

Abstract
The recently described human anion channel Anoctamin (ANO) protein family comprises at least ten members, many of which have been shown to correspond to calcium-activated chloride channels. To date, the only reported human mutations in this family of genes are dominant mutations in ANO5 (TMEM16E, GDD1) in the rare skeletal disorder gnathodiaphyseal dysplasia. We have identified recessive mutations in ANO5 that result in a proximal limb-girdle muscular dystrophy (LGMD2L) in three French Canadian families and in a distal non-dysferlin Miyoshi myopathy (MMD3) in Dutch and Finnish families. These mutations consist of a splice site, one base pair duplication shared by French Canadian and Dutch cases, and two missense mutations. The splice site and the duplication mutations introduce premature-termination codons and consequently trigger nonsense-mediated mRNA decay, suggesting an underlining loss-of-function mechanism. The LGMD2L phenotype is characterized by proximal weakness, with prominent asymmetrical quadriceps femoris and biceps brachii atrophy. The MMD3 phenotype is associated with distal weakness, of calf muscles in particular. With the use of electron microscopy, multifocal sarcolemmal lesions were observed in both phenotypes. The phenotypic heterogeneity associated with ANO5 mutations is reminiscent of that observed with Dysferlin (DYSF) mutations that can cause both LGMD2B and Miyoshi myopathy (MMD1). In one MMD3-affected individual, defective membrane repair was documented on fibroblasts by membrane-resealing ability assays, as observed in dysferlinopathies. Though the function of the ANO5 protein is still unknown, its putative calcium-activated chloride channel function may lead to important insights into the role of deficient skeletal muscle membrane repair in muscular dystrophies.
Main Text
Muscular dystrophies encompass a large and diverse group of inherited diseases defined by skeletal muscle weakness and atrophy. Among these, the limb-girdle muscular dystrophies (LGMD) represent a group of both dominant and recessive disorders, characterized by predominant proximal limb muscle weakness, with 18 known causal genes.1,2 The majority of the proteins involved in LGMD are important for maintaining the integrity of the sarcolemmal membrane, which is susceptible to injury because of the high mechanical stress imposed on muscle fibers during muscle contraction.1,3 Some of the implicated proteins, such as the Sarcoglycans (LGMD2C-2F [MIM 253700, 608099, 604286, 601287]), when mutated disrupt the normal structure and hence the stability of the membrane.4 Another group of mutated proteins, such as Dysferlin (MIM 603009) (LGMD2B [MIM 253601]) and Caveolin-3 [MIM 601253] (LGMD1C [MIM 607801]), play a role in the repair of damaged muscle membrane through a process that requires the recruitment of cytoplasmic vesicles to the membrane wound site and their fusion by exocytosis to form a patch membrane.5–8 The distal myopathies are also a large group of muscular dystrophies with more than ten genes identified to date characterized by initial distal limb weakness.9 In the cases of dysferlinopathies, both a proximal LGMD2B phenotype and a distal Miyoshi myopathy phenotype (MM [MIM 254130]) are caused by mutations in the Dysferlin (DYSF) gene.10–12
The human Anoctamins (ANO) compose a family of at least ten proteins all exhibiting eight transmembrane domains and a DUF590 domain of unknown function.13,14 Recently, ANO1 (MIM 610108), ANO2 (MIM 610109), ANO6 (MIM 608663), ANO8 (MIM 610216), and ANO9 have been recognized to code for the elusive calcium-activated chloride channels (CaCC);15–20 however, the function of ANO5 (MIM 608662) is unknown. In this study we report that recessive mutations in Anoctamin 5 (ANO5) cause both a proximal muscular dystrophy, LGMD2L (MIM 611307), and the distal Miyoshi myopathy MMD3.
A previous study mapped LGMD2L, a novel recessive form of LGMD associated with prominent asymmetrical quadriceps femoris and biceps brachii atrophy, to chromosome 11p12-p13 in a cohort of French Canadian (FC) families.21 SNP genotyping via the Illumina HumanHap300 beadchip (317 503 SNPs; Illumina, San Diego, CA) was performed on the previously reported single large consanguineous FC Family IX21 (Figure 1A) at the McGill University and Genome Québec Innovation Centre genotyping platform. Homozygosity analysis via AutoSNPa22 identified a 4.7 Mb region of homozygosity on chromosome 11p14.3-p15 (616 consecutive SNPs delimited by rs4073508 and rs10834273). This new region shifted telomerically by 11.9 Mb the previous published candidate interval (11p12-p13).21 This region contained 11 annotated genes: NAV2 (MIM 607026), DBX1, HTATIP2 (MIM 605628), PRMT3 (MIM 603190), SLC6A5 (MIM 604159), NELL1 (MIM 602319), ANO5/TMEM16E, FANCF (MIM 603467), GAS2 (MIM 602835), SLC17A6 (MIM 607563), and SVIP. The exons and intron-exon junctions were sequenced for five of these genes: ANO5/TMEM16E, FANCF, GAS2, SLC17A6, and SVIP. Mutations were identified in ANO5/TMEM16E in family IX. Though the sequencing of ANO5 exons and intron-exon borders in the seven remaining families of the original cohort21 did not identify mutations, the screening of two new FC LGMD2L families identified other mutations in ANO5. Informed consent approved by the Centre Hospitalier de l'Université de Montréal ethics committee was obtained for all participants. In Family IX we identified a homozygous nucleotide substitution (c.1295C>G) in genomic DNA, which creates a putative splice donor site within exon 13 as calculated by NetGene2 and Human Splicing Finder v2.4 (Figures 1A and 1B). Amplification and sequencing of patient muscle cDNA confirmed the aberrant splicing of exon 13 that results in the deletion of the last 38 nucleotides of this exon (Figures 1C and 1D), leading to a frameshift and a predicted premature truncation (p.Ala432GlyfsX49). Segregation of this variant in Family IX was confirmed by sequencing the genomic DNA of exon 13 in all siblings (Figure 1A). The same mutation was identified in homozygote state in the two cases belonging to the FC Family XXXI (Figure 1A). The two families are not known to be related and come from different regions, but share a six STR markers haplotype (covering 3.6 Mb), suggesting that it is the same historical mutation that segregates in these families (data not shown). The third LGMD2L FC Family XXIX was heterozygous for two other variants: c.191 dupA in exon 5 (p.Asn64LysfsX15) and c.692G>T in exon 8 (p.G231V) (Figure 1E). The G231 residue is evolutionary conserved (Figure 1F) and is predicted to be located in the putative intracellular N-terminal tail (Figure 2D). Sequencing of the entire 87 kb genomic region of ANO5 is underway to identify mutations in other FC LGMD families.
