

Abstract
Frank-Ter Haar syndrome (FTHS), also known as Ter Haar syndrome, is an autosomal-recessive disorder characterized by skeletal, cardiovascular, and eye abnormalities, such as increased intraocular pressure, prominent eyes, and hypertelorism. We have conducted homozygosity mapping on patients representing 12 FTHS families. A locus on chromosome 5q35.1 was identified for which patients from nine families shared homozygosity. For one family, a homozygous deletion mapped exactly to the smallest region of overlapping homozygosity, which contains a single gene, SH3PXD2B. This gene encodes the TKS4 protein, a phox homology (PX) and Src homology 3 (SH3) domain-containing adaptor protein and Src substrate. This protein was recently shown to be involved in the formation of actin-rich membrane protrusions called podosomes or invadopodia, which coordinate pericellular proteolysis with cell migration. Mice lacking Tks4 also showed pronounced skeletal, eye, and cardiac abnormalities and phenocopied the majority of the defects associated with FTHS. These findings establish a role for TKS4 in FTHS and embryonic development. Mutation analysis revealed five different homozygous mutations in SH3PXD2B in seven FTHS families. No SH3PXD2B mutations were detected in six other FTHS families, demonstrating the genetic heterogeneity of this condition. Interestingly however, dermal fibroblasts from one of the individuals without an SH3PXD2B mutation nevertheless expressed lower levels of the TKS4 protein, suggesting a common mechanism underlying disease causation.
Main Text
In 1973, Frank et al. described a patient with megalocornea, brachycephaly, large anterior fontanels, hypertelorism, anteverted nostrils, thoracolumbar kyphosis, prominent coccyx, short hands, flexion deformity of fingers, club feet, and heart murmur and suggested this to be a hitherto undescribed entity (Figure 1).1 Later, ter Haar et al. described several members of a single family with similar features (see Figure S1 available online).2 This syndrome is now referred to as Frank-Ter Haar syndrome (FTHS [MIM 249420]). A small number of additional FTHS families have been reported, most within families with consanguineous unions, suggestive of a recessive inheritance pattern.3–8 FTHS patients usually die in infancy or in early childhood because of the cardiovascular anomalies, respiratory infections, or unknown causes. To date, the molecular cause of FTHS has not been established.
Figure 1.

Clinical Features of FTHS, Homozygosity Mapping, and Analysis of SH3PXD2B
(A) Left: girl from family 1 showing broad face, megalocornea, low nasal bridge, upturned nares, open mouth appearance, full cheeks, and small chin. Right: the same girl at a later age showing sustained facial features.
(B) Facial feature of patient 9.1 at age 3.5 years demonstrating hypertelorism, large eyes, full cheeks, wide mouth, microretrognathia, and large ears.
(C) Patient 9.2 showing brachycephaly, high forehead, hypertelorism, large eyes, megalocornea, broad mouth, and small chin.
(D) Caudal appendage at coccyx with prominent skin fold (family 6).
(E) Left: hand of patient 9.1 showing short fingers with short and broad terminal phalanges. Camptodactyly was seen only after birth. Right: X-rays showing generalized osteopenia, short tubular bones of the hand.
(F) Increased lower thoracolumbar kyphosis and brachycephaly in patient 9.1.
(G) Poor modeling of long bones including the distal radius and ulna in patient 1.
(H) Schematic representation of the 5q35.1 region that is homozygous in 12 patients and homozygously deleted in family 7. The deletion was identified upon analysis of the 250K SNP array data via the CNAG algorithm, as visualized by the bottom diagram in which the deletion is encircled in blue. The overlap between the deletion and the smallest region of homozygosity harbors only a single gene, SH3PXD2B (gray). An asterisk indicates patients in whom a mutation has been identified. The hg18 UCSC genome build (version 2006) was used for the coordinates in the genetic map.
(I) Schematic representation of TKS4 protein structure with an N-terminal PX domain and four SH3 domains. The position of the mutations is indicated.
(J) Position of the amino acid substitution R43W at a highly conserved region within the PX domain predicts a disruptive effect on its normal function in phosphoinositide binding and membrane association.
(K) Immunoblot analysis with a TKS4-specific antibody detects a 120 kDa band in mouse fibroblasts (3T3), in mouse fibroblasts transformed with Src (Src-3T3), and in fibroblasts from control individuals (control). No such product is seen in fibroblasts from three different patients from families 1 and 2 with an SH3PXD2B mutation. The TKS4 band was also present but at clearly reduced levels in cells from another FTHS patient from family 3 without an SH3PXD2B mutation (unsolved). Tubulin was used as a loading control.
