

Abstract
We identified overlapping homozygous regions within the DFNB25 locus in two Dutch and ten Pakistani families with sensorineural autosomal-recessive nonsyndromic hearing impairment (arNSHI). Only one of the families, W98-053, was not consanguineous, and its sibship pointed toward a reduced critical region of 0.9 Mb. This region contained the GRXCR1 gene, and the orthologous mouse gene was described to be mutated in the pirouette (pi) mutant with resulting hearing loss and circling behavior. Sequence analysis of the GRXCR1 gene in hearing-impaired family members revealed splice-site mutations in two Dutch families and a missense and nonsense mutation, respectively, in two Pakistani families. The splice-site mutations are predicted to cause frameshifts and premature stop codons. In family W98-053, this could be confirmed by cDNA analysis. GRXCR1 is predicted to contain a GRX-like domain. GRX domains are involved in reversible S-glutathionylation of proteins and thereby in the modulation of activity and/or localization of these proteins. The missense mutation is located in this domain, whereas the nonsense and splice-site mutations may result in complete or partial absence of the GRX-like domain or of the complete protein. Hearing loss in patients with GRXCR1 mutations is congenital and is moderate to profound. Progression of the hearing loss was observed in family W98-053. Vestibular dysfunction was observed in some but not all affected individuals. Quantitative analysis of GRXCR1 transcripts in fetal and adult human tissues revealed a preferential expression of the gene in fetal cochlea, which may explain the nonsyndromic nature of the hearing impairment.
Introduction
Autosomal-recessive nonsyndromic hearing impairment (arNSHI [MIM 220700]) is the most common inherited sensory disorder in humans, after color blindness, and exhibits a very high allelic and locus heterogeneity. At present, about 60 loci are known for arNSHI, and they are named as “DFNB” followed by an identification number. For about half of these loci, the causal genes are still unknown. Their identification is hampered by the large size of the critical regions, which are often defined by linkage analysis and/or homozygosity mapping in a small number of consanguineous families or even in a single family. (Hereditary Hearing Loss Homepage).1–3 Critical regions can be significantly delimited by a search for additional consanguineous families with linkage to the same locus. Alternatively, the method of determining regions that are identical by descent (IBD) in affected family members of nonconsanguineous families can be employed. Because the sizes of IBD regions are inversely correlated with the number of generations between the patients and the common ancestors of their parents, analysis of families that are not aware of common ancestry can significantly delimit the critical regions. This is nicely illustrated by the study of Collin et al. and is further supported by the results of Hildebrandt et al.4,5
In the present study, we describe the narrowing of the DFNB25 locus by using homozygosity mapping in consanguineous families of Pakistani and Dutch origin and in a small nonconsanguineous family of Dutch origin. The critical region contains the GRXCR1 gene, and the orthologous gene in mouse was previously reported to carry the causal mutation in the pirouette (pi) mutant, which has hearing loss and circling behavior (Hunker, K.L. et al., 2005, Assoc. Res. Otolaryngol., abstract; see also Odeh et al. in this issue6). GRXCR1 is predicted to have a GRX-like domain; GRX domains are the catalytic domains in glutaredoxins and are involved in the reversible S-glutathionylation of proteins and thereby in the modulation of their activity and/or localization.7 A role in actin organization in hair cells has been suggested because of the localization of GRXCR1 in the hair bundle and stereocilia abnormalities in the pi mutant. (Hunker, K.L. et al., 2006, Assoc. Res. Otolaryngol., abstract).8,9 We demonstrate that mutations in the GRXCR1 gene are responsible for arNSHI that can be accompanied by vestibular dysfunction.
Subjects and Methods
Families
Families W98-053 and W07-0122 are of Dutch origin and have three and one hearing impaired (HI) members, respectively (Figure S1, available online). The parents in the latter family are first cousins. Consanguineous families DEM 4265 and DEM 4349 are from Pakistan and have four and two HI pedigree members, respectively (Figures S2 and S3). For eight additional consanguineous Pakistani families that displayed suggestive linkage to the DFNB25 locus, no potentially causative GRXCR1 mutations were identified: pedigrees DEM 4003A, DEM 4026, DEM 4124A, and DEM 4335; with six, five, four, and four HI family members, respectively, in multiple branches. Pedigrees DEM 4045, DEM 4145, DEM 4171, and DEM 4259 have two, three, two, and two HI family members, respectively, in a single branch.
Family members were examined by otoscopy and pure-tone audiometry in a sound-treated room in accordance with current clinical standards. The patient of family W07-0122 was tested by standardized free-field audiometry. Classification of the hearing loss is in accordance with the GENDEAF guidelines (Hereditary Hearing Loss Homepage). Vestibular function of HI members of families W98-053 and W07-0122 was evaluated by analyzing eye-movement responses to earth-horizontal rotatory stimulation (eyes open) in the dark as described previously.10 This study was approved by the local medical ethics committees in The Netherlands, Pakistan, and the United States. Informed consent was obtained from all participating subjects or, in the case of children, from their parents.
