

Abstract
Genetic risk for multiple sclerosis (MS) is thought to involve both common and rare risk alleles. Recent GWAS and subsequent meta-analysis have established the critical role of the HLA locus and identified new common variants associated to MS. These variants have small odds ratios (ORs) and explain only a fraction of the genetic risk. To expose potentially rare, high-impact alleles, we conducted a GWAS of 68 distantly related cases and 136 controls from a high-risk internal isolate of Finland with increased prevalence and familial occurrence of MS. The top 27 loci with p < 10−4 were tested in 711 cases and 1029 controls from Finland, and the top two findings were validated in 3859 cases and 9110 controls from more heterogeneous populations. SNP (rs744166) within the STAT3 gene was associated to MS (p = 2.75 × 10−10, OR 0.87, confidence interval 0.83–0.91). The protective haplotype for MS in STAT3 is a risk allele for Crohn disease, implying that STAT3 represents a shared risk locus for at least two autoimmune diseases. This study also demonstrates the potential of special isolated populations in search for variants contributing to complex traits.
Main Text
Multiple sclerosis (MS) (MIM #126200) is a complex inflammatory disease of the central nervous system with presumed autoimmune etiology. Both environmental and genetic factors are thought to contribute to the development of MS,1–3 and the genetic risk factors likely include both common and rare risk alleles. Recent GWAS and subsequent meta-analysis have established the critical role of the HLA locus4–6 and identified new MS loci: IL2RA (MIM ∗147730),7 IL7R (MIM ∗146661),7–9 CLEC16A (MIM ∗611303),7,10–13 CD58 (MIM ∗153420),11,12,14 TNFRSF1A (MIM ∗191190),15 IRF8 (MIM ∗601565),15 and TYK2 (MIM ∗176941).12,16,17 These associated variants, except for TYK2, are common, have small odds ratios (ORs), and explain only a fraction of the genetic risk.
The population history of Finland and the province of Southern Ostrobothnia (SO), an internal isolate with increased prevalence of MS,18–22 is compatible with a founder effect.22–24 Previous studies in Finnish MS families originating from this high-risk subisolate have demonstrated linkage and association to the HLA locus (HLA-DRB1 [MIM ∗142857]),25–27 17q22-24,25,28,29 and 5p14-p12.25,30–32 Therefore, we hypothesized that some variants predisposing to MS have either become enriched in SO or can be more easily detected against a homogenous background with a genome-wide, high-density SNP screen. We looked for shared alleles enriched in cases, as well as potential extended homozygous regions and copy number variations (CNVs) enriched in MS cases.
We included in our GWAS 72 cases with either both parents from the high-risk isolate or one parent from the isolate and positive family history of MS and genotyped them with the Illumina HumanHap300 chip. Extensive genealogical research revealed that the majority of the cases could be traced to two large interrelated pedigrees (see Figure S1 available online). A total of 2206 population-based controls were genotyped with either Illumina HumanHap300 chip or with Illumina HumanHap610-quad chip. We excluded samples and SNPs with <95% success rates, leaving 72 cases and 2196 controls for the subsequent analyses, and selected only SNPs present on both Illumina platforms (297,343 SNPs) for analyses. Gender check was performed with X chromosomal SNPs, and no discrepancies between the observed and expected gender were noted. Identity-by-descent (IBD) analysis was performed to study possible close cryptic relatedness between individuals and to identify possible samples with excess relatedness, suggestive of sample contamination. We then performed identity-by-state (IBS) and multidimensional scaling analyses: four cases were initially considered as isolate samples clustered outside the isolate sample set and were excluded from subsequent analyses (Figure S2). We selected the two closest IBS-matched controls for each case, and the final GWAS set (isolate GWAS) consisted of 68 cases and 136 controls. The genomic inflation factor suggested no major inflation (λ 1.078) and a fairly well-matched case-control set, which was also confirmed by quantile-quantile plot analysis of single SNP association results (Figure S3). Because we had parental birthplace data for both the cases and the majority of controls (n = 2174), we could further verify that all cases and 125 of the 136 selected controls had at least one parent born in Southern Ostrobothnia, and of these, 64 cases and 90 controls had both parents born in Southern Ostrobothnia. We have recently shown a correlation between geographical origin of samples (based on parental birthplace information) and genome-wide SNP data in the Finnish population.23 Thus, IBS matching of cases and controls combined with genealogical information should minimize the risk of population substructure in our study set. All patient samples were collected with informed consent, and the study design and the Finnish sample collection have been approved by the Helsinki University Hospital Ethical Committee of Ophthalmology, Otorhinolaryngology, Neurology and Neurosurgery (permit 192/E9/02).