Here we performed homozygosity mapping to localize the genetic defect in 16 patients from 12 apparently unrelated FTHS families with known or suspected consanguinity (Figure S1; Table 1). This included the family originally described by ter Haar,2 for which genealogical studies revealed a common ancestor for the parents of some of the patients eight to ten generations back (Figure S1). Written informed consent was obtained for all patients, and research was approved by the local ethics committee of the Radboud University Nijmegen Medical Centre. Genome-wide homozygosity mapping was carried out with the Affymetrix mapping 250K SNP array, on DNA fragments in the 200 to 1100 bp size range amplified from 250 ng genomic DNA. The data were analyzed by genotyping console. The mapping revealed a region on chromosome 5q35.1 for which 12 out of 16 patients had overlapping regions of homozygosity (Figure 1). To confirm the homozygosity, we used microsatellite markers on chromosome 5q35.1 region. Families 1 and 2, both of Dutch origin, shared the same haplotype, suggesting a founder effect. The common region of homozygosity spanned only 0.27 Mb and comprised the NEURL1B gene and part of the SH3PXD2B gene. Interestingly, analysis of the SNP array data for copy number variation with copy number analyzer for GeneChip (CNAG)9 revealed a homozygous deletion in the patients of family 7, who manifested the typical FTHS phenotype (Table 1).5 Genomic quantitative PCR analysis confirmed the homozygous deletion and mapped the endpoints between STK10 (MIM 603919) and AK026748 (Figure S1). This deletion harbors the UBTD2 (MIM 610174) and SH3PXD2B genes. Thus, the cumulative results of homozygosity mapping and copy number analysis identified SH3PXD2B as the most plausible FTHS candidate gene (Figure 1).
Table 1.
Clinical Features of FTHS Patients in This Study
|
Family |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
11 |
12 |
13 |
||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient | 1 | 1 | 2 | 1 | 1 | 1 | 1 | 1 | 2 | 1 | 1 | 2 | 1 | 2 | 1 | 1 | 1 |
| Gender | M | M | M | M | M | F | F | M | F | F | M | M | M | F | F | M | M |
| Consanguinity | − | + | + | + | + | + | + | + | + | + | + | + | + | + | − | + | + |
| Motor retardation | + | + | + | + | + | + | + | + | + | + | − | + | + | ||||
| Craniofacial Abnormalities | |||||||||||||||||
| Prominent forehead | + | + | + | + | + | + | + | + | + | + | + | + | − | − | + | + | + |
| Brachycephaly | + | + | + | + | + | + | + | + | + | − | + | + | − | − | − | + | + |
| Wide anterior fontanel | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | |
| Hypertelorism | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | ||
| Congenital glaucoma | + | + | + | − | + | − | + | + | −a | − | − | − | − | + | −b | − | |
| Large cornea | + | + | + | + | + | + | + | + | − | + | + | + | + | + | + | + | |
| Prominent eyes | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| Full cheeks | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | |
| Anteverted nostrils | + | + | + | + | − | + | + | − | − | − | + | + | + | + | + | ||
| Broad mouth | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | |
| Broad alveolar ridges | + | + | − | + | + | + | − | − | − | + | + | + | + | ||||
| Micrognathia | + | + | + | + | + | + | − | + | + | + | + | + | + | + | − | + | + |
| Protruding ears | + | + | + | + | + | − | − | − | + | + | − | + | |||||
| Skeletal Abnormalities | |||||||||||||||||
| Kyphosis | + | + | − | + | + | + | − | − | + | − | − | − | − | + | + | ||
| Prominent coccyx | + | + | + | + | + | + | + | + | − | − | + | + | − | + | + | ||
| Bowing of long bones | + | + | + | − | + | − | + | + | − | − | − | − | − | − | +c | + | |
| Short hands | + | + | + | − | + | + | + | + | − | + | + | − | − | − | + | + | |
| Flexion deformity of fingers | + | − | − | + | + | + | + | − | − | + | − | − | + | + | − | − | + |
| Club feet | + | + | − | + | − | − | PA | + | + | − | − | − | + | −d | + | ||
| Cardiac Manifestations | |||||||||||||||||
| Mitral valve anomaly | + | + | MVP | + | − | − | − | − | − | − | +e | − | |||||
| Double right outlet | + | + | − | − | − | − | − | − | |||||||||
| Ventricular septal defect | − | + | + | − | + | − | − | + | HCM | ASD | + | +f | |||||
| Other abnormalities | gall stones | brain MRIg | auricular pits | ||||||||||||||
| SH3PXD2B mutation | + | + | + | − | − | − | + | + | + | − | + | + | − | − | − | + | + |
The following abbreviations are used: ASD, atrial septal defect; VSD, ventricular septal defect; MVP, mitral valve prolapsed; PA, pes adductus; HCM, hypertrophic cardiomyopathy. + denotes that the feature is present and – denotes that the feature was not detected in the patients. Family 2 was described by ter Haar et al.2 and Hamel et al.,3 family 3 by Wallerstein et al.,7 families 4 and 6 by Maas et al.,4 and family 7 by Megarbane et al.5
Bilateral iris and retinal coloboma.