Figure 1.
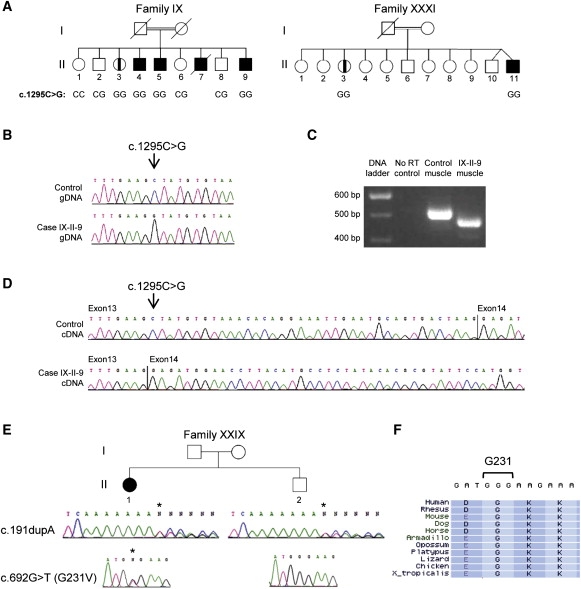
Identification of ANO5 Mutations in French Canadian LGMD2L Families
(A) Pedigrees of LGMD2L families IX and XXXI. Parents of the two families are first-degree cousins. Genotypes for the c.1295C>G variant are indicated below the individuals for whom DNA was available. Family IX has been described earlier.21
(B) Genomic sequence chromatogram of exon 13 in patient IX-II-9 reveal an exonic substitution (c.1295C>G). Sequencing of ANO5 in the two families in (A) confirmed that affected individuals IX-II-3, IX-II-4, IX-II-5, XXXI-II-3, and XXXI-II-11 were also homozygous for the mutation.
(C) cDNA amplicons of ANO5 exons 10 to 15 from muscle RNA samples for patient IX-II-9 and a control individual.
(D) Sequence chromatograms of the cDNA amplicons in (C) in the region of the nucleotide c.1295.
(E) Pedigree of LGMD2L family XXIX. Genomic sequence chromatograms show that patient XXIX-II-1 is compound heterozygote for two exonic variants: a 1 bp duplication in exon 5 (c.191 dupA) and a missense mutation in exon 8 (c.692G>T/G231V). Her unaffected brother is a carrier for the c.191 dupA.
(F) Protein alignment of the G231 residue in ANO5 via Multiz.
Figure 2.
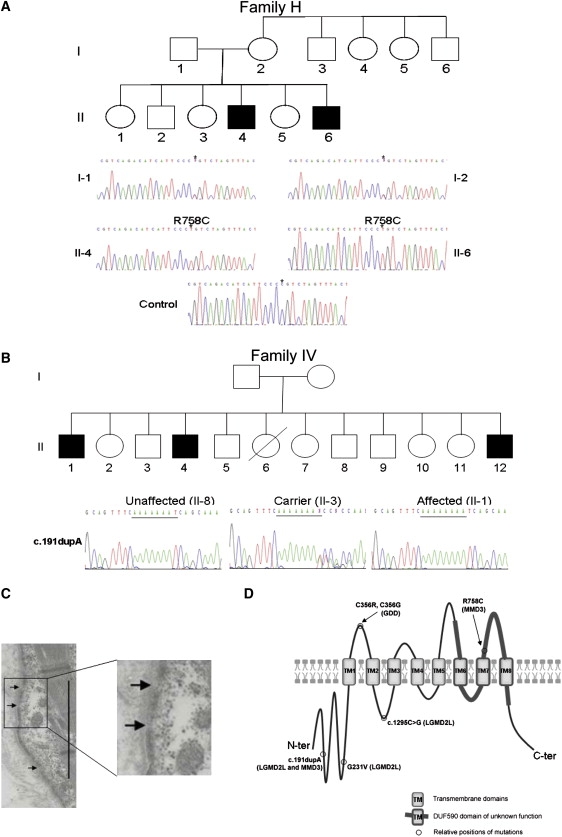
Identification of ANO5 Mutations in Non-Dysferlin Miyoshi Myopathy Families
(A) Genomic sequence chromatograms of exon 20 of ANO5 show that in the non-dysferlin MM Finnish family H that is linked with defective membrane repair,24 there is a homozygous nucleotide substitution that would result in an amino acid substitution (c.2272C>T/R758C).
(B) In the MMD3 Dutch family IV, the mutation identified is c.191 dupA in exon 5 inherited as homozygous. The same mutation is present as heterozygous in the LGMD2L Family XXIX.