Homozygosity Mapping and Linkage Analysis
Genomic DNA was isolated from peripheral-blood lymphocytes by standard procedures. DNA samples from families W98-053 and W07-0122 were genotyped by use of the Affymetrix mapping 250K SNP array. Total genomic DNA (250 ng) was digested with NspI. Generic primers that recognize the adaptor sequence are used to amplify adaptor-ligated DNA fragments. PCR conditions were optimized to preferentially amplify fragments in the 200–1100 bp size range. The amplified DNA was then fragmented with the Affymetrix fragmentation reagent, in accordance with the manufacturer's protocol, to a size of 50–200 bp and was then labeled and hybridized to a GeneChip Human Mapping 250K array. Genotype calling and calculation of the regions of homozygosity was performed with the Genotyping Console software (Affymetrix), with the default settings used. The families from Pakistan were genotyped with several platforms. Pedigrees DEM 4265 and DEM 4349 were genotyped with the Illumina linkage Panels IVb and 12, respectively. Both of these linkage panels contain ∼6000 SNP loci. Of those pedigrees that displayed (suggestive) linkage to DFNB25 but lacked potentially causative GRXCR1 mutations, DEM 4259 and DEM 4345 were genotyped with the Illumina linkage panels IVb and 12, respectively. For pedigrees DEM 4003A, DEM 4026, DEM 4045, DEM 4124A, DEM 4145, and DEM 4171, a genome scan was carried out with the use of ∼400 fluorescently labeled, short tandem repeat markers covering both autosomes and sex chromosomes at a spacing of ∼10 cM. PEDCHECK was used to identify Mendelian inconsistencies, and the MERLIN program was utilized to detect potential genotyping errors that did not produce a Mendelian inconsistency.11,12 Two-point linkage analysis was carried out with the MLINK program of the FASTLINK computer package, and multipoint linkage analysis was performed with ALLEGRO.13,14 An autosomal-recessive mode of inheritance with complete penetrance and a disease-allele frequency of 0.001 were assumed. Marker-allele frequencies were estimated from the founders, as well as from these founders' reconstructed genotypes both from these pedigrees and from additional pedigrees that underwent a genome scan at the same time. Genetic-map distances were then derived from the Rutgers combined linkage-physical map of the human genome, either directly or by interpolation.15 For identification of regions of homozygosity, haplotypes were constructed via SIMWALK2.16,17
Mutation Analysis
Primers for amplification of exons and exon-intron boundaries of GRXCR1 and exon 1 of ATP8A1 (MIM 609542) were designed with ExonPrimer and the reference sequence NT_006238.10. Primer sequences and PCR conditions are provided in Table S1. Amplification by PCR was performed on 40 ng of genomic DNA with Taq DNA polymerase (Invitrogen). Because the PCR product of exon 1 of ATP8A1 has a GC content of 72%, dimethyl sulfoxide was added to the PCR mixture for a final concentration of 10%. PCR fragments were purified with the use of NucleoFast 96 PCR plates (Clontech) in accordance with the manufacturer's protocol. Sequence analysis was performed with the ABI PRISM Big Dye Terminator Cycle Sequencing V3.1 Ready Reaction Kit and the ABI PRISM 3730 DNA Analyzer (Applied Biosystems). NT_006238.10 was used as a reference sequence for both GRXCR1 and ATP8A1.
For the mutations detected in the Dutch and Pakistani families, 180 and 240 ethnically matched healthy controls were tested, respectively. For determining the presence of the c.628-9C>A transversion, exon 3 was amplified and PCR products were purified as described for sequencing. Digestion of the PCR products with BfaI (New England Biolabs) was performed in accordance with the manufacturer's protocol, and restriction fragments were analyzed on 2% agarose gels. The mutation introduces a restriction site. The c.627+19A>T transversion was tested via an amplification refractory mutation system (ARMS) approach; primer sequences are provided in Table S1. Sequence analysis was used to test for the presence of the additional variants in healthy controls.
Bioinformatic Analysis of Sequence Variants
For prediction of the effect of the c.628-9C>A and c.627+19A>T transversions on splicing efficiency, we used the following software tools: NNSPLICE version 0.9, NetGene2, and GeneSplicer.18–20 For prediction of exonic splice enhancers (ESEs), the ESEfinder was used.21,22 We employed Polymorphism Phenotyping (Polyphen) and Sorting Intolerant from Tolerant (SIFT) to predict the possible pathogenic effect of the p.Arg138Cys substitution. In addition, we applied Grantham's chemical-difference matrix and the Blosum62 and PAM120 matrices.23–25
Experimental Evaluation of Sequence Variants for Effects on Splicing
Total RNA was isolated from Epstein-Barr-virus-transformed lymphoblastoid cells of affected individuals with the NucleoSpin RNA II Kit (Machery Nagel) in accordance with the manufacturer's protocol. Subsequently, cDNA synthesis was performed with 1.5 μg RNA as starting material with the use of the iScript cDNA Synthesis Kit (Bio-Rad Laboratories),in accordance with the manufacturer's protocol. For evaluation of the effect of the c.628-9C>A and c.627+19A>T mutations on splicing, 2 μl of cDNA was amplified via a seminested PCR. For the second PCR, 1 μl of the first PCR was used with an alternative reverse primer in a total number of 35 PCR cycles. Primer sequences are provided in Table S1. PCR products were analyzed via agarose gel electrophoresis, purified, and sequenced as described above.
GRXCR1 Expression Profile
RNA derived from adult heart, prostate, liver, lung, retina, brain, duodenum, kidney, spleen, and testis as well as from fetal liver was purchased from Clontech. RNA derived from fetal brain, colon, kidney, stomach, spleen, and thymus was purchased from Stratagene. In addition, RNA was isolated from fetal cochlea, heart, muscle, and lung as described previously.26 cDNA was synthesized as described above and purified with NucleoSpin Extract II columns (Macherey-Nagel) in accordance with the manufacturer's protocol. For PCR, specific primers (Table S1) were designed with Primer3Plus and reference sequence NM_001080476.1. PCRs were performed with the Applied Biosystem Fast 7900 System in accordance with the manufacturer's protocol. The human beta glucuronidase gene (GUSB [MIM 611499]) was employed as an internal control. PCR mixtures were prepared with the Power Syber Green Master Mix (Applied Biosystems) in accordance with the manufacturer's protocol. Temperatures and reaction times for PCR were as follows: 10 min at 95°C, followed by 40 cycles of 15 s at 95°C and 30 s at 60°C. All reactions were performed in duplicate. Relative gene expression levels were determined with the delta delta Ct method as described previously.27
Results
Homozygosity Mapping Confines the Critical Region of DFNB25
For the identification of genes that cause arNSHI, homozygosity mapping was performed in a large series of 59 familial and 23 isolated arNSHI patients of Dutch origin. In family W98-053, with three affected siblings, the largest overlapping homozygous region, 3.1 Mb, was located on chromosome 2q11.1-11.2. The second-largest overlapping homozygous region, 2.0 Mb, was identified on chromosome 4p13, which was located within the deafness locus DFNB25, presented on the Hereditary Hearing Loss Homepage, and for which the causative gene had not yet been identified. In the single HI individual of W07-0122, multiple large homozygous regions were detected. However, the largest homozygous region (38.6 Mb), located on 4p13-q21.21, partially overlapped with the homozygous region found in family W98-053. Linkage analysis in families of Pakistani origin revealed linkage or an indication of linkage to the chromosomal region associated with DFNB25 in ten families (Table 1). The homozygous region shared by all families contained the first exon of the ATP8A1 gene, the complete GRXCR1 gene, and four additional uncharacterized expressed sequence tags (ESTs) (Figure 1). The haplotypes of the shared homozygous interval differed among the families, suggesting that different mutations are present (Table S2).