Taking advantage of the distant relatedness in the subisolate, we conducted homozygosity analyses with PLINK.33 First we searched for extended regions of homozygosity (ROHs), the signature features of isolated populations, enriched in MS cases to identify loci that could influence MS susceptibility in a recessive manner. ROHs with at least 50 consecutive SNPs and a minimum length of 500 kb were identified in each individual. On average, we identified 149 (standard deviation: 12 in cases, 10 in controls) ROHs per individual with an average length of 1030 kb (500 kb–31.3 Mb) in cases and 1018 kb (500 kb–49.6 Mb) in controls. We then evaluated which overlapping homozygous regions were enriched in cases by permuting the group (case-control) labels 10,000 times. The analysis revealed three putative regions with empirical p < 10−3: 1q42.12 (242 kb, 24 SNPs, p = 3 × 10−4), 2q24.3 (512 kb, 39 SNPs, p = 8 × 10−5), and 12q24.33 (573 kb, 48 SNPs, p = 3 × 10−4) (Table S1 and Figure S4). Although the cases and controls are matched on the basis of their genome-wide IBS sharing and are augmented by parental birthplace information, the permutation-based approach is susceptible to population substructure, and obtained p values should be interpreted with caution. Excess homozygous sharing was observed with the same haplotype for 13% (9 of MS cases) and 7% (10 of controls) for 1q42.12 and 37% (25 of MS cases) and 20% (27 of controls) for 2q24.3. For the 12q24 region, we observed multiple different haplotypes (Table S1). These regions have not been previously implicated in MS except for suggestive linkage in 12q23-24,34 and their putative role in MS susceptibility requires further validation. Haplotype sharing outside of the isolate in the population control samples (n = 2194) was similar to the GWAS internal isolate control population (frequencies 5.8% for 1q42.12 and 20% for 2q24.3 haplotypes). This indicates that the homozygous haplotypes have been enriched in the subisolate MS cases, but not in the isolate controls, although the IBD analysis showed the isolate controls to be as related to each other as the isolate MS cases (Table S2).
The Illumina HumanHap300 platform has relatively sparse coverage and is void of probes in the most common CNV regions but could be suitable for detecting rare, large CNVs, potentially enriched in the internal isolate population. We used the QuantiSNP software35 for CNV detection (GC content correction option, restricted to CNVs with log Bayesian factor > 10 and length ≥ 3 SNPs) and verified these results visually with Bead Studio 3.3. All CNVs in centromeric regions were excluded. We identified altogether 106 CNV regions in 68 cases (Table S3); all but 6 of the 106 CNVs have been previously reported. Furthermore, all novel CNVs were found in only one case each. Hypothesizing that genes mapping next to the 106 CNVs identified in cases could belong to a common pathway involved in MS etiology, we used Ingenuity Pathway Analysis to search for connecting pathways. One pathway potentially regulating oligodendrocyte differentiation and myelin sheet formation36–41 involving NRG3 (MIM ∗605533), ERBB4 (MIM ∗600543), DLG2 (MIM ∗603583), UTRN (MIM ∗128240), and LARGE (MIM ∗603590) (all CNVs previously reported) was identified (Figure S5), but CNV deletions in these genes were observed to have similar frequency in MS cases compared to controls with Fisher's exact test (ERBB4: 11% of cases and 12% of controls, p = 0.388; NRG3: 4% of cases and 4% of controls, p = 0.90; and DLG2: 1% of cases and 0% of controls, p = 0.404) when genotyped in an independent set of 703 cases and 1051 controls with an in-house-developed PCR-based fragment analysis method.42
Southern Ostrobothnia is an old isolate, and thus the expected shared haplotypes are of modest length. We therefore performed single SNP standard χ2 allelic association analysis with PLINK.33 Because of the limited power, we analyzed all 27 loci (28 of the 37 initial SNPs) showing nominal association in the GWAS analysis (p < 10−4; Table S4) in a larger independent Finnish sample set of 711 cases and 1029 controls, of which 83 MS cases and 365 controls were from the isolate (Table 1). Population-stratified Cochran-Mantel-Haenszel (CMH) association analysis provided evidence for three SNPs: rs3135338 in the HLA region (p = 1.6 × 10−25), rs744166 in first intron of STAT3 (MIM ∗102582) in chromosome 17q21.1 (p = 0.0012), and rs1364194 in chromosome 16 (p = 0.0047) (Table S4). The non-HLA SNPs were then analyzed in an international sample of 3859 MS cases and 9110 controls from six different populations (Table 1). The combined evidence for association to STAT3 (rs744166) was significant (p = 2.75 × 10−10 and OR 0.87 [95% confidence interval (CI) 0.83–0.91]) (Figure 1; Table S5). The Breslow-Day analysis of heterogeneity of odds ratios revealed no significant heterogeneity (p = 0.34). When the combined replication data set was analyzed by logistic regression for additive, dominant, and recessive models with study set as a covariate in the analyses, the statistically most significant p value was obtained for the additive model (Table S6). We obtained no additional support for the chromosome 16q region.
Table 1.