(C) EM micrograph of patient II-6 muscle from Family H highlights multifocal loss of sarcolemmal membrane. Magnification ×5000. Scale bar represents 5 μm.
(D) Predicted structure of human ANO5 and the relative position of the GDD1, LGMD2L, and MMD3 mutations. ANO5 is predicted to contain eight transmembrane (TM) domains with both the N-terminal and the C-terminal regions located intracellularly. The DUF590 domain contains at last three TM domains (TM6-8). The MMD3 R758C mutation that is associated with defective membrane repair is located extracellularly as are the two GDD mutations. The LGMD2L mutations and one MMD3 mutation are located to intracellular regions.
Further evidence that ANO5 mutations cause muscular dystrophy came from an independent study of families presenting with a distal non-dysferlin Miyoshi myopathy. Informed consents approved by the Kainuu Central Hospital (Finland), the Academic Medical Centre (The Netherlands), and the University of Durham (United Kingdom) ethical committees were obtained for these families. Two Miyoshi myopathy families, in which mutations in DYSF and linkage to the 10p MMD2 locus had been excluded,23 were genotyped via the Affymetrix GeneChip Human Mapping 50k (performed by AROS Applied Biotechnology, Aarhus Nord, Denmark). They were linked to a Miyoshi myopathy (MMD3) locus on chromosome 11p14.3-cen (rs722490 to rs509244, cumulative LOD score of >2.5) overlapping with the LGMD2L candidate region21 (data not shown). Sequencing of the ANO5 gene identified mutations in both families. In the Finnish Family H,24 a homozygous nucleotide substitution (c.2272C>T) was identified in exon 20 that leads to the substitution of a conserved arginine to a cysteine residue (R758C; Figure 2A). The same heterozygote exon 5 mutation (c.191 dupA) observed in the FC Family XXIX (Figure 1E) was found in a homozygous state in the Dutch Family IV23 (Figure 2B). Both variants were shown to segregate with the disease in the two families. None of the mutations have been reported as variants in the following databases: the National Center for Biotechnology Information (NCBI) database of genetic variation (dbSNP build 130), the Human Genome Diversity Project in collaboration with the Centre d'Étude du Polymorphisme Humain (HGDP-CEPH), and the International HapMap Project (Merged phases 1, 2, & 3). Considering that the five ANO5 families have diverse origins (three FC, one Dutch, and one Finnish), we tested their carrier frequencies in these three populations, as well as in panels of United Kingdom (UK) Caucasian controls (from the Health Protection Agency of United Kingdom) and CEPH controls. The c.1295C>G variant was absent in 210 FC and 162 CEPH control chromosomes, and the c.692G>T (G231V) variant was absent in 210 FC control chromosomes. The c.2272C>T (R758C) variant was not detected in 100 UK, nor in 208 FC, control chromosomes, but was detected in one out of 368 Finnish control chromosomes, indicating that this variant is present in the Finnish population at low frequency. The c.191 dupA mutation was absent in 210 FC and in 152 CEPH control chromosomes, but was identified in 1 out of 100 UK and in 2 out of 210 Dutch control chromosomes. The observed absence or very low population frequencies of the ANO5 mutations strongly support our conclusion that these cause LGMD2L and MMD3.
ANO5 is predicted to produce several alternatively spliced isoforms, with the major isoform in muscle retaining all 22 exons.25 Based on the predicted full coding sequence (NCBI reference sequence NM_213599.2), it is expected that the mutations c.1295C>G (LGMD2L) and c.191 dupA (LGMD2L and MMD3) lead to a frameshift and premature truncation (p.Ala432GlyfsX49 and p.Asn64LysfsX15, respectively; Figure 2D). As is often the case with mRNAs containing premature termination codons, a translation-coupled nonsense-mediated RNA decay (NMD) mechanism may lead to their more rapid degradation.26 To test whether transcripts carrying the mutations c.1295C>G and c.191 dupA are subject to NMD, lymphoblastoid cell lines isolated from EBV-transformed lymphocytes were grown in IMDM medium (Invitrogen, Carlsbad, CA) supplemented with 10% Fetal Bovine serum (Invitrogen) and incubated for 9 hr with 0.1% DMSO (vehicle) or 100 μg/mL of cycloheximide (Sigma, St. Louis, MO), a translation inhibitor. Quantitative RT-PCR were performed in triplicate on extracted RNA, and expression of ANO5 was normalized to a set of three control genes (GAPDH [MIM 138400], PUM1 [MIM 607204], and RPL13A) via QBase, a modified ΔΔCt method,27 at the Génome Québec and Université de Sherbrooke RNomics Centre. All controls and cases demonstrated an increase in the relative ANO5 expression as a consequence of cycloheximide treatment (Figure 3A); however, the differential ANO5 expression between cycloheximide-treated and untreated cells was significantly greater in patients homozygous for c.1295C>G (n = 2) versus controls (n = 3, p < 0.001, Student's t test; Figure 3B), suggesting that the mutated transcripts were protected from NMD when translation was inhibited. The approximate 5-fold increase observed for case XXIX-II-1 (Figure 3A), as compared to the 8- and 9-fold increases observed for the two c.1295C>G aberrant splicing mutation homozygote cases, is in agreement with her heterozygote state for a premature termination (c.191 dupA) and missense (c.692G>T, G231V) mutations. These results strongly suggest that the two variants c.1295C>G and c.191 dupA are associated with a loss of ANO5 function. Previously, dominant ANO5 mutations have been reported in two families with gnathodiaphyseal dysplasia (GDD [MIM 166260]), a rare skeletal syndrome characterized by bone fragility, cement-osseous lesions of the maxilla and mandible, and diaphyseal sclerosis of tubular bones.28 In these patients no other abnormalities in nonskeletal tissues have been reported.28 The missense mutations in GDD patients affect a conserved cysteine located in an extracellular loop of ANO5, similar to the R758C mutation found in MMD3 patients (Figure 2D). However, because GDD is a dominant disease, it was hypothesized that the mutations in these patients cause a gain-of-function effect predominantly in skeletal tissues.28 The only two available polyclonal antibodies generated against N- and C-terminal epitopes of mouse Ano529 failed to recognize the normal human protein on western blots generated from transfected HeLa and Cos-1 cell lysates. By immunofluorescence, nonspecific perimembranous and cytoplasmic staining was detected in skeletal muscle (data not shown), thereby precluding the testing of the impact of the mutations on the expression and localization of ANO5 in patient muscle.