Table 1.
GRXCR1 Mutations and Maximum LOD Scores for the DFNB25 Region
| Family ID | No. of HI Individuals | Maximum LOD Score | Sequence Variants | No. of Alleles Observed in Ethnically Matched Chromosomes |
|---|---|---|---|---|
| W98-053a | 3 | - | c.628-9C>A | 0/360 |
| W07-0122a | 1 | - | c.627+19A>T | 0/360 |
| DEM 4003Ab | 6 | 3.42 | - | - |
| DEM 4026b | 5 | 1.29 | - | - |
| DEM 4045b | 2 | 1.68 | - | - |
| DEM 4124Ab | 4 | 2.87 | - | - |
| DEM 4145b | 3 | 1.01 | - | - |
| DEM 4171b | 2 | 1.20 | - | - |
| DEM 4259c | 2 | 1.93 | - | - |
| DEM 4265c | 4 | 3.30 | c.229C>T (p.Gln77X) | 0/480 |
| DEM 4335c | 4 | 1.93 | - | - |
| DEM 4349c | 2 | 1.93 | c.412C>T (p.Arg138Cys) | 0/480 |
These families were studied by homozygosity mapping.
These families were genotyped via a genome scan with ∼400 fluorescently labeled short tandem repeat markers.
These families were genotyped via a linkage panel containing approximately 6000 SNPs.
Figure 1.
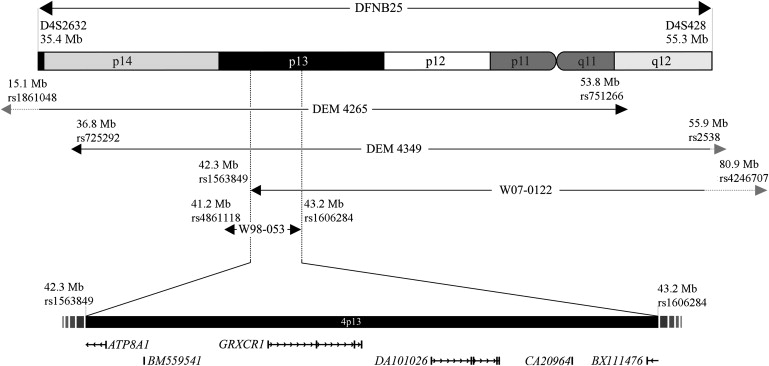
Homozygous Regions and Linkage Intervals within DFNB25
Schematic representation of chromosomal region 4p14-q12 showing the homozygous regions in families W98-053 and W07-0122 and the linkage intervals for families DEM 4265 and DEM 4349. The overlapping region contains the GRXCR1 gene and exon 1 of the ATP8A1 gene. In addition, four ESTs are localized in the region, which are not further characterized. The Mb positions and chromosome bands were determined with the UCSC Genome Browser (NCBI build 36.1; hg18 annotation tracks, March 2006).
Mutations in GRXCR1 Are Causative of DFNB25
Mutation analysis was performed for exon 1 of ATP8A1 and all exons of GRXCR1 including all exon-intron boundaries. No sequence variants were found in the first exon of ATP8A1. However, in both Dutch families and in two of the Pakistani families, potentially pathogenic variants were detected in GRXCR1, including intronic variants in splice sites, a missense mutation, and a nonsense mutation. In family W98-053, a homozygous sequence variant, c.628-9C>A, was present in the splice acceptor site of exon 3, and in family W07-0122, the variant c.627+19A>T was found in the homozygous state in the splice donor site of exon 2 (Figure 2).
Figure 2.
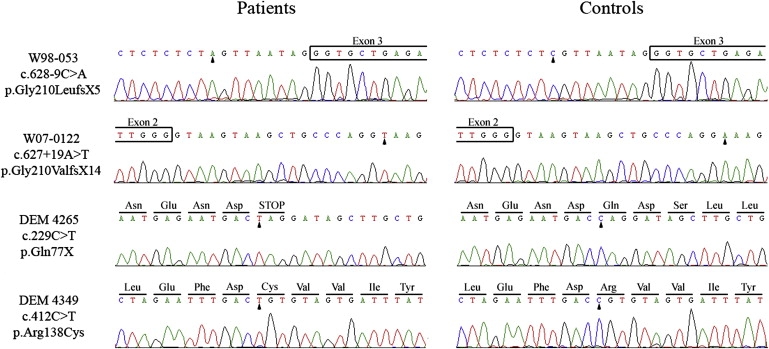
Mutation Analysis of GRXCR1 in arNSHI Patients
Partial sequences are shown of GRXCR1 from affected members of the different families and healthy controls. The predicted amino acid changes and the surrounding amino acids are indicated above the sequence. Mutated nucleotides are marked by an arrowhead. Reference sequences are NT_006238.10 for the nucleotide sequence and NP_001073945.1 for the amino acid sequence.
In family DEM 4265, of Pakistani origin, we identified a homozygous nonsense mutation, c.229C>T, which leads to premature protein truncation after residue 76 (p.Gln77X). Furthermore, in family DEM 4349, a homozygous missense mutation, c.412C>T (p.Arg138Cys), was detected in exon 2. Arg138 is fully conserved in GRXCR1 orthologs (Figure 3B and Figure S4). The p.Arg138Cys substitution introduces a hydrophic residue for a hydrophilic, positively charged residue and was scored as “probably damaging” for protein function with PolyPhen and as “not tolerated” by SIFT. Also, the Blosum62 matrix, the PAM120 matrix, and the Grantham score for this amino acid change indicate that it is potentially damaging for protein function.23–25 Analysis of the c.412C>T alteration with the ESEfinder showed that a SC35 exonic splicing enhancer motif is present in the wild-type sequence but is replaced by a SRp40 recognition motif in the mutated sequence. This may have an effect on splicing efficiency.