Description of Sample Sets in GWAS and Replication Studies
| Study Population | Number of MS Cases | Number of Controls | Genotyping Platform |
|---|---|---|---|
| Southern Ostrobothnia (SO) isolate GWASa | 68 | 136 | Illumina HumanHap300 or Illumina Human610-quad |
| Finland SO replicationb | 83 | 365 | Sequenom iPlex Gold |
| Finlandc | 628 | 668 | Sequenom iPlex Gold |
| Norwayd | 607 | 816 | Sequenom iPlex Gold |
| Denmarke | 628 | 1074 | Sequenom iPlex Gold |
| Gene MSA Switzerlandf | 253 | 208 | Illumina HumanHap300 |
| Gene MSA Netherlandsf | 230 | 232 | Illumina HumanHap300 |
| Gene MSA USf | 486 | 431 | Illumina HumanHap300 |
| IMSGC UKg | 453 | 2950 | Affymetrix 500K |
| IMSGC USg | 342 | 1679 | Affymetrix 500K |
| BWHg | 860 | 1720 | Affymetrix 6.0 |
| Total | 4638 | 10,279 |
All samples have been diagnosed with clinically definite MS according to either Poser's or McDonald's criteria.
MS samples included in the genome-wide analysis originated from the MS high-risk isolate located on the western coast of Finland (Southern Ostrobothnia, SO) with ∼2-fold prevalence and higher familial clustering of MS compared to other regions of Finland. Most of the cases were distantly related, and no closer than second-degree relatives were included (Table S2). The cases were genotyped with Illumina HumanHap300 at The Broad Institute of MIT and Harvard. Control samples (n = 136) were selected by utilizing identical-by-state (IBS)-sharing and parental birthplace information from a pool of population-based controls (total n = 2206) genotyped either with Illumina HumanHap300-duo chips at the Institute for Molecular Medicine Finland (FIMM) Technology Centre or with Illumina Human610-quad chips at the Sanger Institute.
The MS cases have at least one parent born within SO, and anonymous population controls were collected from the Central Hospital of Seinajoki in Southern Ostrobothnia. The samples were genotyped in the FIMM Technology Centre.
Finnish MS patients (excluding samples from the SO region) from various regions (Tampere, Helsinki, Kuopio, Oulu) and anonymous population controls collected from Kuopio and Helsinki University Hospitals. The samples were genotyped in the FIMM Technology Centre.
Norwegian samples have been described in more detail in Lorentzen et al.54 The samples were genotyped in the FIMM Technology Centre.
The Danish nationwide study set cases have been diagnosed with clinically definite MS according to the McDonald criteria. The controls are healthy blood donors and hospital workers residing in the same region as patients. Experimental protocols (KF 01314 009) were approved by the local ethics board, and informed consent was obtained from all participants. The samples were genotyped in the FIMM Technology Centre.
Figure 1.
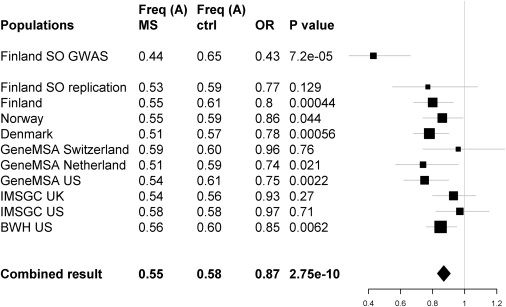
Population-Specific Association for the STAT3 rs744166 A Allele
The rs744166 A allele that tags a putative MS protective haplotype associated to MS and shows consistent reduced risk in all studied populations with available genotypes. The results are presented here by study set. Each line represents one study set showing the name of the set, A allele frequencies for cases and controls, ORs, p value for association, and graphic illustration of the odds ratio (square, size relative to study set size) with the 95% confidence intervals for the odds ratio (thin lines).
Evaluation of the STAT3 linkage disequilibrium (LD) block that contains the associated SNP rs744166 in Hapmap2 (build 23a) samples43 with Haploview 4.044 showed that rs744166 non-risk-associated A allele completely tags the most common haplotype in Southern Utah residents of European descent (CEU) (56%), Han Chinese from Beijing (CHB 65%), and Tokyo Japanese (JPT 57%), but the G allele is present on four different haplotypes (Table S7). In the Yoruban population from Nigeria (YRI), the A allele is present on four different haplotype backgrounds, and the most common A haplotype in CEU, CHB, and JPT populations has the frequency of 7% in the YRI population. We speculate that this notable enrichment of a single haplotype in non-African populations might suggest positive selection of the putative MS protective haplotype outside Africa, although this locus did not reach genome-wide significance in an analysis of signs of recent positive selection.45 The rs744166 A allele also shows changes in frequency distribution in the Human Genome Diversity Panel (Figure S6).45 The LD block carrying the haplotype is 54 kb in length in the CEU population and contains the beginning of STAT3 and its immediate promoter region (Figure 2).
Figure 2.
