and XVII in the mouse and the position of the light-ear locus. J. Hered. 1967;58:21–24. doi: 10.1093/oxfordjournals.jhered.a107531. [DOI] [PubMed] [Google Scholar]
- 31.Bixby K.A., Nanao M.H., Shen N.V., Kreusch A., Bellamy H., Pfaffinger P.J., Choe S. Zn2+-binding and molecular determinants of tetramerization in voltage-gated K+ channels. Nat. Struct. Biol. 1999;6:38–43. doi: 10.1038/4911. [DOI] [PubMed] [Google Scholar]
- 32.Holmgren A. Antioxidant function of thioredoxin and glutaredoxin systems. Antioxid. Redox Signal. 2000;2:811–820. doi: 10.1089/ars.2000.2.4-811. [DOI] [PubMed] [Google Scholar]
- 33.Holmgren A. Thioredoxin and glutaredoxin systems. J. Biol. Chem. 1989;264:13963–13966. [PubMed] [Google Scholar]
- 34.Bellí G., Polaina J., Tamarit J., De La Torre M.A., Rodríguez-Manzaneque M.T., Ros J., Herrero E. Structure-function analysis of yeast Grx5 monothiol glutaredoxin defines essential amino acids for the function of the protein. J. Biol. Chem. 2002;277:37590–37596. doi: 10.1074/jbc.M201688200. [DOI] [PubMed] [Google Scholar]
- 35.Katti S.K., Robbins A.H., Yang Y., Wells W.W. Crystal structure of thioltransferase at 2.2 A resolution. Protein Sci. 1995;4:1998–2005. doi: 10.1002/pro.5560041005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matthews J.M., Sunde M. Zinc fingers—folds for many occasions. IUBMB Life. 2002;54:351–355. doi: 10.1080/15216540216035. [DOI] [PubMed] [Google Scholar]
- 37.Bartles J.R. Parallel actin bundles and their multiple actin-bundling proteins. Curr. Opin. Cell Biol. 2000;12:72–78. doi: 10.1016/s0955-0674(99)00059-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fernandes A.P., Holmgren A. Glutaredoxins: glutathione-dependent redox enzymes with functions far beyond a simple thioredoxin backup system. Antioxid. Redox Signal. 2004;6:63–74. doi: 10.1089/152308604771978354. [DOI] [PubMed] [Google Scholar]
- 39.Rhee S.G., Chang T.S., Bae Y.S., Lee S.R., Kang S.W. Cellular regulation by hydrogen peroxide. J. Am. Soc. Nephrol. 2003;14(8, Suppl 3):S211–S215. doi: 10.1097/01.asn.0000077404.45564.7e. [DOI] [PubMed] [Google Scholar]
- 40.Rhee S.G. Redox signaling: hydrogen peroxide as intracellular messenger. Exp. Mol. Med. 1999;31:53–59. doi: 10.1038/emm.1999.9. [DOI] [PubMed] [Google Scholar]
- 41.Finkel T. Reactive oxygen species and signal transduction. IUBMB Life. 2001;52:3–6. doi: 10.1080/15216540252774694. [DOI] [PubMed] [Google Scholar]
- 42.Board P.G., Coggan M., Chelvanayagam G., Easteal S., Jermiin L.S., Schulte G.K., Danley D.E., Hoth L.R., Griffor M.C., Kamath A.V. Identification, characterization, and crystal structure of the Omega class glutathione transferases. J. Biol. Chem. 2000;275:24798–24806. doi: 10.1074/jbc.M001706200. [DOI] [PubMed] [Google Scholar]
- 43.Jeong D., Kim J.M., Cha H., Oh J.G., Park J., Yun S.H., Ju E.S., Jeon E.S., Hajjar R.J., Park W.J. PICOT attenuates cardiac hypertrophy by disrupting calcineurin-NFAT signaling. Circ. Res. 2008;102:711–719. doi: 10.1161/CIRCRESAHA.107.165985. [DOI] [PubMed] [Google Scholar]
- 44.Kaltenbach J.A., Falzarano P.R., Simpson T.H. Postnatal development of the hamster cochlea. II. Growth and differentiation of stereocilia bundles. J. Comp. Neurol. 1994;350:187–198. doi: 10.1002/cne.903500204. [DOI] [PubMed] [Google Scholar]
- 45.Zine A., Romand R. Development of the auditory receptors of the rat: a SEM study. Brain Res. 1996;721:49–58. doi: 10.1016/0006-8993(96)00147-3. [DOI] [PubMed] [Google Scholar]
- 46.Manor U., Kachar B. Dynamic length regulation of sensory stereocilia. Semin. Cell Dev. Biol. 2008;19:502–510. doi: 10.1016/j.semcdb.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brown S.D., Hardisty-Hughes R.E., Mburu P. Quiet as a mouse: dissecting the molecular and genetic basis of hearing. Nat. Rev. Genet. 2008;9:277–290. doi: 10.1038/nrg2309. [DOI] [PubMed] [Google Scholar]