Figure 3.
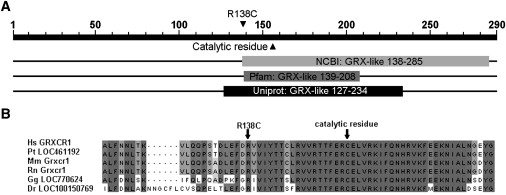
Alignment of the GRXCR1 Proteins of Different Species
(A) Overview of the conserved domains in GRXCR1 as predicted by different software tools. The GRX-like domain is predicted to be present by all three programs. However, the exact positions differ.
(B) An alignment of GRXCR1 orthologs was made with ClustalW and Jalview,39,40 and part of the GRXCR1 protein is shown. Abbreviations are as follows: Hs, Homo sapiens; Pt, Pan troglodytes; Mm, Mus musculus; Rn, Rattus norvegicus; Gg, Gallus gallus; Dr, Danio rerio. Fully conserved amino acids are marked in dark gray, and less-conserved amino acids are in lighter colors. Accession numbers of the various protein sequences are as follows: Hs GRXCR1, NP_001073945.1; Pt LOC461192, XP_517170.2; Mm Grxcr1, NP_001018019.1; Rn Grxcr1, XP_573590.2; Gg LOC770624, XP_001233963.1; Dr LOC100150769, XP_001919567.1.
Three bioinformatic tools, Genesplicer, NetGene2, and NNSPLICE, were used to predict the effect of the mutations on splicing efficiency (Table 2). For the c.628-9C>A change, a novel splice acceptor site is predicted to replace the original one. For the c.627+19A>T variation, a novel splice donor site is predicted in addition to the original site. Usage of these novel splice sites would result in a frameshift and a premature-stop codon; pGly210LeufsX5 and pGly210ValfsX14, respectively. Neither additional splice donor or acceptor sites nor changes in existing sites were predicted to result from the other mutations.
Table 2.
Overview of the Predicted Splicing Effects of the GRXCR1 Mutations
| Family | Mutation | NNsplice | NetGene2 | GeneSplicer | ESEfinder |
|---|---|---|---|---|---|
| W98-053 | c.628-9C>Aa | original splice acceptor site: 0.94 | original splice acceptor site: 0.50 | original acceptor splice site: 8.58 | no changes |
| novel splice acceptor site: 0.98 | novel splice acceptor site: 0.28 | novel splice acceptor site: 5.73 | |||
| W07-0122 | c.627+19A>Tb | original splice donor site: 1.00 | original splice donor site: 1.00 | original splice donor site: 3.08 | no changes |
| novel splice donor site: 1.00 | novel splice donor site: 0.00 | novel splice donor site: 5.26 | |||
| DEM 4265 | c.299C>T | no changes | no changes | no changes | no changes |
| DEM 4349 | c.412C>T | no changes | no changes | no changes | SC35 motif replaced by a SRp40 motif |
With all splice-prediction programs, the original splice acceptor site is replaced by a novel splice acceptor site as a result of the c.628-9C>A mutation.
With all splice-prediction programs, the original splice donor site is predicted with an unchanged score, and a second splice donor site is predicted to be introduced by the c.627+19A>T mutation.
All four presumably pathogenic GRXCR1 mutations cosegregated with hearing impairment in the families (Figure S3) and were not present in ethnically matched controls; i.e., 180 Dutch or 240 Pakistani individuals with normal hearing.
To evaluate the prevalence of GRXCR1 mutations in arNSHI in The Netherlands, we screened this gene for mutations in 79 unrelated familial and isolated patients of Dutch origin. This revealed no putatively pathogenic variants. However, two SNPs were detected that were not present in any of the public SNP databases. The c.25G>A variant (p.Glu9Lys; dbSNP ss182258860) was found heterozygously in 12 Dutch index patients and in 14 of 360 Dutch control alleles. This variant was also observed in a homozygous state in several members of family DEM 4349, including an unaffected parent and unaffected child, and in 19 of 480 Pakistani control alleles. One additional alteration, c.272G>T (p.Gly91Val; dbSNP ss182258861), was found in the heterozygous state in one index patient. Although this alteration was not present in 180 Dutch control individuals, it was present in the heterozygous state and in the homozygous state in 13 of 240 and in 1 of 240 normal-hearing Pakistani controls, respectively.
c.628-9C>A Alters Splicing of GRXCR1
RNA was isolated from lymphoblastoid cells and cDNA was synthesized for affected members of families W98-053 and W07-0122 for assessment of the effect of the putative mutations on splicing. In two siblings of family W98-053 (II.2 and II.3), amplification of GRXCR1 cDNA was successful and sequence analysis demonstrated the presence of seven additional base pairs between exons 2 and 3 leading to a frameshift and a premature stopcodon, p.Gly210LeufsX5 (Figure 4). Amplification of GRXCR1 cDNA for the third affected individual of this family and for the patient of family W07-0122 was unsuccessful in several attempts, which is likely a result of the low expression level of GRXCR1 in lymphoblastoid cells.
Figure 4.
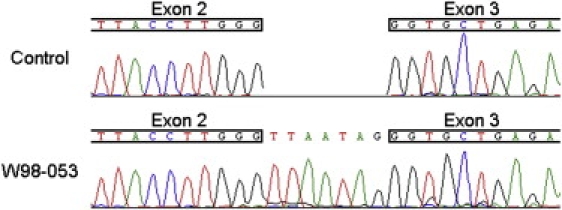
Splicing Effect of the c.628-9C>A Mutation
Partial sequence of GRXCR1 cDNA from an affected member of family W98-053 and a healthy control. Additional nucleotides are present between exons 2 and 3, leading to a frameshift and a premature-stop codon, p.Gly210LeufsX5.