Figure 3.
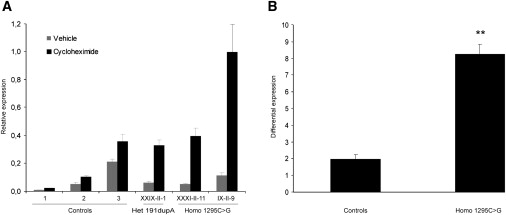
Analysis of ANO5 Expression after Cycloheximide Treatment in Patients Carrying Mutations Producing Premature Terminations
(A) ANO5 relative expression in lymphoblastoid cell lines treated with 100 μg/ml of the translation inhibitor cycloheximide for 9 hr as compared to cells treated with vehicle (0.1% DMSO). A greater increase was observed for all three patients (9-, 8-, and 5.5-fold) compared to controls (2-fold). Data represent means ± standard errors of the experimental triplicates. Patients IX-II-9 and XXXI-II-11 are homozygous for c.1295C>G, causing an aberrant splicing in exon 13, a frameshift, and a premature termination codon. Patient XXIX-II-1 is heterozygote for c.191 dupA, causing a frameshift and a premature stop codon, and for c.692G>T, predicted to result in a missense.
(B) The differential expression represents the ANO5 expression ratio of cycloheximide-treated versus untreated cells. Cases homozygous for c.1295C>G (n = 2) showed an 8-fold increase, which was significantly greater than the 2-fold differential increase observed in controls (n = 3, p < 0.001, Student's t test). Data represent the mean differential expression ± standard error of, respectively, three control samples and two case samples.
Table 1 summarizes the clinical findings for patients carrying ANO5 mutations that are also presented in greater details in Table S1 available online. LGMD2L patients are characterized by late-onset proximal scapular and pelvic girdle muscle weakness (mean age 34.4, 20–55), accompanied by asymmetrical atrophy of the quadriceps femoris and biceps brachii.21 Though calf hypertrophy can be observed at presentation, they may develop later in the course asymmetrical mild calf atrophy usually not associated with weakness. No distal weakness in the upper limbs was observed. None of the LGMD2L patients lost walking, though the proximal weakness led to difficulties climbing stairs. The intrafamilial variability is striking with two patients reporting no symptoms at ages 68 (IX-II-3) and 61 (XXXI-II-3), though mild iliopsoas weakness was documented on examination and the creatine kinases (CK) levels were known to be elevated in the past years for both cases (Table 1). The phenotype is quite different for MMD3 patients. The Dutch patients (family IV) showed early calf weakness, along with difficulties of walking on tiptoes, without atrophy.23,24 In the Finnish patients (family H), calf hypertrophy was seen in both cases during the early stages of their disease, but case H-II-6 later developed asymmetric calf atrophy. Despite the hypertrophy, calf weakness has always been an initial manifestation of all MMD3 cases. Asymmetric involvement of the proximal muscles of the lower and upper limb-girdles is a later manifestation with quadriceps atrophy being observed with time in two out of five MMD3 patients (Table 1 and Table S1). The FC case XXIX-II-1, which shares the exon 5 c.191 dupA mutation with the MMD3 Dutch Family IV, had a somewhat overlapping LGMD2L and MMD3 phenotype. At age 63, she had concomitant late-onset asymmetrical proximal upper limb (deltoid, biceps, triceps) and ilopsoas weakness and distal upper limb (wrist and finger extensors) and tibialis anterior weakness, while having asymmetrical normal strength gastrocnemius hypertrophy. CK were elevated in all patients (1032-15860IU, mean 5514IU). No cardiac abnormalities were detected on electrocardiomyogram (ECG), Holter ECG, and echocardiography for the FC and the Dutch patients. Electron microscopy performed on muscle from the LGMD2L patient IX-II-921 and MMD3 patient H-II-6 (Figure 2C) both showed multifocal disruption of the sarcolemmal membrane but no subsarcolemmal vesicle accumulation, as has been reported in dysferlinopathies.30,31 Furthermore, in the MMD3 Family H, membrane repair capability was found to be defective in patient fibroblasts by testing membrane resealing ability via either multiphoton laser irradiation or glass-bead-mediated wounding in the presence of fluorescent dyes, whereas lysosomal and enlargeosomal exocytosis was shown to occur normally, indicating that the conventional membrane repair pathways were not disrupted.24
Table 1.