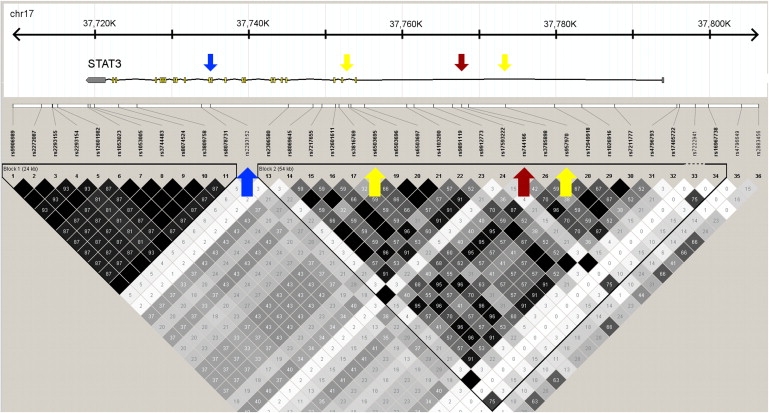
Description of the Associated LD Region in STAT3
The associated SNP and haplotypes are in a 54 kb LD block covering the beginning and immediate 5′ region of the STAT3 gene. The associated rs744166 SNP is marked with a red arrow, and the other two SNPs, rs6503695 and rs957970, used in the haplotype analysis are marked with yellow arrows. The SNP marked with the blue arrow (rs2293152) was listed among the 100 top SNPs suggestively associated to MS in a previous meta-analysis.15
We tagged the haplotypes with three SNPs (rs744166, rs6503695, and rs957970) with Haploview 4.0 tagging option. These SNPs were genotyped in the Finnish sample set, and the data for the same SNPs were available from four other populations from a recent meta-analysis.7,15,46 We phased the haplotypes with PLINK and performed a CMH analysis with populations as clusters. We could define both a putative predisposing haplotype (30.9% in MS, 27.1% in controls, OR 1.18, 95% CI 1.11–1.27) with CMH p = 1.29 × 10−6 and a tentative protective haplotype (55.0% MS, 58.7% controls, OR 0.86, 95% CI 0.81–0.91) (Figure 1) with CMH combined p = 1.19 × 10−6 (Table 2; Tables S7 and S8). The Breslow-Day test revealed no significant heterogeneity of odds ratios (p = 0.271 and p = 0.301, respectively). Further studies, including resequencing, will be needed to identify the true affecting variants segregating in one or both of these haplotypes.
Table 2.
Summary of the 54 kb STAT3 Haplotype Data Showing One Putative Predisposing and One Putative Protective Haplotype
| Haplotype |
Frequency MS (n = 3255) |
Frequency Control (n = 8133) |
P Value | OR | 95% CI |
|---|---|---|---|---|---|
| CGGa | 0.309 | 0.271 | 1.29 × 10−6 | 1.18 | (1.11–1.27) |
| TAAb | 0.550 | 0.587 | 1.19 × 10−6 | 0.86 | (0.81–0.91) |
| TGG | 0.082 | 0.080 | 0.439 | 1.06 | (0.94–1.17) |
| CGA | 0.057 | 0.059 | 0.591 | 0.98 | (0.85–1.10) |
The haplotypes were constructed with SNPs rs6503695, rs744166, and rs957970 and phased with PLINK. Only phased haplotypes with posterior probability of 1 were included in the analysis. Each haplotype was analyzed separately and showed no evidence for heterogeneity of odds ratios between populations in the Breslow-Day test, which allowed us to combine the haplotype results with CMH. The analysis included a total of 3255 MS cases and 8133 controls from Finnish, BWH, IMSGC UK, IMSGC US, Gene MSA US, Gene MSA CH, and Gene MSA NL sample sets. The results for individual populations are provided in Table S5.
The predisposing haplotype CGG is significantly overrepresented in the MS cases.
The protective haplotype TAA is significantly underrepresented in the MS cases.
STAT3 codes for a transcription factor that is involved in multiple pathways and functions, including the Jak-STAT pathway, neuron axonal guidance, apoptosis, activation of immune responses, and Th17 cell differentiation.47 Interestingly, the A allele of rs744166 tagging the MS-protective haplotype is associated with Crohn disease,48 and mutations in STAT3 are known to cause hyperimmunoglobulin E recurrent infection syndrome (HIES [MIM #147060]),49,50 a rare autosomal-dominant disorder characterized by elevated immunoglobulin E levels and inflammation. Additionally, mouse studies have shown that targeted deletion of Stat3 in CD4+ T cells prevents the development of experimental autoimmune encephalomyelitis (EAE), the rodent model of MS,51 and that Treg-specific ablation of Stat3 resulted in the development of a fatal intestinal inflammation due to unstrained TH17 response.52 Recent meta-analysis of GWAS in MS listed STAT3 as one of the genes with a suggestive role in at least two autoimmune disorders15 but failed to replicate the initial STAT3 association. The failure to replicate the initial association was probably due to selecting the most significantly associated regional SNP (rs2293152), which resides just outside of the rs744166 containing LD region and has only limited LD with the rs744166 (r2 0.35 in HapMap2 CEU population), for the replication analysis (Figure 2). These observations support a wider role for STAT3 in autoimmunity and adds this gene to the growing list of MS-susceptibility genes with validated or substantial evidence for association in at least two inflammatory diseases.48–50 All of these together suggest a significant role of this locus in immune system and autoimmune disease pathogenesis.