GRXCR1 Is Preferentially Expressed in the Cochlea
The expression of GRXCR1 relative to GUSB was studied via quantitative PCR in human fetal cochlea and compared to that in ten adult and ten fetal-stage human tissues (Figure 5). Because this was performed for adult and fetal tissues in two separate experiments, fetal cochlea was included in both in order to enable comparison of the expression levels. In adult tissues, there was relatively low expression of GRXCR1 in lung, brain, and duodenum, and a relatively moderate expression was seen in testis. The expression in fetal cochlea was 5.8–163 times higher than in the adult tissues that express GRXCR1. In six of the adult tissues—heart, prostate, liver, retina, kidney, and spleen—expression of GRXCR1 was below the detection level. Furthermore, a low relative GRXCR1 expression was observed in fetal heart and fetal spleen, but the overall highest relative expression by far was seen in fetal cochlea. The expression in fetal cochlea is 15.7 times higher than that in fetal heart and is over 300 times higher than that in fetal spleen. In the other fetal tissues, including fetal muscle, liver, lung, brain, colon, kidney, stomach, and thymus, expression was below the detection level.
Figure 5.
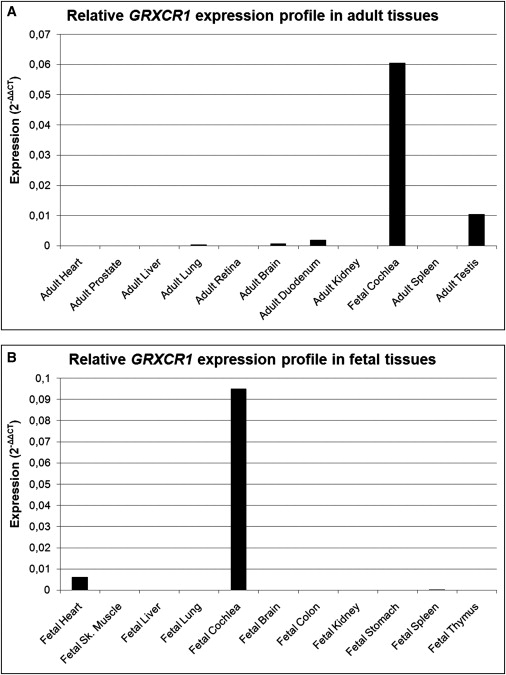
GRXCR1 Expression Profile
Relative GRXCR1 mRNA expression determined by quantitative PCR in adult (A) and fetal (B) tissues. The relative expression values were determined with the delta delta Ct method. There is a highly preferential expression in fetal cochlea.
Characteristics of Hearing Loss and Vestibular Dysfunction Associated with GRXCR1 Mutations
Affected individuals of families W98-053 and W07-0122 exhibited normal development of speech despite the very early-onset or even congenital nature of the disease. The HI child in W07-0122 did not pass neonatal hearing screening, and brainstem-evoked response audiometry (BERA) at the age of 2.5 mo showed a threshold of 40 dB at both sides. For family W98-053, BERA was performed for individuals II.2 and II.4 at the ages of 3.5 mo and 6 mo, respectively, which revealed a threshold of 55–65 dB for II.4 and no responses for II.2. For individual II.1, hearing impairment was diagnosed at the age of 19 mo. In families W98-053 and W07-0122, the hearing loss is bilateral, symmetric, and sensorineural. Otoacoustic emissions were examined only for the affected subject of family W07-0122 and were found to be absent. Representative audiograms of all four HI subjects are presented in Figure 6.
Figure 6.
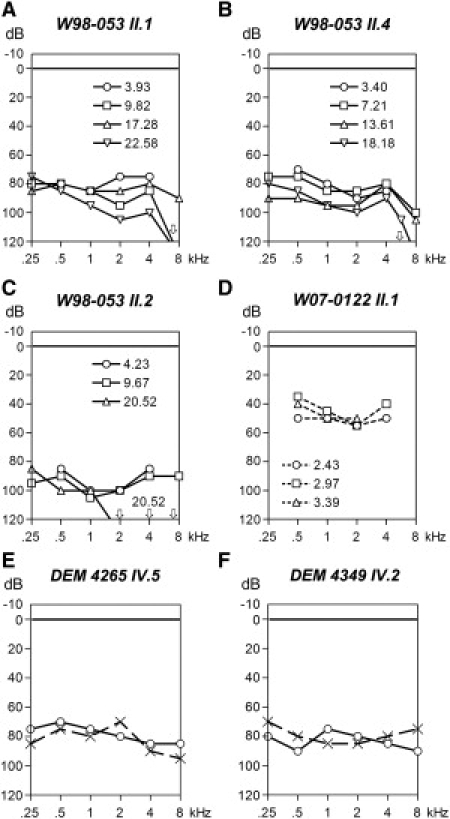
Clinical Characterization of DFNB25 Families
(A–D) Representative audiograms of the right ear of affected individuals of families W98-053 and W07-0122 at different ages. Dashed lines indicate free-field measurements.
(E and F) Representative audiograms of affected individuals of families DEM 4265 and DEM 4349, obtained at the ages of 19 and 26 yrs, respectively. Circles and crosses represent the right and left ear, respectively.
Linear-regression analyses of longitudinal series of pure-tone audiograms showed significant progression of hearing loss in the HI members of family W98-053. The hearing loss progressed from moderate or severe to severe or profound in this family. The annual threshold deterioration was in the range of 0.3–3 dB. The HI child of family W07-0122 is now 4 yrs old and has moderate hearing loss. Future measurements will reveal whether the hearing loss in this child is progressive.
All subjects of family W98-053 had normal motor development. However, symptoms of vestibular dysfunction were noticed for individuals II.1 and II.2 around the ages of 4 and 6 yrs, respectively, and no responses could be registered upon rotatory stimulation. Normal motor development in these patients suggests that the vestibular dysfunction was not congenital. So far, there were no symptoms of vestibular dysfunction in the HI individual of W07-0122, and motor development was normal until now.