Retinal detachment, sudden vision loss at age 7.
Severe genu valgum, dislocated left knee, bilateral hips, both elbows.
Pes valgus.
Cardiomegaly, severe mitral and aortic valve prolapsed.
VSD, ASD, and patent ductus arteriosis.
Slightly prominent ventricles on MRI.
Next, we screened the SH3PXD2B gene in probands representing 13 FTHS families from our cohort by direct DNA sequencing. Intron-specific primers were designed for the amplification of exons 1–13 of the SH3PXD2B gene (Table S1; data not shown). The segregation of DNA variants was verified in the corresponding families. In addition, we analyzed all variants in 50 unrelated control individuals to exclude benign polymorphisms. A total of four additional homozygous mutations were identified in six families (Figure 1). In families 1 and 2, a homozygous insertion c.147insT was detected, which predicted the creation of an immediate stop in the same codon (p.F49X), in all three affected individuals available for testing. In families 6 and 9, which are of unrelated ethnicity, we detected a homozygous 1 bp deletion c.969delG, which predicts a frameshift followed by a premature stop codon (p.G323fsX19; Figure 1; Figure S1). In family 12, we identified a substitution c.129C>T (p.R43W), which disrupts a highly conserved amino acid within the phox homology (PX) domain of the protein (Figure 1) and which is predicted to abolish binding to phosphoinositides.10,11 Finally, a c.76-2A>C splice site mutation was identified in an additional patient who was not included for homozygosity mapping (family 13), which disrupts the strictly conserved adenosine of the splice acceptor site. All mutations segregated within the respective families in a pattern compatible with autosomal-recessive inheritance (Figure S1). None of the truncating mutations were detected in a minimum of 50 control individuals. In addition, none of 210 control chromosomes contained the nontruncating mutations. Together, these data establish that SH3PXD2B gene mutations are causative for a subset (7 of 13) of FTHS families. The phenotype of patients with an intragenic SH3PXD2B mutation does not appear to be notably different from that of the patients of family 7, who carry a deletion that also disrupts UBTD2 (Table 1). Apparently, the function of UBTD2 is at least partially redundant in humans. The clinical features in FTHS patients in whom no mutations were detected are highly similar to those of patients with SH3PXD2B mutations, indicating genetic heterogeneity in FTHS. Indeed, the result of homozygosity mapping analysis excludes the 5q35.1 locus in four families and supports the involvement of multiple other loci.
The protein product of the SH3PXD2B gene is known as TKS4 (tyrosine kinase substrate with four Src homology 3 [SH3] domains). It is an adaptor protein with a PX domain, four SH3 domains, and a substrate of the tyrosine kinase Src.10 In Src-transformed murine fibroblasts, Tks4 is required for the formation of podosomes/invadopodia, which are dynamic, actin-rich plasma membrane protrusions involved in cell adhesion, migration, and matrix degradation.12 In these cells, Tks4 recruits to podosomes the membrane type-1 matrix metalloprotease (MT1-MMP),10 an obligate podosome/invadopodia protein involved in pericellular proteolysis of extracellular matrix.13 A paralog of Tks4, Tks5, encoded by SH3PXD2A on chromosome 10, is also an adaptor protein involved in podosome/invadopodia formation,14 but none of the unexplained FTHS patients showed homozygosity at this locus (data not shown). Podosomes are necessary for the adhesion and migration of a variety of cell types, including macrophages, dendritic cells, osteoclasts, vascular smooth muscle, and endothelial cells,12,13,15 several of which may be relevant to the FTHS phenotype.
The TKS4 protein levels in human primary dermal fibroblasts were analyzed by immunoblot of total cell lysates as previously described.10 A Tks4 signal was observed in control fibroblasts, whereas no signal could be detected in cells from patients of families 1 and 2 with an SH3PXD2B mutation, demonstrating that FTHS can be caused by homozygous loss-of-function mutation of SH3PXD2B (Figure 1). Quantitative RT-PCR analysis was conducted to analyze the SH3PXD2B transcript in these fibroblasts. Normal levels of SH3PXD2B transcript were detected for three affected individuals with the c.147insT (F49X) mutation, even though no TKS4 protein was detected. This indicates that the premature stop codon introduced by this mutation does not result in nonsense-mediated RNA decay and likely results instead in a truncated unstable protein. Fibroblasts from other affected individuals with SH3PXD2B mutations were not available, precluding a more detailed analysis.