Most of the currently validated (IL2RA, IL7R, CD58, CLEC16A, IRF8, TNFRSF1A, TYK2)7,9,12–17,53 and suggested (C7 [MIM ∗217070], CD6 [MIM ∗186720], IL12A [MIM ∗161560], OLIG3 [MIM ∗609323]–TNFAIP3 [MIM ∗191163], PTGER4 [MIM ∗601586], RGS1 [MIM ∗600323])15,30 non-HLA MS susceptibility loci have known functions in the immune system and particularly in T cells. Although their independent ORs are modest, their combined effect might be larger, and a large-scale international study would be required to estimate their combined effect toward disease predisposition. The present study demonstrates the power of the founder population study design to complement large-scale GWAS in identifying genes and pathways of general significance, not only rare high-impact alleles.
Acknowledgments
We wish to thank all participating MS patients and families. Elli Kempas, Liisa Arala, Anne Vikman, Anne Nyberg, Minna Suvela, and Marja-Leena Sairanen are acknowledged for their invaluable assistance and technical support. We sincerely thank Ida Surakka, Samuli Ripatti, and Carl Anderson for their valuable assistance and advice in statistical analyses and Nicole Soranzo for guidance on Haplotter. The International Multiple Sclerosis Genetics Consortium (IMSGC) and Gene MSA Consortium are acknowledged for the data utilized in the replication phase. The Danish Multiple Sclerosis Society is acknowledged for supporting the Danish sample collection, and the Norwegian Bone Marrow Donor Registry is acknowledged for collaboration in establishment of the Norwegian control material. The Institute for Molecular Medicine Finland (FIMM) Technology Center (previously Finnish Genome Center) and the Broad Institute Center for Genotyping and Analysis are acknowledged for conducting genotyping on the Illumina GWA platform. The Health 2000 project is thanked for providing population-based controls for this GWAS study. This work was supported by the National Institutes of Health (grant RO1 NS 43559), the Center of Excellence for Disease Genetics of the Academy of Finland (grants 213506 and 129680), the Sigrid Juselius Foundation, the Biocentrum Helsinki Foundation, Helsinki University Central Hospital Research Foundation, the Neuropromise EU project (grant LSHM-CT-2005-018637), and The Wellcome Trust (grant 089061/Z/09/Z). The Broad Institute Center for Genotyping and Analysis is supported by the National Center for Research Resources (grant U54 RR020278). The genotyping of the Health 2000 controls was funded by the SGENE EU project (LSHM-CT-2006-037761) and Simons Foundation (R01MH71425-01A1). P.L.D.J. is a Harry Weaver Neuroscience Scholar of the National MS Society. L.P. is a member of the Board of the Orion Pharma Limited, Helsinki, Finland.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
PLINK: Whole Genome Association Analysis Toolset, http://pngu.mgh.harvard.edu/∼purcell/plink/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
Ingenuity Pathway Analysis (IPA) Software, http://www.ingenuity.com
Database of Genomic Variants (DGV), http://projects.tcag.ca/variation/
International HapMap Project, http://www.hapmap.org/
The Human Genome Diversity Project (HGDP) Selection Browser, http://hgdp.uchicago.edu/
References
- 1.Ebers G.C., Sadovnick A.D., Risch N.J., Canadian Collaborative Study Group A genetic basis for familial aggregation in multiple sclerosis. Nature. 1995;377:150–151. doi: 10.1038/377150a0. [DOI] [PubMed] [Google Scholar]
- 2.Compston A. Genetic epidemiology of multiple sclerosis. J. Neurol. Neurosurg. Psychiatry. 1997;62:553–561. doi: 10.1136/jnnp.62.6.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mumford C.J., Wood N.W., Kellar-Wood H., Thorpe J.W., Miller D.H., Compston D.A. The British Isles survey of multiple sclerosis in twins. Neurology. 1994;44:11–15. doi: 10.1212/wnl.44.1.11. [DOI] [PubMed] [Google Scholar]
- 4.Yeo T.W., De Jager P.L., Gregory S.G., Barcellos L.F., Walton A., Goris A., Fenoglio C., Ban M., Taylor C.J., Goodman R.S. A second major histocompatibility complex susceptibility locus for multiple sclerosis. Ann. Neurol. 2007;61:228–236. doi: 10.1002/ana.21063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jersild C., Fog T. Histocompatibility (HL-A) antigens associated with multiple sclerosis. Acta Neurol. Scand. Suppl. 1972;51:377. [PubMed] [Google Scholar]
- 6.Dyment D.A., Willer C.J., Scott B., Armstrong H., Ligers A., Hillert J., Paty D.W., Hashimoto S., Devonshire V., Hooge J. Genetic susceptibility to MS: A second stage analysis in Canadian MS families. Neurogenetics. 2001;3:145–151. doi: 10.1007/s100480100113. [DOI] [PubMed] [Google Scholar]