The affected individuals of families DEM 4265 and DEM 4349 did not develop speech, which suggests a more-severe hearing loss in the critical period as compared to that in families W98-053 and W07-0122. Representative audiograms for affected subjects of the Pakistani families DEM 4265 and DEM 4349 show a pattern similar to those of HI individuals of family W98-053. The audiograms are flat, slightly U-shaped, or slightly downsloping, and the hearing loss is moderate to severe. There is no evidence for progression of the hearing loss or vestibular dysfunction in families DEM 4265 and DEM 4349.
On the basis of the current data, we can conclude that mutations in GRXCR1 cause congenital or early-onset moderate to severe hearing loss and that the hearing impairment can progress to profound. Also, the hearing loss can be accompanied by vestibular dysfunction with a childhood onset.
Discussion
Here, we describe the DFNB25 locus and present evidence that mutations in GRXCR1 underlie arNSHI associated with this locus, which was indicated previously at the Hereditary Hearing Loss Homepage. In four families, we identified three putatively truncating mutations and a missense mutation. The latter is not associated with a less-severe hearing loss as compared to the truncating mutations. No GRCXR1 mutations were detected in eight families in this study with suggestive linkage to the DFNB25 locus, and the original DFNB25 family did not reveal any GRXCR1 mutations. Also, in the latter family the LOD score did not reach statistical significance (Richard Smith, personal communication). These families may have mutations in regions of the gene that have not been analyzed; the regulatory and intronic regions. Alternatively, the disease in these families may be caused by defects in a second deafness gene in the region or by a gene in a different locus, given that a statistically significant LOD score was reached for only one of these families.
GRXCR1 (glutaredoxin, cysteine rich 1) is a 290-residue protein with a cystein-rich C-terminal region and a predicted GRX domain. The p.R138C substitution is at the N terminus of the GRX domain or just outside, depending on the prediction tool (Figure 3A) and therefore may also affect its function. The nonsense mutation p.Gln77X truncates the protein at the N terminus of the predicted GRX domain. Aberrant splicing due to both splice-site mutations may result in the absence of part of this domain unless nonsense-mediated decay (NMD) occurs. NMD will lead to absence or strongly reduced amounts of a truncated protein, which may well be unstable or mislocalized. NMD eliminates mRNAs that contain premature-translation termination codons.28 In mammalian cells, a termination codon is recognized as premature if it is located more than 50–54 nt 5′ to the final exon-exon junction.29 When one applies this 50–54 nt boundary rule, the nonsense mutation and one of the splice-site mutations, c.628-9C>A, may result in NMD of the GRXCR1 mRNA. Whether NMD indeed occurs for these mutations has not been investigated.
The GRX domain is the catalytic domain in glutaredoxins, enzymes that utilize the reducing power of glutathione to catalyze disulfide reductions in the presence of NADPH and glutathione reductase.6,30 As such, glutaredoxins are involved in the reversible S-glutathionylation of proteins and thereby in the modulation of activity and/or localization of these proteins.6 Actin is one of the well-known examples of S-glutathionylated proteins, and the modification is likely to decrease the rate of polymerization and affect localization.6,31
A role of glutaredoxins, and possibly GRXCR1, in actin organization is interesting in light of the phenotype of the pi mouse mutant. A defective mouse ortholog of GRXCR1, Grxcr1, was reported to underlie the deafness and circling behavior of this pi mouse. (Hunker, K.L. et al., 2005, Assoc. Res. Otolaryngol., abstract; Hunker, K.L. et al., 2006, Assoc. Res. Otolaryngol., abstract; Kohrman, D.C. et al., 2008, Assoc. Res. Otolaryngol., abstract). In the homozygous pi mutant mice, stereocilia arise normally but their maturation is altered, resulting in somewhat disorganized and abnormally short and thin sterecilia of cochlear and vestibular hair cells.8,9 In addition, an actin-rich bundle called cytocaud is present in inner hair cells and type I vestibular hair cells. The cytocaud spans the whole cell, from the cuticular plate to the basal part, and extends further, toward the basement membrane.8 Cytocauds are also described in hair cells of shaker 2 mice with a mutated Myo15a and in the Tasmanian devil (tde) and tg-370 transgenic lines.8,9,32 Both the tde and tg-370 strains have inner-ear dysfunction comparable to that in the pi strain, and they are described to be allelic to pi on the basis of complementation tests. Defects in Grxcr1 have so far not been described for these strains.
The abnormally short and thin stereocilia in combination with the formation of cytocauds due to defects in Grxcr1 indeed indicate a role in the regulation of actin polymerization or organization in hair cells. This is supported by the observations of Hunker et al. (Hunker KL et al., 2006, Assoc. Res. Otolaryngol., abstract) of Grxcr1's localization to stereocilia of hair cells and microvilli of nonsensory cells when expressed in cochlear explants. Association of Grxcr1 with actin-filament-rich structures is also seen upon expression in cultured fibroblasts and other epithelial cells. (Hunker KL et al., 2006, Assoc. Res. Otolaryngol., abstract).
Our results indicate that defects in GRXCR1 can be associated with both progressive and stable hearing loss. So far, progression of arNSHI has been associated only with mutations in a few other genes; namely, MYO3A (MIM 606808), TMPRSS3 (MIM 605511), PJVK (MIM 610219), and LOXHD1(MIM 613072)33–37 and, recently, with a recessive mutation in TMC1 (MIM 606706) (C.C., P.H., H.K., and J.O., unpublished data). In patients with mutations in these genes, the onset of progressive hearing loss is during childhood, whereas in family W98-053, it is congenital or has an onset in early infancy. An association with both progressive and stable hearing impairment has previously been reported for TMPRSS334,35 and is also known for TMC1 (C.C., P.H., H.K., and J.O., unpublished data).38
In conclusion, defects in GRXCR1/Grxcr1 in humans and mice lead to early-onset hearing loss and can be associated with vestibular dysfunction. The latter is so far diagnosed in one of the families. Residual hearing has been described for the pi strain,9 and in families DEM 4265 and DEM 4349, residual hearing is present. This is also the case in family W98-053, although progression in this family may lead to complete deafness.