No SH3PXD2B mutations were identified in six FTHS families, including families 3, 4, and 5, for which patients showed homozygosity at the 5q35.1 locus. It is possible that mutations in cis-regulatory elements have been missed in these three families. In keeping with this hypothesis, although fibroblasts derived from the patient from family 37 with no mutation in the SH3PXD2B coding sequence did contain detectable Tks4 protein, its abundance was sharply reduced compared to wild-type cells. Quantitative PCR did not reveal any differences at the mRNA level (data not shown), so it is possible that a mutation affecting TKS4 protein synthesis or stability has been missed. Alternatively, this patient carries a mutation in another gene which regulates TKS4 protein homeostasis. Genetic heterogeneity of FTHS would also be consistent with the observation that there are several other homozygous regions in patients from each of six unsolved families. The results of homozygosity mapping also support the involvement of multiple other loci.
Murine Tks4 is 87% identical to the human paralog and has a similar domain structure. To gain more insight into where Tks4 may play a role during development, we performed cRNA in situ hybridization analysis of embryonic mouse cryosections with probes specific for Sh3pxd2b. Spatiotemporally restricted Sh3pxd2b expression was observed at various embryonic development stages in tissues that are affected in FTHS patients: heart, bone, and eye (Figure S2). At embryonic day (E) 10.5, a speckled expression of Sh3pxd2b was observed in heart and hindbrain, which appeared to have increased at E12.5, and involved a subset of cells on the luminal side of the left ventricular wall in the case of the heart and neuroepithelium in the case of the brain. At E14.5, expression was present in developing bones (e.g., the proximal ribs, lower jaw, clavicle), but the expression in the heart was no longer detectable. At stages E16.5 and E18.5, strong expression was seen in the long bones of the limbs, particularly in the growth plates, as well as in the facial and cranial bones and the primordial incisor. Expression in the ribs was seen in the proximal regions in those areas where the transition from cartilage to bone is expected to occur. Disruption of expression during rib and vertebral bone development could explain the chest deformities and kyphosis observed in FTHS. Expression in the eye at E16.5 was highly specific for the ganglion cell layer. The significance of this observation for the occurrence of glaucoma in FTHS patients and in Sh3pxd2b mice (see below) is presently not clear. Overall, the fact that Sh3pxd2b expression can be found in tissues that are affected in FTHS suggests that it is required for the correct patterning and development of these organs. However, expression is also observed in areas that do not directly relate to the FTHS phenotype, for example the hindbrain. In the future it will be interesting to determine whether this is relevant to the motor retardation frequently observed in affected individuals (Table 1).
To investigate the function of Tks4 in vivo, we analyzed Sh3pxd2b mutant mice that were generated via a gene-trap strategy by Lexicon Pharmaceuticals (embryonic stem cell clone ID GST_1527_E5). Inactivation of Sh3pxd2b in these mice is caused by insertion of the gene-trap vector VICTR 48 between exons 3 and 4 of the Sh3pxd2b gene on chromosome 11. Mice were kept in a mixed genetic background (C57BL6/a129SvJ). All animal experiments were approved by the Burnham Institute's Institutional Animal Research Committee. Disruption of the Sh3pxd2b gene was assessed by PCR of genomic DNA and immunoblot on total lysates from murine embryonic fibroblasts.10 No Tks4 protein was detected in fibroblasts derived from E12.5 homozygous mutant embryos, suggesting that the gene trap caused a complete loss of gene expression from this locus; therefore, these mice will be referred to as Sh3pxd2b null. We noted that Sh3pxd2b null mice were born at Mendelian ratios, but approximately 20% of the null mice died in the first weeks of life of undetermined causes. For those that survived, lifespan did not appear to be compromised (Figure 2). At birth, Sh3pxd2b null mice were on average the same size as their wild-type and heterozygous littermates. However, the null mice were markedly smaller by weaning, and this growth retardation was maintained during their lives (Figure 2; Figure S3).
Figure 2.
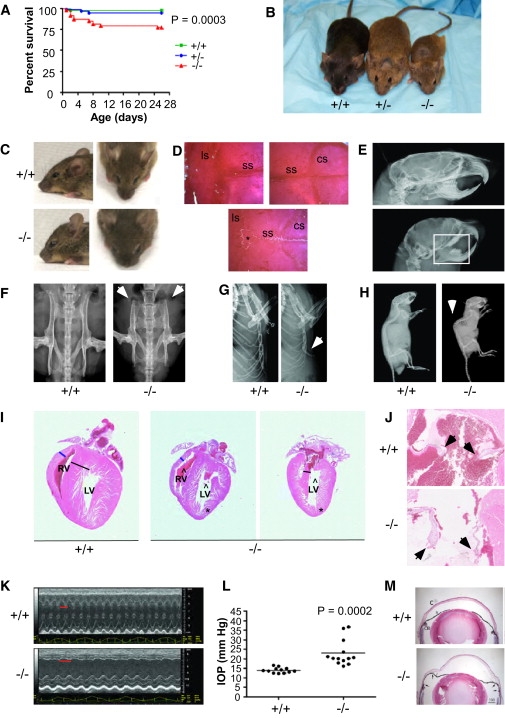
Developmental Defects Due to Sh3pxd2b Inactivation in the Mouse
(A) Mice in which the Sh3pxd2b gene is inactivated because of a gene-trap insertion have increased mortality during the first 3 weeks of life.