- 7.Hafler D.A., Compston A., Sawcer S., Lander E.S., Daly M.J., De Jager P.L., de Bakker P.I., Gabriel S.B., Mirel D.B., Ivinson A.J., International Multiple Sclerosis Genetics Consortium Risk alleles for multiple sclerosis identified by a genomewide study. N. Engl. J. Med. 2007;357:851–862. doi: 10.1056/NEJMoa073493. [DOI] [PubMed] [Google Scholar]
- 8.Lundmark F., Duvefelt K., Hillert J. Genetic association analysis of the interleukin 7 gene (IL7) in multiple sclerosis. J. Neuroimmunol. 2007;192:171–173. doi: 10.1016/j.jneuroim.2007.08.015. [DOI] [PubMed] [Google Scholar]
- 9.Gregory S.G., Schmidt S., Seth P., Oksenberg J.R., Hart J., Prokop A., Caillier S.J., Ban M., Goris A., Barcellos L.F., Multiple Sclerosis Genetics Group Interleukin 7 receptor alpha chain (IL7R) shows allelic and functional association with multiple sclerosis. Nat. Genet. 2007;39:1083–1091. doi: 10.1038/ng2103. [DOI] [PubMed] [Google Scholar]
- 10.Zoledziewska M., Costa G., Pitzalis M., Cocco E., Melis C., Moi L., Zavattari P., Murru R., Lampis R., Morelli L. Variation within the CLEC16A gene shows consistent disease association with both multiple sclerosis and type 1 diabetes in Sardinia. Genes Immun. 2009;10:15–17. doi: 10.1038/gene.2008.84. [DOI] [PubMed] [Google Scholar]
- 11.Rubio J.P., Stankovich J., Field J., Tubridy N., Marriott M., Chapman C., Bahlo M., Perera D., Johnson L.J., Tait B.D. Replication of KIAA0350, IL2RA, RPL5 and CD58 as multiple sclerosis susceptibility genes in Australians. Genes Immun. 2008;9:624–630. doi: 10.1038/gene.2008.59. [DOI] [PubMed] [Google Scholar]
- 12.Australia and New Zealand Multiple Sclerosis Genetics Consortium (ANZgene) Genome-wide association study identifies new multiple sclerosis susceptibility loci on chromosomes 12 and 20. Nat. Genet. 2009;41:824–828. doi: 10.1038/ng.396. [DOI] [PubMed] [Google Scholar]
- 13.International Multiple Sclerosis Genetics Consortium (IMSGC) The expanding genetic overlap between multiple sclerosis and type I diabetes. Genes Immun. 2009;10:11–14. doi: 10.1038/gene.2008.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Jager P.L., Baecher-Allan C., Maier L.M., Arthur A.T., Ottoboni L., Barcellos L., McCauley J.L., Sawcer S., Goris A., Saarela J. The role of the CD58 locus in multiple sclerosis. Proc. Natl. Acad. Sci. USA. 2009;106:5264–5269. doi: 10.1073/pnas.0813310106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Jager P.L., Jia X., Wang J., de Bakker P.I., Ottoboni L., Aggarwal N.T., Piccio L., Raychaudhuri S., Tran D., Aubin C., International MS Genetics Consortium Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat. Genet. 2009;41:776–782. doi: 10.1038/ng.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ban M., Goris A., Lorentzen A.R., Baker A., Mihalova T., Ingram G., Booth D.R., Heard R.N., Stewart G.J., Bogaert E., Wellcome Trust Case-Control Consortium (WTCCC) Replication analysis identifies TYK2 as a multiple sclerosis susceptibility factor. Eur. J. Hum. Genet. 2009;17:1309–1313. doi: 10.1038/ejhg.2009.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burton P.R., Clayton D.G., Cardon L.R., Craddock N., Deloukas P., Duncanson A., Kwiatkowski D.P., McCarthy M.I., Ouwehand W.H., Samani N.J. Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat. Genet. 2007;39:1329–1337. doi: 10.1038/ng.2007.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wikström J. Studies on the clustering of multiple sclerosis in Finland II: Microepidemiology in one high-risk county with special reference to familial cases. Acta Neurol. Scand. 1975;51:173–183. doi: 10.1111/j.1600-0404.1975.tb07598.x. [DOI] [PubMed] [Google Scholar]
- 19.Wikström J., Palo J. Studies on the clustering of multiple sclerosis in Finland I: Comparison between the domiciles and places of birth in selected subpopulations. Acta Neurol. Scand. 1975;51:85–98. doi: 10.1111/j.1600-0404.1975.tb01362.x. [DOI] [PubMed] [Google Scholar]
- 20.Sumelahti M.L., Tienari P.J., Wikström J., Palo J., Hakama M. Regional and temporal variation in the incidence of multiple sclerosis in Finland 1979-1993. Neuroepidemiology. 2000;19:67–75. doi: 10.1159/000026241. [DOI] [PubMed] [Google Scholar]
- 21.Sumelahti M.L., Tienari P.J., Wikström J., Palo J., Hakama M. Increasing prevalence of multiple sclerosis in Finland. Acta Neurol. Scand. 2001;103:153–158. doi: 10.1034/j.1600-0404.2001.103003153.x. [DOI] [PubMed] [Google Scholar]