Supplemental Data
Supplemental Data include four figures and two tables and can be found with this article online at http://www.ajhg.org.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
ESE finder, http://rulai.cshl.edu/cgi-bin/tools/ESE3/esefinder.cgi
ExonPrimer, http://ihg2.helmholtz-muenchen.de/ihg/ExonPrimer.html
Genesplicer, http://www.tigr.org/tdb/GeneSplicer/gene_spl.html
Hereditary Hearing Loss Homepage, http://webh01.ua.ac.be/hhh/
Jalview (a java multiple alignment editor), http://www.jalview.org
NNSPLICE version 0.9, http://www.fruitfly.org/seq_tools/splice.html
NetGene2, http://www.cbs.dtu.dk/services/NetGene2/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim
Polymorphism Phenotyping (PolyPhen), http://genetics.bwh.harvard.edu/pph/
Primer3Plus, http://www.bioinformatics.nl/cgi-bin/primer3plus/primer3plus.cgi
Sorting Intolerant from Tolerant (SIFT), http://sift.jcvi.org/
UCSC Human Genome Database, Build hg18, March 2006, http://www.genome.ucsc.edu
Acknowledgments
We thank Saskia van der Velde-Visser, Christel Beumer, and Suzanne Keijzers-Vloet for excellent technical assistance and A. Beynon and K.P. Krommenhoek for evaluation of vestibular function. This work was financially supported by RNID (grant GR36 to H.K.), Fonds NutsOhra (project SNO-T-0702-102 to H.K.), EUROHEAR (contract no. LSHG-CT-2004-512063 to H.K. and C.C.), The Janivo Stichting, The Heinsius Houbolt Foundation (to H.K.), the Higher Education Commission (HEC) of the Government of Pakistan (to W.A.), and the National Institutes of Health (NIH)-National Institute of Deafness and Other Communication Disorders (grant DC03594 to S.M.L.). Genotyping services were provided by the Center for Inherited Disease Research (CIDR) to S.M.L. CIDR is fully funded through a federal contract from the NIH to Johns Hopkins University, contract no. N01-HG-65403.
References
- 1.Hilgert N., Smith R.J., Van Camp G. Forty-six genes causing nonsyndromic hearing impairment: which ones should be analyzed in DNA diagnostics? Mutat. Res. 2009;681:189–196. doi: 10.1016/j.mrrev.2008.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tranebaerg L. Genetics of congenital hearing impairment: a clinical approach. Int. J. Audiol. 2008;47:535–545. doi: 10.1080/14992020802249259. [DOI] [PubMed] [Google Scholar]
- 3.Morton C.C., Nance W.E. Newborn hearing screening—a silent revolution. N. Engl. J. Med. 2006;354:2151–2164. doi: 10.1056/NEJMra050700. [DOI] [PubMed] [Google Scholar]
- 4.Collin R.W., Littink K.W., Klevering B.J., van den Born L.I., Koenekoop R.K., Zonneveld M.N., Blokland E.A., Strom T.M., Hoyng C.B., den Hollander A.I., Cremers F.P. Identification of a 2 Mb human ortholog of Drosophila eyes shut/spacemaker that is mutated in patients with retinitis pigmentosa. Am. J. Hum. Genet. 2008;83:594–603. doi: 10.1016/j.ajhg.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hildebrandt F., Heeringa S.F., Ruschendorf F., Attanasio M., Nurnberg G., Becker C., Seelow D., Huebner N., Chernin G., Vlangos C.N. A systematic approach to mapping recessive disease genes in individuals from outbred populations. PLoS Genet. 2009;5:1–10. doi: 10.1371/journal.pgen.1000353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Odeh H., Hunker K.L., Belyantseva I.A., Azaiez H., Avenarius M.R., Zheng L., Peters L.M., Gagnon L.H., Hagiwara N., Skynner M.J. Mutations in Grxcr1 are the basis for inner ear dysfunction in the pirouette mouse. Am. J. Hum. Gen. 2010;86:148–160. doi: 10.1016/j.ajhg.2010.01.016. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shelton M.D., Chock P.B., Mieyal J.J. Glutaredoxin: role in reversible protein s-glutathionylation and regulation of redox signal transduction and protein translocation. Antioxid. Redox Signal. 2005;7:348–366. doi: 10.1089/ars.2005.7.348. [DOI] [PubMed] [Google Scholar]
- 8.Beyer L.A., Odeh H., Probst F.J., Lambert E.H., Dolan D.F., Camper S.A., Kohrman D.C., Raphael Y. Hair cells in the inner ear of the pirouette and shaker 2 mutant mice. J. Neurocytol. 2000;29:227–240. doi: 10.1023/a:1026515619443. [DOI] [PubMed] [Google Scholar]
- 9.Odeh H., Hagiwara N., Skynner M., Mitchem K.L., Beyer L.A., Allen N.D., Brilliant M.H., Lebart M.C., Dolan D.F., Raphael Y., Kohrman D.C. Characterization of two transgene insertional mutations at pirouette, a mouse deafness locus. Audiol. Neurootol. 2004;9:303–314. doi: 10.1159/000080701. [DOI] [PubMed] [Google Scholar]
- 10.Kunst H., Marres H., Huygen P., Van Camp G., Joosten F., Cremers C. Autosomal dominant non-syndromal low-frequency sensorineural hearing impairment linked to chromosome 4p16 (DFNA14): statistical analysis of hearing threshold in relation to age and evaluation of vestibulo-ocular functions. Audiology. 1999;38:165–173. doi: 10.3109/00206099909073018. [DOI] [PubMed] [Google Scholar]
- 11.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 12.O'Connell J.R., Weeks D.E. PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am. J. Hum. Genet. 1998;63:259–266. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cottingham R.W., Idury R.M., Schäffer A.A. Faster sequential genetic linkage computations. Am. J. Hum. Genet. 1993;53:252–263. [PMC free article] [PubMed] [Google Scholar]
- 14.Gudbjartsson D.F., Jonasson K., Frigge M.L., Kong A. Allegro, a new computer program for multipoint linkage analysis. Nat. Genet. 2000;25:12–13. doi: 10.1038/75514. [DOI] [PubMed] [Google Scholar]