Clinical Data of Patients Carrying ANO5 Mutations
| Phenotype | Fam-IDa | Ethnic Bckg | Sex | ANO5 Mutations (gDNA) | Age (2009) | Age of Onset | Distal Weakness Arm | Iliops. MRC | Quad. Atrophy | Quad. MRC | Calf Muscles | Distal Weakness Leg | Loss of Walking | Maximum CK (IU) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LGMD2L | IX-II-321 | FC | F | c.1295C>Gb | 68 | AS | none | 4/5 | + | 5/5 | none | none | no | 1,649 |
| LGMD2L | IX-II-421 | FC | M | c.1295C>Gb | 67 | 40 | none | 4/5 | + | 4.5/5 | mild atrophy | none | no | 1,032 |
| LGMD2L | IX-II-521 | FC | M | c.1295C>Gb | 65 | 37 | none | 4/5 | + | 4/5 | mild atrophy | none | no | 1,156 |
| LGMD2L | IX-II-921 | FC | M | c.1295C>Gb | 47 | 20 | none | 4/5 | + | 5/5 | mild atrophy | none | no | 6,000 |
| LGMD2L | XXXI-II-11 | FC | M | c.1295C>Gb | 43 | 20 | none | 4/5 | + | 5/5 | atrophy | calf muscles 4/5 | no | 8,081 |
| LGMD2L | XXXI-II-3 | FC | F | c.1295C>Gb | 61 | AS | none | 4.5/5 | - | 5/5 | hypertrophy | none | no | 4,061 |
| LGMD2L | XXIX-II-1 | FC | F | c.191 dupA/c.692G>T | 64 | 55 | none | 3.5/5 | + | 4.5/5 | hypertrophy | tibialis ant.4/5 | no | 1,339 |
| MMD3 | H-II-624 | Fin | M | c.2272C>Tb | 44 | 20 | none | Sin 4/5 | + | 4/5/2/5 | hypertrophy | calf muscles 2/5 | no | 15,860 |
| MMD3 | H-II-424 | Fin | M | c.2272C>Tb | 50 | 25 | none | 5/5 | - | 5/5 | hypertrophy | calf muscles 4/5 | no | 12,290 |
| MMD3 | IV-II-123 | Du | M | c.191 dupAb | 72 | 51 | none | 4/5 | - | 4/5 | none | calf muscles 2/5 | no | 5,400 |
| MMD3 | IV-II-423 | Du | M | c.191 dupAb | 67 | 45 | none | 5/5 | + | 5/5 | none | calf muscles 2/5 | no | 4,500 |
| MMD3 | IV-II-1223 | Du | M | c.191 dupAb | 53 | 39 | none | 5/5 | - | 5/5 | none | calf muscles 2/5 | no | 4,800 |
| Means | 37 | 5,514 |
Abbreviations: Ethnic bckg, ethnic background; FC, French Canadian; Fin, Finnish; Du, Dutch; AS, asymptomatic; Quad., quadriceps; Iliops., iliopsoas; MRC, Medical Research Council muscle strength scale; Tibialis ant., Tibialis anterior
Families IX, H, and IV refer to original articles (see 21,23,24).
Patients homozygous for the mutation.
Until recently the function of the Anoctamins was unknown. The ten human ANO proteins share an eight transmembrane domain structure, which led to their earlier classification as the TransMEMbrane Protein 16 (TMEM16) family, and a DUF590 domain of unknown function.13,32 Three recent reports have demonstrated that ANO1 corresponds to the elusive calcium-activated chloride channel (CaCC).15–17 In one of these studies, Schroeder et al. demonstrated that Ano1 coded for the CaCC responsible for polyspermia blockade in Xenopus oocytes.16 Since then several other independent studies confirmed that ANO1 and ANO2 function as CaCC in mouse and in human.18–20,33,34 ANO6, ANO8, and ANO9 were also confirmed to be chloride channels implicated in the control of cellular volume.20 Because of the significant structural similarity between members of the Anoctamin family, it is predicted that all these proteins, including ANO5, are likely to function as CaCC. The physiological roles attributed to CaCC, ranging from epithelial transport of electrolytes, cell volume control, olfactory and photoreceptor transduction, and cardiac membrane excitability to smooth muscle contraction, still remain unclear in many tissues.35 Until now no CaCC conductance has been reported in the plasma membrane of skeletal muscles, but the high sarcolemmal chloride conductance owing to the voltage-dependant chloride channel (CLCN1 [MIM 118425]) may have precluded the electrophysiological observation of such activity.36 Calcium-activated chloride channels have also been reported in sarcoplasmic reticulum vesicles from rabbit skeletal muscles37 but appeared to respond to much higher calcium concentrations than the one observed for ANO1.17 A chloride current has been observed after membrane wounding of sea urchin embryos,38 which raises the possibility that ANO5 may be responsible for a chloride current needed during membrane repair in human muscle. Several additional lines of evidence suggest that ANO5 may be important in the development and maintenance of skeletal muscle: ANO5 during embryogenesis is first expressed in the somites, particularly in the myotomal cells, and then in the muscle progenitor cells;29 it is mostly expressed in skeletal muscle tissue in adult mice25 and in skeletal muscle and cardiac tissues in human;28 its expression is increased in dystrophin-deficient mdx mice muscle;29 and lastly ANO5 is upregulated during myogenic differentiation of cultured cells (C2C12, Sol8, L6).29 The identification of recessive ANO5 mutations in patients with muscular dystrophies associated with sarcolemmal membrane lesions on electron microscopy and defective membrane repair documented on one MMD3 family raise the possibility that ANO5 may play an important role in the dysferlin-dependent muscle membrane repair pathway. By biochemical fractionation ANO5 has been shown to be present at the membrane and in vesicles in L6 myotubes and mouse skeletal muscle.29 Dysferlin shows a similar distribution in C2C12 myotubes and muscle tissue.39,40 Dysferlin was identified as the first protein component of the sarcolemmal repair machinery and is predicted to function as a fusogen in the formation of the patch membrane required for membrane resealing.8 Other proteins mediating with dysferlin in muscle membrane repair have been identified and include Tripartite motif-containing 72 (TRIM72),41 Annexins (MIM 151690, 151740),42,43 AHNAK (MIM 103390),44 and Caveolin-3.6,45 It could be hypothesized that ANO5 present in vesicular membranes may respond to the intracellular calcium influx known to occur with membrane damage46 by leading to a chloride influx in the vesicle that modifies its conformation so that it is recruited to the damage site.8 The observation that ANO5 mutations, as in dysferlinopathy, can lead to both a proximal and a distal muscular dystrophy further suggests that anoctaminopathies may also cause skeletal muscle demise through defective membrane repair.