- 22.Tienari P.J., Sumelahti M.L., Rantamäki T., Wikström J. Multiple sclerosis in western Finland: Evidence for a founder effect. Clin. Neurol. Neurosurg. 2004;106:175–179. doi: 10.1016/j.clineuro.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 23.Jakkula E., Rehnström K., Varilo T., Pietiläinen O.P., Paunio T., Pedersen N.L., deFaire U., Järvelin M.R., Saharinen J., Freimer N. The genome-wide patterns of variation expose significant substructure in a founder population. Am. J. Hum. Genet. 2008;83:787–794. doi: 10.1016/j.ajhg.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Varilo T. National Public Health Institute; Helsinki, Finland: 1999. The Age of the Mutations in the Finnish Disease Heritage; A Genealogical and Linkage Equilibrium Study. [Google Scholar]
- 25.Kuokkanen S., Gschwend M., Rioux J.D., Daly M.J., Terwilliger J.D., Tienari P.J., Wikström J., Palo J., Stein L.D., Hudson T.J. Genomewide scan of multiple sclerosis in Finnish multiplex families. Am. J. Hum. Genet. 1997;61:1379–1387. doi: 10.1086/301637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lincoln M.R., Montpetit A., Cader M.Z., Saarela J., Dyment D.A., Tiislar M., Ferretti V., Tienari P.J., Sadovnick A.D., Peltonen L. A predominant role for the HLA class II region in the association of the MHC region with multiple sclerosis. Nat. Genet. 2005;37:1108–1112. doi: 10.1038/ng1647. [DOI] [PubMed] [Google Scholar]
- 27.Tienari P.J., Wikström J., Koskimies S., Partanen J., Palo J., Peltonen L. Reappraisal of HLA in multiple sclerosis: Close linkage in multiplex families. Eur. J. Hum. Genet. 1993;1:257–268. doi: 10.1159/000472423. [DOI] [PubMed] [Google Scholar]
- 28.Saarela J., Kallio S.P., Chen D., Montpetit A., Jokiaho A., Choi E., Asselta R., Bronnikov D., Lincoln M.R., Sadovnick A.D. PRKCA and multiple sclerosis: Association in two independent populations. PLoS Genet. 2006;2:e42. doi: 10.1371/journal.pgen.0020042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saarela J., Schoenberg Fejzo M., Chen D., Finnilä S., Parkkonen M., Kuokkanen S., Sobel E., Tienari P.J., Sumelahti M.L., Wikström J. Fine mapping of a multiple sclerosis locus to 2.5 Mb on chromosome 17q22-q24. Hum. Mol. Genet. 2002;11:2257–2267. doi: 10.1093/hmg/11.19.2257. [DOI] [PubMed] [Google Scholar]
- 30.Kallio S.P., Jakkula E., Purcell S., Suvela M., Koivisto K., Tienari P.J., Elovaara I., Pirttilä T., Reunanen M., Bronnikov D. Use of a genetic isolate to identify rare disease variants: C7 on 5p associated with MS. Hum. Mol. Genet. 2009;18:1670–1683. doi: 10.1093/hmg/ddp073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Riise Stensland H.M., Saarela J., Bronnikov D.O., Parkkonen M., Jokiaho A.J., Palotie A., Tienari P.J., Sumelahti M.L., Elovaara I., Koivisto K. Fine mapping of the multiple sclerosis susceptibility locus on 5p14-p12. J. Neuroimmunol. 2005;170:122–133. doi: 10.1016/j.jneuroim.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 32.Kuokkanen S., Sundvall M., Terwilliger J.D., Tienari P.J., Wikström J., Holmdahl R., Pettersson U., Peltonen L. A putative vulnerability locus to multiple sclerosis maps to 5p14-p12 in a region syntenic to the murine locus Eae2. Nat. Genet. 1996;13:477–480. doi: 10.1038/ng0896-477. [DOI] [PubMed] [Google Scholar]
- 33.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haines J.L., Bradford Y., Garcia M.E., Reed A.D., Neumeister E., Pericak-Vance M.A., Rimmler J.B., Menold M.M., Martin E.R., Oksenberg J.R., Multiple Sclerosis Genetics Group Multiple susceptibility loci for multiple sclerosis. Hum. Mol. Genet. 2002;11:2251–2256. doi: 10.1093/hmg/11.19.2251. [DOI] [PubMed] [Google Scholar]
- 35.Colella S., Yau C., Taylor J.M., Mirza G., Butler H., Clouston P., Bassett A.S., Seller A., Holmes C.C., Ragoussis J. QuantiSNP: An objective Bayes Hidden-Markov model to detect and accurately map copy number variation using SNP genotyping data. Nucleic Acids Res. 2007;35:2013–2025. doi: 10.1093/nar/gkm076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Colognato H., Baron W., Avellana-Adalid V., Relvas J.B., Baron-Van Evercooren A., Georges-Labouesse E., ffrench-Constant C. CNS integrins switch growth factor signalling to promote target-dependent survival. Nat. Cell Biol. 2002;4:833–841. doi: 10.1038/ncb865. [DOI] [PubMed] [Google Scholar]
- 37.Colognato H., Galvin J., Wang Z., Relucio J., Nguyen T., Harrison D., Yurchenco P.D., Ffrench-Constant C. Identification of dystroglycan as a second laminin receptor in oligodendrocytes, with a role in myelination. Development. 2007;134:1723–1736. doi: 10.1242/dev.02819. [DOI] [PubMed] [Google Scholar]