(B) Sh3pxd2b null (−/−) mice experience severe growth retardation as compared to wild-type and heterozygous mutants. The image shown is of mice at 3 months of age.
(C) Lateral and dorsal view of wild-type (WT) and Sh3pxd2b−/− mouse heads from 10-week-old mice. Sh3pxd2b−/− mice have craniofacial defects characterized by a shorter distance between nose and forehead and hypertelorism. A depressed nasal bridge and small chin can also be seen.
(D) Alizarin red stained skulls of 5-month-old WT and Sh3pxd2b−/− mice. Mutants have open sagittal sutures, as well as wormian bones (∗). The following abbreviations are used: ls, lambdoid suture; ss, sagittal suture; cs, coronal suture.
(E) Other skeletal defects in Sh3pxd2b−/− adult (2-month-old) mice visible by X-ray imaging include the teeth (reduced size), neurocranium (decreased anterior-posterior diameter), and viscerocranium (short maxilla, short mandibula, and more vertical orientation of skull base).
(F) View of the iliac crest showing shortening and widening with decreased flaring of iliac wings in the null mice at 2 months of age.
(G) Prominence of the xyphoid part of the sternum is noted in the null mice at 2 months of age.
(H) The vertebral column, showing marked thoracic kyphosis and mild lumbosacral lordosis in null mice at 2 months of age.
(E–H) Defects indicated by arrowheads and square.
(I) H&E-stained coronal heart sections from 4-month-old WT and old Sh3pxd2b−/− mice. The ventricular septum thickness is indicated with a black line. The asterisks indicate apical indentation. Carats mark the dysmorphic chambers, and the blue line shows the ventricular wall thickness. The following abbreviations are used: RV, right ventricle; LV, left ventricle.
(J) Coronal H&E sections of 4-month-old WT and Sh3pxd2b−/− mice show the mitral valves, which are indicated by the black arrowheads.
(K) Echocardiogram of 2.5-month-old WT and Sh3pxd2b−/− mice. Sh3pxd2b−/− mice were bradycardic, as shown by the longer interval (red line) between two consecutive systoles.
(L) IOP measurements of eyes from WT and Sh3pxd2b−/−. The ages ranged from 4 to 16 months in both groups.
(M) H&E staining of anterior segments of eyes from WT and Sh3pxd2b−/− mice reveal the structure of the cornea and iris. Note that the angle between the cornea (C) and the iris (Ir) extends back to the ciliary body (CB) in the wild-type eye. In contrast, homozygous null mice had apposition of the peripheral iris to the corneal endothelium (between arrows). In the homozygous null mice, the cornea bulges anteriorly. Scale bar represents 300 μm.
Alizarin red staining of Sh3pxd2b null skulls demonstrated severe craniofacial defects characterized by a shorter nasal bone, micrognathia, hypertelorism, sagittal suture agenesis, and the presence of wormian bones (Figure 2; Figure S4). Mouse skeletons were also analyzed by X-ray with a FAXITRON MX-20 machine. Mutant mice displayed marked kyphosis, poorly aligned teeth, anomalies in the iliac crest, and a prominent xiphisternum when compared to heterozygotes and wild-types (Figure 2). Most of these developmental abnormalities bear a remarkable similarity to the skeletal defects of FTHS patients (Table 2).
Table 2.
Comparison of Features Present in FTHS Patients and Sh3pxd2b−/− Mice
| FTHS Patients | Sh3pxd2b−/− Mice | |
|---|---|---|
| Craniofacial | ||
| Brachycephaly | + | − |
| Suture agenesis | + | + |
| Wormian bones | + | + |
| Prominent forehead | + | + |
| Hypertelorism | + | + |
| High palate | + | + |
| Dentition abnormalities | +a | + |
| Micrognathia | + | + |
| Skeletal | ||
| Prominent sternum/xyphoid | − | + |
| Pectus excavatum | + | − |
| Increased thoracic kyphosis | + | + |
| Pelvic anomalies | +b | + |
| Bowing of long bones | + | − |
| Extremity abnormalities | +c | ND |
| Prominent coccyx | + | NA |
| Eye | ||
| Glaucoma | + | + |
| Megalocornea | + | +d |
| Buphthalmos | + | + |
| Cardiac | ||
| Ventricular defectse | + | + |
| Other | ||
| Growth retardationf | + | + |
| Adiposity | ?g | + |
The following abbreviations are used: ND, not determined; NA, not applicable. + denotes that the feature is present and – denotes that the feature was not detected in the patients or mice.