Acknowledgments
We are grateful to all the patients and their family for their participation in this study. Special thanks to Professor Louise F. Charron for her original referral. We thank Dr. Anna-Kaisa Anttonen (Department of Genetics, University of Helsinki, Finland) for extracting the DNA from the Finnish family and Prof. Anna-Elina Lehesjoki (Folkhälsan Institute of Genetics, University of Helsinki, Finland) who carried mutation testing in controls. We thank Dr. Guy A. Rouleau for providing CEPH controls. We would also like to thank the Sequencing and Genotyping platforms of the McGill University/Génome Québec Innovation Centre, and the Sequencing Facility at Durham University. Many thanks to the Génome Québec and Université de Sherbrooke RNomics Centre who carried out the qPCR for the NMD assays, in particular Philippe Thibault. This study was supported by the Muscular Dystrophy Association of United States (MDA 4001 and MDA131863), Muscular Dystrophy Campaign (UK), and the Jain Foundation. V.B. is a recipient of a Canadian Institutes of Health Research (CIHR) scholarship.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
AutoSNPa, http://dna.leeds.ac.uk/autosnpa/
Centre d'Étude du Polymorphisme Humain, http://www.cephb.fr/
Génome Québec and Université de Sherbrooke RNomics Centre, http://lgfus.ca
Health Protection Agency of the United Kingdom, http://www.hpacultures.org.uk/
Human Genome Diversity Project in collaboration with the Centre d’Étude du Polymorphisme Humain (HGDP-CEPH), http://hgdp.uchicago.edu/
Human Splicing Finder, http://www.umd.be/HSF/HSF.html
The International HapMap Project, http://hapmap.ncbi.nlm.nih.gov/index.html.en
McGill University and Genome Québec Innovation Centre, http://www.genomequebecplatforms.com/mcgill/home/index.aspx
Multiz, http://genome.ucsc.edu
National Centre for Biotechnology information (NCBI) database of genetic variation (dbSNP), http://www.ncbi.nlm.nih.gov/snp/
NetGene2 server, http://www.cbs.dtu.dk/services/NetGene2/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
References
- 1.Guglieri M., Straub V., Bushby K., Lochmuller H. Limb-girdle muscular dystrophies. Curr. Opin. Neurol. 2008;21:576–584. doi: 10.1097/WCO.0b013e32830efdc2. [DOI] [PubMed] [Google Scholar]
- 2.Norwood F., de Visser M., Eymard B., Lochmuller H., Bushby K. EFNS guideline on diagnosis and management of limb girdle muscular dystrophies. Eur. J. Neurol. 2007;14:1305–1312. doi: 10.1111/j.1468-1331.2007.01979.x. [DOI] [PubMed] [Google Scholar]
- 3.Cohn R.D., Campbell K.P. Molecular basis of muscular dystrophies. Muscle Nerve. 2000;23:1456–1471. doi: 10.1002/1097-4598(200010)23:10<1456::aid-mus2>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 4.Allikian M.J., McNally E.M. Processing and assembly of the dystrophin glycoprotein complex. Traffic. 2007;8:177–183. doi: 10.1111/j.1600-0854.2006.00519.x. [DOI] [PubMed] [Google Scholar]
- 5.Bansal D., Miyake K., Vogel S.S., Groh S., Chen C.C., Williamson R., McNeil P.L., Campbell K.P. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature. 2003;423:168–172. doi: 10.1038/nature01573. [DOI] [PubMed] [Google Scholar]
- 6.Cai C., Weisleder N., Ko J.K., Komazaki S., Sunada Y., Nishi M., Takeshima H., Ma J. Membrane repair defects in muscular dystrophy are linked to altered interaction between MG53, caveolin-3, and dysferlin. J. Biol. Chem. 2009;284:15894–15902. doi: 10.1074/jbc.M109.009589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Glover L., Brown R.H., Jr. Dysferlin in membrane trafficking and patch repair. Traffic. 2007;8:785–794. doi: 10.1111/j.1600-0854.2007.00573.x. [DOI] [PubMed] [Google Scholar]
- 8.Han R., Campbell K.P. Dysferlin and muscle membrane repair. Curr. Opin. Cell Biol. 2007;19:409–416. doi: 10.1016/j.ceb.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Malicdan M.C., Nonaka I. Distal myopathies a review: Highlights on distal myopathies with rimmed vacuoles. Neurol. India. 2008;56:314–324. doi: 10.4103/0028-3886.43450. [DOI] [PubMed] [Google Scholar]
- 10.Bashir R., Britton S., Strachan T., Keers S., Vafiadaki E., Lako M., Richard I., Marchand S., Bourg N., Argov Z. A gene related to Caenorhabditis elegans spermatogenesis factor fer-1 is mutated in limb-girdle muscular dystrophy type 2B. Nat. Genet. 1998;20:37–42. doi: 10.1038/1689. [DOI] [PubMed] [Google Scholar]
- 11.Liu J., Aoki M., Illa I., Wu C., Fardeau M., Angelini C., Serrano C., Urtizberea J.A., Hentati F., Hamida M.B. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat. Genet. 1998;20:31–36. doi: 10.1038/1682. [DOI] [PubMed] [Google Scholar]