- 48.Zheng L., Sekerková G., Vranich K., Tilney L.G., Mugnaini E., Bartles J.R. The deaf jerker mouse has a mutation in the gene encoding the espin actin-bundling proteins of hair cell stereocilia and lacks espins. Cell. 2000;102:377–385. doi: 10.1016/s0092-8674(00)00042-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rzadzinska A., Schneider M., Noben-Trauth K., Bartles J.R., Kachar B. Balanced levels of Espin are critical for stereociliary growth and length maintenance. Cell Motil. Cytoskeleton. 2005;62:157–165. doi: 10.1002/cm.20094. [DOI] [PubMed] [Google Scholar]
- 50.Salles F.T., Merritt R.C., Manor U., Dougherty G.W., Sousa A.D., Moore J.E., Yengo C.M., Dosé A.C., Kachar B. Myosin IIIa boosts elongation of stereocilia by transporting espin 1 to the plus ends of actin filaments. Nat. Cell Biol. 2009;11:443–450. doi: 10.1038/ncb1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Daudet N., Lebart M.C. Transient expression of the t-isoform of plastins/fimbrin in the stereocilia of developing auditory hair cells. Cell Motil. Cytoskeleton. 2002;53:326–336. doi: 10.1002/cm.10092. [DOI] [PubMed] [Google Scholar]
- 52.Flock A., Bretscher A., Weber K. Immunohistochemical localization of several cytoskeletal proteins in inner ear sensory and supporting cells. Hear. Res. 1982;7:75–89. doi: 10.1016/0378-5955(82)90082-x. [DOI] [PubMed] [Google Scholar]
- 53.Revenu C., Athman R., Robine S., Louvard D. The co-workers of actin filaments: from cell structures to signals. Nat. Rev. Mol. Cell Biol. 2004;5:635–646. doi: 10.1038/nrm1437. [DOI] [PubMed] [Google Scholar]
- 54.Probst F.J., Fridell R.A., Raphael Y., Saunders T.L., Wang A., Liang Y., Morell R.J., Touchman J.W., Lyons R.H., Noben-Trauth K. Correction of deafness in shaker-2 mice by an unconventional myosin in a BAC transgene. Science. 1998;280:1444–1447. doi: 10.1126/science.280.5368.1444. [DOI] [PubMed] [Google Scholar]
- 55.Karolyi I.J., Probst F.J., Beyer L., Odeh H., Dootz G., Cha K.B., Martin D.M., Avraham K.B., Kohrman D., Dolan D.F. Myo15 function is distinct from Myo6, Myo7a and pirouette genes in development of cochlear stereocilia. Hum. Mol. Genet. 2003;12:2797–2805. doi: 10.1093/hmg/ddg308. [DOI] [PubMed] [Google Scholar]
- 56.Belyantseva I.A., Boger E.T., Naz S., Frolenkov G.I., Sellers J.R., Ahmed Z.M., Griffith A.J., Friedman T.B. Myosin-XVa is required for tip localization of whirlin and differential elongation of hair-cell stereocilia. Nat. Cell Biol. 2005;7:148–156. doi: 10.1038/ncb1219. [DOI] [PubMed] [Google Scholar]
- 57.Wang A., Liang Y., Fridell R.A., Probst F.J., Wilcox E.R., Touchman J.W., Morton C.C., Morell R.J., Noben-Trauth K., Camper S.A., Friedman T.B. Association of unconventional myosin MYO15 mutations with human nonsyndromic deafness DFNB3. Science. 1998;280:1447–1451. doi: 10.1126/science.280.5368.1447. [DOI] [PubMed] [Google Scholar]
- 58.Naz S., Griffith A.J., Riazuddin S., Hampton L.L., Battey J.F., Khan S.N., Riazuddin S., Wilcox E.R., Friedman T.B. Mutations of ESPN cause autosomal recessive deafness and vestibular dysfunction. J. Med. Genet. 2004;41:591–595. doi: 10.1136/jmg.2004.018523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schraders M., Lee K., Oostrik J., Huygen P.L.M., Ali G., Hoefsloot L.H., Veltman J.A., Cremers F.B.M., Basit S., Ansar M. Homozygosity Mapping Reveals Mutations of GRXCR1 as a cause of autosomal- recessive nonsyndromic hearing impairment. Am. J. Hum. Genet. 2010;86:138–147. doi: 10.1016/j.ajhg.2009.12.017. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Halleck M.S., Pradhan D., Blackman C., Berkes C., Williamson P., Schlegel R.A. Multiple members of a third subfamily of P-type ATPases identified by genomic sequences and ESTs. Genome Res. 1998;8:354–361. doi: 10.1101/gr.8.4.354. [DOI] [PubMed] [Google Scholar]
- 61.Tang B.L., Ong Y.S., Huang B., Wei S., Wong E.T., Qi R., Horstmann H., Hong W. A membrane protein enriched in endoplasmic reticulum exit sites interacts with COPII. J. Biol. Chem. 2001;276:40008–40017. doi: 10.1074/jbc.M106189200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.