Patients present broad alveolar ridges.
Patients present hip dysplasia, whereas mice have malformation of iliac crest.
Patients present short hands, flexion deformity of hands, and club feet.
Unlike megalocornea in humans, the enlargement of the cornea increases with age.
Defects in patients include mitral valve anomalies, double-outlet right ventricle, and ventricular septal defects. In the mice, dysmorphia of the septal anterior wall, ventricles, and mitral valve have been observed.
Patients have been described to be in the lower weight and height centiles as they grow.
One patient reported to have “little or no subcutaneous fat.”
Many FTHS patients have cardiac deficiencies. To investigate possible heart defects in the Sh3pxd2b null mice, we dissected, fixed, embedded, and sectioned hearts at 5 μm for hematoxylin and eosin (H&E) staining. Coronal sectioning of hearts showed that all Sh3pxd2b null mice examined had cardiac abnormalities (8 of 8), although there was variability in the range of phenotypes seen. Anomalies included dysmorphic ventricular chambers (8 of 8), thin mitral valves (4 of 8) that may be causative of prolapse, and immature and disarrayed trabeculae (6 of 8) with frequent apical indentation (5 of 8) (Figure 2). Echocardiographic analyses were also performed in male mice with a Visual Sonic Vevo 770 fitted with an 8–15 MHz linear array transducer, as previously described.16,17 This analysis revealed reduced thickness of the septal anterior wall and bradycardia (300–400 bpm compared to >500 for wild-types) (3 of 3) (Figure 2). It is of interest to note that FTHS individuals also showed septal, ventricular, and mitral valve defects (Table 2). Further analysis will be required to determine the molecular basis for these observations.
Because one of the most highly penetrant features of the FTHS patients involves the eye, we also looked for eye phenotypes in the mice. Intraocular pressure (IOP) was directly measured in both eyes within the first 7 min after injecting ketamine and xylazine anesthesia as previously described.18 Data were analyzed via Student's t test. This analysis demonstrated that the Sh3pxd2b null mice had glaucoma, which was not detected in either wild-type or heterozygous littermates (Figure 2). Analysis of H&E-stained fixed sections of eyes revealed substantial variability in the structure of Sh3pxd2b null mouse eyes. Apposition of the peripheral iris to the corneal endothelium and enlargement of the cornea was generally noted (Figure 2). The corneal enlargement was similar to human megalocornea except that in the mice the cornea was largest in older animals, suggesting continued enlargement with increasing age. Aqueous humor drains from the anterior chamber of the mouse eye via trabecular outflow and uveoscleral outflow, which both originate between the iris and cornea adjacent to the ciliary body.19,20 Hence, the peripheral apposition of the iris and cornea in the Sh3pxd2b null eyes likely obstructed outflow and induced elevated intraocular pressure in a manner analogous to closed angle glaucoma.21,22 Furthermore, corneal thickening, increased opacity, and phtisis bulbi with partial exudation of the lens through a central corneal perforation were sometimes noted.
Taken together, many of the abnormalities we observed in the Sh3pxd2b null mice are highly reminiscent of phenotypes characteristic of FTHS patients, particularly the skeletal, cardiac, and eye phenotypes, and confirm a role for the TKS4 protein in the generation of FTHS (Table 2). Two other striking features of the Sh3pxd2b null mice are their decreased size and marked loss of adipose tissue (Figure 2; Figures S3 and S5). Although these features are not currently part of a FTHS diagnosis, we note that many affected individuals are of short stature,2,4,7 and lack of adiposity has been noted in at least one case.7 Very recently, a mutant mouse with skeletal, eye, and adipose abnormalities was shown to express a truncated form of Tks4:23 glaucoma and cardiac defects were not noted, suggesting that complete loss of Tks4 may give rise to different phenotypes than truncation of the protein product.
Our results establish a crucial role for Tks4 in bone, heart, and eye development, as well as a variety of other tissues. To date, the only known function of Tks4 is in podosome formation,10 leading us to speculate that defective podosome formation may contribute to a human developmental disorder. FTHS was originally reported as an atypical form of Melnick-Needles syndrome, which is usually caused by mutation of the FLNA gene (MIM 300017).24 In osteoclasts, filamin A is present in the podosome belt, and its cleavage by calpain is necessary for osteoclast spreading and motility during bone development and homeostasis.25 Given the apparent genetic heterogeneity of FTHS, it is likely that other FTHS cases and related disorders such as megalocornea mental retardation syndrome (MIM 249310) and serpentine fibula-polycystic kidney syndrome (MIM 600330) have defects in genes that might interact with TKS4 and/or in genes involved in podosome formation or function, which may facilitate the identification of causative genes for these conditions.