- 12.Urtizberea J.A., Bassez G., Leturcq F., Nguyen K., Krahn M., Levy N. Dysferlinopathies. Neurol. India. 2008;56:289–297. doi: 10.4103/0028-3886.43447. [DOI] [PubMed] [Google Scholar]
- 13.Galindo B.E., Vacquier V.D. Phylogeny of the TMEM16 protein family: Some members are overexpressed in cancer. Int. J. Mol. Med. 2005;16:919–924. [PubMed] [Google Scholar]
- 14.Hartzell H.C., Yu K., Xiao Q., Chien L.T., Qu Z. Anoctamin/TMEM16 family members are Ca2+-activated Cl- channels. J. Physiol. 2009;587:2127–2139. doi: 10.1113/jphysiol.2008.163709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caputo A., Caci E., Ferrera L., Pedemonte N., Barsanti C., Sondo E., Pfeffer U., Ravazzolo R., Zegarra-Moran O., Galietta L.J. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science. 2008;322:590–594. doi: 10.1126/science.1163518. [DOI] [PubMed] [Google Scholar]
- 16.Schroeder B.C., Cheng T., Jan Y.N., Jan L.Y. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell. 2008;134:1019–1029. doi: 10.1016/j.cell.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang Y.D., Cho H., Koo J.Y., Tak M.H., Cho Y., Shim W.S., Park S.P., Lee J., Lee B., Kim B.M. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature. 2008;455:1210–1215. doi: 10.1038/nature07313. [DOI] [PubMed] [Google Scholar]
- 18.Stohr H., Heisig J.B., Benz P.M., Schoberl S., Milenkovic V.M., Strauss O., Aartsen W.M., Wijnholds J., Weber B.H., Schulz H.L. TMEM16B, a novel protein with calcium-dependent chloride channel activity, associates with a presynaptic protein complex in photoreceptor terminals. J. Neurosci. 2009;29:6809–6818. doi: 10.1523/JNEUROSCI.5546-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stephan A.B., Shum E.Y., Hirsh S., Cygnar K.D., Reisert J., Zhao H. ANO2 is the cilial calcium-activated chloride channel that may mediate olfactory amplification. Proc. Natl. Acad. Sci. USA. 2009;106:11776–11781. doi: 10.1073/pnas.0903304106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Almaca J., Tian Y., Aldehni F., Ousingsawat J., Kongsuphol P., Rock J.R., Harfe B.D., Schreiber R., Kunzelmann K. TMEM16 proteins produce volume regulated chloride currents that are reduced in mice lacking TMEM16A. J. Biol. Chem. 2009;284:28571–28578. doi: 10.1074/jbc.M109.010074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jarry J., Rioux M.F., Bolduc V., Robitaille Y., Khoury V., Thiffault I., Tetreault M., Loisel L., Bouchard J.P., Brais B. A novel autosomal recessive limb-girdle muscular dystrophy with quadriceps atrophy maps to 11p13-p12. Brain. 2007;130:368–380. doi: 10.1093/brain/awl270. [DOI] [PubMed] [Google Scholar]
- 22.Carr I.M., Flintoff K.J., Taylor G.R., Markham A.F., Bonthron D.T. Interactive visual analysis of SNP data for rapid autozygosity mapping in consanguineous families. Hum. Mutat. 2006;27:1041–1046. doi: 10.1002/humu.20383. [DOI] [PubMed] [Google Scholar]
- 23.Linssen W.H., de Visser M., Notermans N.C., Vreyling J.P., Van Doorn P.A., Wokke J.H., Baas F., Bolhuis P.A. Genetic heterogeneity in Miyoshi-type distal muscular dystrophy. Neuromuscul. Disord. 1998;8:317–320. doi: 10.1016/s0960-8966(98)00020-0. [DOI] [PubMed] [Google Scholar]
- 24.Jaiswal J.K., Marlow G., Summerill G., Mahjneh I., Mueller S., Hill M., Miyake K., Haase H., Anderson L.V., Richard I. Patients with a non-dysferlin Miyoshi myopathy have a novel membrane repair defect. Traffic. 2007;8:77–88. doi: 10.1111/j.1600-0854.2006.00505.x. [DOI] [PubMed] [Google Scholar]
- 25.Tsutsumi S., Inoue H., Sakamoto Y., Mizuta K., Kamata N., Itakura M. Molecular cloning and characterization of the murine gnathodiaphyseal dysplasia gene GDD1. Biochem. Biophys. Res. Commun. 2005;331:1099–1106. doi: 10.1016/j.bbrc.2005.03.226. [DOI] [PubMed] [Google Scholar]
- 26.Brogna S., Wen J. Nonsense-mediated mRNA decay (NMD) mechanisms. Nat. Struct. Mol. Biol. 2009;16:107–113. doi: 10.1038/nsmb.1550. [DOI] [PubMed] [Google Scholar]
- 27.Hellemans J., Mortier G., De Paepe A., Speleman F., Vandesompele J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007;8:R19. doi: 10.1186/gb-2007-8-2-r19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsutsumi S., Kamata N., Vokes T.J., Maruoka Y., Nakakuki K., Enomoto S., Omura K., Amagasa T., Nagayama M., Saito-Ohara F. The novel gene encoding a putative transmembrane protein is mutated in gnathodiaphyseal dysplasia (GDD) Am. J. Hum. Genet. 2004;74:1255–1261. doi: 10.1086/421527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mizuta K., Tsutsumi S., Inoue H., Sakamoto Y., Miyatake K., Miyawaki K., Noji S., Kamata N., Itakura M. Molecular characterization of GDD1/TMEM16E, the gene product responsible for autosomal dominant gnathodiaphyseal dysplasia. Biochem. Biophys. Res. Commun. 2007;357:126–132. doi: 10.1016/j.bbrc.2007.03.108. [DOI] [PubMed] [Google Scholar]