- 38.Garcia R.A., Vasudevan K., Buonanno A. The neuregulin receptor ErbB-4 interacts with PDZ-containing proteins at neuronal synapses. Proc. Natl. Acad. Sci. USA. 2000;97:3596–3601. doi: 10.1073/pnas.070042497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Longman C., Brockington M., Torelli S., Jimenez-Mallebrera C., Kennedy C., Khalil N., Feng L., Saran R.K., Voit T., Merlini L. Mutations in the human LARGE gene cause MDC1D, a novel form of congenital muscular dystrophy with severe mental retardation and abnormal glycosylation of alpha-dystroglycan. Hum. Mol. Genet. 2003;12:2853–2861. doi: 10.1093/hmg/ddg307. [DOI] [PubMed] [Google Scholar]
- 40.Carteron C., Ferrer-Montiel A., Cabedo H. Characterization of a neural-specific splicing form of the human neuregulin 3 gene involved in oligodendrocyte survival. J. Cell Sci. 2006;119:898–909. doi: 10.1242/jcs.02799. [DOI] [PubMed] [Google Scholar]
- 41.Huang Y.Z., Won S., Ali D.W., Wang Q., Tanowitz M., Du Q.S., Pelkey K.A., Yang D.J., Xiong W.C., Salter M.W., Mei L. Regulation of neuregulin signaling by PSD-95 interacting with ErbB4 at CNS synapses. Neuron. 2000;26:443–455. doi: 10.1016/s0896-6273(00)81176-9. [DOI] [PubMed] [Google Scholar]
- 42.Sulonen A.M., Kallio S.P., Ellonen P., Suvela M., Elovaara I., Koivisto K., Pirttilä T., Reunanen M., Tienari P.J., Palotie A. No evidence for shared etiology in two demyelinative disorders, MS and PLOSL. J. Neuroimmunol. 2009;206:86–90. doi: 10.1016/j.jneuroim.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frazer K.A., Ballinger D.G., Cox D.R., Hinds D.A., Stuve L.L., Gibbs R.A., Belmont J.W., Boudreau A., Hardenbol P., Leal S.M., International HapMap Consortium A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barrett J.C., Fry B., Maller J., Daly M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 45.Pickrell J.K., Coop G., Novembre J., Kudaravalli S., Li J.Z., Absher D., Srinivasan B.S., Barsh G.S., Myers R.M., Feldman M.W., Pritchard J.K. Signals of recent positive selection in a worldwide sample of human populations. Genome Res. 2009;19:826–837. doi: 10.1101/gr.087577.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baranzini S.E., Galwey N.W., Wang J., Khankhanian P., Lindberg R., Pelletier D., Wu W., Uitdehaag B.M., Kappos L., Polman C.H., GeneMSA Consortium Pathway and network-based analysis of genome-wide association studies in multiple sclerosis. Hum. Mol. Genet. 2009;18:2078–2090. doi: 10.1093/hmg/ddp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Egwuagu C.E. STAT3 in CD4+ T helper cell differentiation and inflammatory diseases. Cytokine. 2009;47:149–156. doi: 10.1016/j.cyto.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barrett J.C., Hansoul S., Nicolae D.L., Cho J.H., Duerr R.H., Rioux J.D., Brant S.R., Silverberg M.S., Taylor K.D., Barmada M.M., NIDDK IBD Genetics Consortium. Belgian-French IBD Consortium. Wellcome Trust Case Control Consortium Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat. Genet. 2008;40:955–962. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Holland S.M., DeLeo F.R., Elloumi H.Z., Hsu A.P., Uzel G., Brodsky N., Freeman A.F., Demidowich A., Davis J., Turner M.L. STAT3 mutations in the hyper-IgE syndrome. N. Engl. J. Med. 2007;357:1608–1619. doi: 10.1056/NEJMoa073687. [DOI] [PubMed] [Google Scholar]
- 50.Minegishi Y., Saito M., Tsuchiya S., Tsuge I., Takada H., Hara T., Kawamura N., Ariga T., Pasic S., Stojkovic O. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. 2007;448:1058–1062. doi: 10.1038/nature06096. [DOI] [PubMed] [Google Scholar]
- 51.Liu X., Lee Y.S., Yu C.R., Egwuagu C.E. Loss of STAT3 in CD4+ T cells prevents development of experimental autoimmune diseases. J. Immunol. 2008;180:6070–6076. doi: 10.4049/jimmunol.180.9.6070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chaudhry A., Rudra D., Treuting P., Samstein R.M., Liang Y., Kas A., Rudensky A.Y. CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science. 2009;326:986–991. doi: 10.1126/science.1172702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brynedal B., Bomfim I.L., Olsson T., Duvefelt K., Hillert J. Differential expression, and genetic association, of CD58 in Swedish multiple sclerosis patients. Proc. Natl. Acad. Sci. USA. 2009;106 doi: 10.1073/pnas.0904338106. E58, author reply E59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lorentzen A.R., Smestad C., Lie B.A., Oturai A.B., Akesson E., Saarela J., Myhr K.M., Vartdal F., Celius E.G., Sørensen P.S. The SH2D2A gene and susceptibility to multiple sclerosis. J. Neuroimmunol. 2008;197:152–158. doi: 10.1016/j.jneuroim.2008.04.037. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.