Acknowledgments
We thank the families for their cooperation in this study; Hulya Kayserili (Istanbul), Helen Stewart (London), Emma Wakeling (London), Valerie Cormier-Daire (Paris), and Charles Scott Jr. (Wilmington) for providing patient material; Ehud Banne for clinical investigations in family 13; Karen X. Duong-Polk for assistance with the experiments on the mouse eyes; Saskia van der Velde-Vissers and Christel Beumer for fibroblast culture; Irene Janssen for carrying out the SNP microarray analysis; and Jeroen Bakkers and Evelyn Kouwenhoven for animal studies and scientific discussion. This research in the Courtneidge laboratory was funded by the National Cancer Institute (CA098383) and the Mathers Foundation, and by Beatriu de Pinos fellowship support (Comissionat per a Universitats I Recerca del Departament d'Innovacio, Universitats I Empresa de la Generalitat de Catalunya) to P.C.-M. P.R.-L. and J.L.M. are supported by the National Institutes of Health, and M.C.S. is a fellow of the California Institute of Regenerative Medicine. Z.I. was supported by a fellowship from the Higher Education Commission (HEC) of Pakistan. The authors declare no conflicting financial interests.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Ensembl, http://www.ensembl.org/
UCSC Genome Browser, http://genome.ucsc.edu/
Polymorphism Phenotyping (Polyphen), http://genetics.bwh.harvard.edu/pph
Simple Modular Architecture Research Tool (SMART), http://smart.embl-heidelberg.de/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
ExPASy Proteomics Server (ExPASy), http://www.expasy.ch/
Pfam Database, http://pfam.sanger.ac.uk/
References
- 1.Frank Y., Ziprkowski M., Romano A., Stein R., Katznelson M.B., Cohen B., Goodman R.M. Megalocornea associated with multiple skeletal anomalies: A new genetic syndrome? J. Genet. Hum. 1973;21:67–72. [PubMed] [Google Scholar]
- 2.ter Haar B., Hamel B., Hendriks J., de Jager J. Melnick-Needles syndrome: Indication for an autosomal recessive form. Am. J. Med. Genet. 1982;13:469–477. doi: 10.1002/ajmg.1320130418. [DOI] [PubMed] [Google Scholar]
- 3.Hamel B.C., Draaisma J.M., Pinckers A.J., Boetes C., Hoppe R.L., Ropers H.H., Brunner H.G. Autosomal recessive Melnick-Needles syndrome or ter Haar syndrome? Report of a patient and reappraisal of an earlier report. Am. J. Med. Genet. 1995;56:312–316. doi: 10.1002/ajmg.1320560320. [DOI] [PubMed] [Google Scholar]
- 4.Maas S.M., Kayserili H., Lam J., Apak M.Y., Hennekam R.C. Further delineation of Frank-ter Haar syndrome. Am. J. Med. Genet. A. 2004;131:127–133. doi: 10.1002/ajmg.a.30244. [DOI] [PubMed] [Google Scholar]
- 5.Mégarbané A., Tomey K., Wakim G. Congenital glaucoma, limb deformities, skeletal dysplasia, and facial anomalies: Report of another family. Am. J. Med. Genet. 1997;73:67–71. doi: 10.1002/(sici)1096-8628(19971128)73:1<67::aid-ajmg13>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 6.Rosser E.M., Mann N.P., Hall C.M., Winter R.M. Serpentine fibula syndrome: Expansion of the phenotype with three affected siblings. Clin. Dysmorphol. 1996;5:105–113. doi: 10.1097/00019605-199604000-00002. [DOI] [PubMed] [Google Scholar]
- 7.Wallerstein R., Scott C.I., Jr., Nicholson L. Extended survival in a new case of ter Haar syndrome: Further delineation of the syndrome. Am. J. Med. Genet. 1997;70:267–272. doi: 10.1002/(sici)1096-8628(19970613)70:3<267::aid-ajmg10>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 8.Dundar M., Saatci C., Tasdemir S., Akcakus M., Caglayan A.O., Ozkul Y. Frank-ter Haar syndrome with unusual clinical features. Eur. J. Med. Genet. 2009;52:247–249. doi: 10.1016/j.ejmg.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 9.Nannya Y., Sanada M., Nakazaki K., Hosoya N., Wang L., Hangaishi A., Kurokawa M., Chiba S., Bailey D.K., Kennedy G.C., Ogawa S. A robust algorithm for copy number detection using high-density oligonucleotide single nucleotide polymorphism genotyping arrays. Cancer Res. 2005;65:6071–6079. doi: 10.1158/0008-5472.CAN-05-0465. [DOI] [PubMed] [Google Scholar]