- 30.Selcen D., Stilling G., Engel A.G. The earliest pathologic alterations in dysferlinopathy. Neurology. 2001;56:1472–1481. doi: 10.1212/wnl.56.11.1472. [DOI] [PubMed] [Google Scholar]
- 31.Cenacchi G., Fanin M., De Giorgi L.B., Angelini C. Ultrastructural changes in dysferlinopathy support defective membrane repair mechanism. J. Clin. Pathol. 2005;58:190–195. doi: 10.1136/jcp.2004.018978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rock J.R., Harfe B.D. Expression of TMEM16 paralogs during murine embryogenesis. Dev. Dyn. 2008;237:2566–2574. doi: 10.1002/dvdy.21676. [DOI] [PubMed] [Google Scholar]
- 33.Ousingsawat J., Martins J.R., Schreiber R., Rock J.R., Harfe B.D., Kunzelmann K. Loss of TMEM16A causes a defect in epithelial Ca2+ dependent chloride transport. J. Biol. Chem. 2009;284:28698–28703. doi: 10.1074/jbc.M109.012120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rock J.R., O'Neal W.K., Gabriel S.E., Randell S.H., Harfe B.D., Boucher R.C., Grubb B.R. Transmembrane protein 16A (TMEM16A) is a Ca2+-regulated Cl- secretory channel in mouse airways. J. Biol. Chem. 2009;284:14875–14880. doi: 10.1074/jbc.C109.000869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hartzell C., Putzier I., Arreola J. Calcium-activated chloride channels. Annu. Rev. Physiol. 2005;67:719–758. doi: 10.1146/annurev.physiol.67.032003.154341. [DOI] [PubMed] [Google Scholar]
- 36.Aromataris E.C., Rychkov G.Y. ClC-1 chloride channel: Matching its properties to a role in skeletal muscle. Clin. Exp. Pharmacol. Physiol. 2006;33:1118–1123. doi: 10.1111/j.1440-1681.2006.04502.x. [DOI] [PubMed] [Google Scholar]
- 37.Kourie J.I., Laver D.R., Ahern G.P., Dulhunty A.F. A calcium-activated chloride channel in sarcoplasmic reticulum vesicles from rabbit skeletal muscle. Am. J. Physiol. 1996;270:C1675–C1686. doi: 10.1152/ajpcell.1996.270.6.C1675. [DOI] [PubMed] [Google Scholar]
- 38.Fein A., Terasaki M. Rapid increase in plasma membrane chloride permeability during wound resealing in starfish oocytes. J. Gen. Physiol. 2005;126:151–159. doi: 10.1085/jgp.200509294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klinge L., Laval S., Keers S., Haldane F., Straub V., Barresi R., Bushby K. From T-tubule to sarcolemma: Damage-induced dysferlin translocation in early myogenesis. FASEB J. 2007;21:1768–1776. doi: 10.1096/fj.06-7659com. [DOI] [PubMed] [Google Scholar]
- 40.Ampong B.N., Imamura M., Matsumiya T., Yoshida M., Takeda S. Intracellular localization of dysferlin and its association with the dihydropyridine receptor. Acta Myol. 2005;24:134–144. [PubMed] [Google Scholar]
- 41.Cai C., Masumiya H., Weisleder N., Matsuda N., Nishi M., Hwang M., Ko J.K., Lin P., Thornton A., Zhao X. MG53 nucleates assembly of cell membrane repair machinery. Nat. Cell Biol. 2009;11:56–64. doi: 10.1038/ncb1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lennon N.J., Kho A., Bacskai B.J., Perlmutter S.L., Hyman B.T., Brown R.H., Jr. Dysferlin interacts with annexins A1 and A2 and mediates sarcolemmal wound-healing. J. Biol. Chem. 2003;278:50466–50473. doi: 10.1074/jbc.M307247200. [DOI] [PubMed] [Google Scholar]
- 43.McNeil A.K., Rescher U., Gerke V., McNeil P.L. Requirement for annexin A1 in plasma membrane repair. J. Biol. Chem. 2006;281:35202–35207. doi: 10.1074/jbc.M606406200. [DOI] [PubMed] [Google Scholar]
- 44.Huang Y., Laval S.H., van Remoortere A., Baudier J., Benaud C., Anderson L.V., Straub V., Deelder A., Frants R.R., den Dunnen J.T. AHNAK, a novel component of the dysferlin protein complex, redistributes to the cytoplasm with dysferlin during skeletal muscle regeneration. FASEB J. 2007;21:732–742. doi: 10.1096/fj.06-6628com. [DOI] [PubMed] [Google Scholar]
- 45.Hernandez-Deviez D.J., Howes M.T., Laval S.H., Bushby K., Hancock J.F., Parton R.G. Caveolin regulates endocytosis of the muscle repair protein, dysferlin. J. Biol. Chem. 2008;283:6476–6488. doi: 10.1074/jbc.M708776200. [DOI] [PubMed] [Google Scholar]
- 46.McNeil P.L., Kirchhausen T. An emergency response team for membrane repair. Nat. Rev. Mol. Cell Biol. 2005;6:499–505. doi: 10.1038/nrm1665. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.