- 10.Buschman M.D., Bromann P.A., Cejudo-Martin P., Wen F., Pass I., Courtneidge S.A. The novel adaptor protein Tks4 (SH3PXD2B) is required for functional podosome formation. Mol. Biol. Cell. 2009;20:1302–1311. doi: 10.1091/mbc.E08-09-0949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wishart M.J., Taylor G.S., Dixon J.E. Phoxy lipids: Revealing PX domains as phosphoinositide binding modules. Cell. 2001;105:817–820. doi: 10.1016/s0092-8674(01)00414-7. [DOI] [PubMed] [Google Scholar]
- 12.Gimona M., Buccione R., Courtneidge S.A., Linder S. Assembly and biological role of podosomes and invadopodia. Curr. Opin. Cell Biol. 2008;20:235–241. doi: 10.1016/j.ceb.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 13.Gimona M., Buccione R. Adhesions that mediate invasion. Int. J. Biochem. Cell Biol. 2006;38:1875–1892. doi: 10.1016/j.biocel.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 14.Seals D.F., Azucena E.F., Jr., Pass I., Tesfay L., Gordon R., Woodrow M., Resau J.H., Courtneidge S.A. The adaptor protein Tks5/Fish is required for podosome formation and function, and for the protease-driven invasion of cancer cells. Cancer Cell. 2005;7:155–165. doi: 10.1016/j.ccr.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 15.Linder S. Invadosomes at a glance. J. Cell Sci. 2009;122:3009–3013. doi: 10.1242/jcs.032631. [DOI] [PubMed] [Google Scholar]
- 16.Tanaka N., Dalton N., Mao L., Rockman H.A., Peterson K.L., Gottshall K.R., Hunter J.J., Chien K.R., Ross J., Jr. Transthoracic echocardiography in models of cardiac disease in the mouse. Circulation. 1996;94:1109–1117. doi: 10.1161/01.cir.94.5.1109. [DOI] [PubMed] [Google Scholar]
- 17.Hinton R.B., Jr., Alfieri C.M., Witt S.A., Glascock B.J., Khoury P.R., Benson D.W., Yutzey K.E. Mouse heart valve structure and function: Echocardiographic and morphometric analyses from the fetus through the aged adult. Am. J. Physiol. Heart Circ. Physiol. 2008;294:H2480–H2488. doi: 10.1152/ajpheart.91431.2007. [DOI] [PubMed] [Google Scholar]
- 18.Aihara M., Lindsey J.D., Weinreb R.N. Reduction of intraocular pressure in mouse eyes treated with latanoprost. Invest. Ophthalmol. Vis. Sci. 2002;43:146–150. [PubMed] [Google Scholar]
- 19.Smith R.S., Zabaleta A., Savinova O.V., John S.W. The mouse anterior chamber angle and trabecular meshwork develop without cell death. BMC Dev. Biol. 2001;1:3. doi: 10.1186/1471-213X-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lindsey J.D., Weinreb R.N. Identification of the mouse uveoscleral outflow pathway using fluorescent dextran. Invest. Ophthalmol. Vis. Sci. 2002;43:2201–2205. [PubMed] [Google Scholar]
- 21.Tello C., Tran H.V., Liebmann J., Ritch R. Angle closure: Classification, concepts, and the role of ultrasound biomicroscopy in diagnosis and treatment. Semin. Ophthalmol. 2002;17:69–78. doi: 10.1076/soph.17.2.69.14722. [DOI] [PubMed] [Google Scholar]
- 22.Aihara M., Lindsey J.D., Weinreb R.N. Experimental mouse ocular hypertension: Establishment of the model. Invest. Ophthalmol. Vis. Sci. 2003;44:4314–4320. doi: 10.1167/iovs.03-0137. [DOI] [PubMed] [Google Scholar]
- 23.Mao M., Thedens D.R., Chang B., Harris B.S., Zheng Q.Y., Johnson K.R., Donahue L.R., Anderson M.G. The podosomal-adaptor protein SH3PXD2B is essential for normal postnatal development. Mamm. Genome. 2009;20:462–475. doi: 10.1007/s00335-009-9210-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robertson S.P., Twigg S.R., Sutherland-Smith A.J., Biancalana V., Gorlin R.J., Horn D., Kenwrick S.J., Kim C.A., Morava E., Newbury-Ecob R., OPD-spectrum Disorders Clinical Collaborative Group Localized mutations in the gene encoding the cytoskeletal protein filamin A cause diverse malformations in humans. Nat. Genet. 2003;33:487–491. doi: 10.1038/ng1119. [DOI] [PubMed] [Google Scholar]
- 25.Marzia M., Chiusaroli R., Neff L., Kim N.Y., Chishti A.H., Baron R., Horne W.C. Calpain is required for normal osteoclast function and is down-regulated by calcitonin. J. Biol. Chem. 2006;281:9745–9754. doi: 10.1074/jbc.M513516200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.