- 15.Matise T.C., Chen F., Chen W., De La Vega F.M., Hansen M., He C., Hyland F.C., Kennedy G.C., Kong X., Murray S.S. A second-generation combined linkage physical map of the human genome. Genome Res. 2007;17:1783–1786. doi: 10.1101/gr.7156307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sobel E., Lange K. Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker-sharing statistics. Am. J. Hum. Genet. 1996;58:1323–1337. [PMC free article] [PubMed] [Google Scholar]
- 17.Weeks D.E., Sobel E., O'Connell J.R., Lange K. Computer programs for multilocus haplotyping of general pedigrees. Am. J. Hum. Genet. 1995;56:1506–1507. [PMC free article] [PubMed] [Google Scholar]
- 18.Reese M.G., Eeckman F.H., Kulp D., Haussler D. Improved splice site detection in Genie. J. Comput. Biol. 1997;4:311–323. doi: 10.1089/cmb.1997.4.311. [DOI] [PubMed] [Google Scholar]
- 19.Brunak S., Engelbrecht J., Knudsen S. Prediction of human mRNA donor and acceptor sites from the DNA sequence. J. Mol. Biol. 1991;220:49–65. doi: 10.1016/0022-2836(91)90380-o. [DOI] [PubMed] [Google Scholar]
- 20.Hebsgaard S.M., Korning P.G., Tolstrup N., Engelbrecht J., Rouzé P., Brunak S. Splice site prediction in Arabidopsis thaliana pre-mRNA by combining local and global sequence information. Nucleic Acids Res. 1996;24:3439–3452. doi: 10.1093/nar/24.17.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith P.J., Zhang C., Wang J., Chew S.L., Zhang M.Q., Krainer A.R. An increased specificity score matrix for the prediction of SF2/ASF-specific exonic splicing enhancers. Hum. Mol. Genet. 2006;15:2490–2508. doi: 10.1093/hmg/ddl171. [DOI] [PubMed] [Google Scholar]
- 22.Cartegni L., Wang J., Zhu Z., Zhang M.Q., Krainer A.R. ESEfinder: A web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003;31:3568–3571. doi: 10.1093/nar/gkg616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dayhoff, M.O., Schwartz, R.M., Orcutt, B.C. (1978). A model of evolutionary change in proteins. vol. 5 Suppl. 3., 345–352.
- 24.Henikoff S., Henikoff J.G. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grantham R. Amino acid difference formula to help explain protein evolution. Science. 1974;185:862–864. doi: 10.1126/science.185.4154.862. [DOI] [PubMed] [Google Scholar]
- 26.Luijendijk M.W., van de Pol T.J., van Duijnhoven G., den Hollander A.I., ten Caat J., van Limpt V., Brunner H.G., Kremer H., Cremers F.P. Cloning, characterization, and mRNA expression analysis of novel human fetal cochlear cDNAs. Genomics. 2003;82:480–490. doi: 10.1016/s0888-7543(03)00150-2. [DOI] [PubMed] [Google Scholar]
- 27.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 28.Behm-Ansmant I., Kashima I., Rehwinkel J., Sauliere J., Wittkopp N., Izaurralde E. mRNA quality control: An ancient machinery recognizes and degrades mRNAs with nonsense codons. FEBS Lett. 2007;19:2845–2853. doi: 10.1016/j.febslet.2007.05.027. [DOI] [PubMed] [Google Scholar]
- 29.Inácio A., Silva A.L., Pinto J., Ji X., Morgado A., Almeida F., Faustino P., Lavinha J., Liebhaber S.A., Romão L. Nonsense mutations in close proximity to the initiation codon fail to trigger full nonsense-mediated mRNA decay. J. Biol. Chem. 2004;279:32170–32180. doi: 10.1074/jbc.M405024200. [DOI] [PubMed] [Google Scholar]
- 30.Fernandes A.P., Holmgren A. Glutaredoxins: glutathione-dependent redox enzymes with functions far beyond a simple thioredoxin backup system. Antioxid. Redox Signal. 2004;6:63–74. doi: 10.1089/152308604771978354. [DOI] [PubMed] [Google Scholar]
- 31.Wang J., Boja E.S., Tan W., Tekle E., Fales H.M., English S., Mieyal J.J., Chock P.B. Reversible glutathionylation regulates actin polymerization in A431 cells. J. Biol. Chem. 2001;276:47763–47766. doi: 10.1074/jbc.C100415200. [DOI] [PubMed] [Google Scholar]
- 32.Erven A., Skynner M.J., Okumura K., Takebayashi S., Brown S.D., Steel K.P., Allen N.D. A novel stereocilia defect in sensory hair cells of the deaf mouse mutant Tasmanian devil. Eur. J. Neurosci. 2002;16:1433–1441. doi: 10.1046/j.1460-9568.2002.02213.x. [DOI] [PubMed] [Google Scholar]
- 33.Walsh T., Walsh V., Vreugde S., Hertzano R., Shahin H., Haika S., Lee M.K., Kanaan M., King M.C., Avraham K.B. From flies' eyes to our ears: mutations in a human class III myosin cause progressive nonsyndromic hearing loss DFNB30. Proc. Natl. Acad. Sci. USA. 2002;99:7518–7523. doi: 10.1073/pnas.102091699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Elbracht M., Senderek J., Eggermann T., Thürmer C., Park J., Westhofen M., Zerres K. Autosomal recessive postlingual hearing loss (DFNB8): compound heterozygosity for two novel TMPRSS3 mutations in German siblings. J. Med. Genet. 2007;44:e81. doi: 10.1136/jmg.2007.049122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scott H.S., Kudoh J., Wattenhofer M., Shibuya K., Berry A., Chrast R., Guipponi M., Wang J., Kawasaki K., Asakawa S. Insertion of beta-satellite repeats identifies a transmembrane protease causing both congenital and childhood onset autosomal recessive deafness. Nat. Genet. 2001;27:59–63. doi: 10.1038/83768. [DOI] [PubMed] [Google Scholar]
- 36.Schwander M., Sczaniecka A., Grillet N., Bailey J.S., Avenarius M., Najmabadi H., Steffy B.M., Federe G.C., Lagler E.A., Banan R. A forward genetics screen in mice identifies recessive deafness traits and reveals that pejvakin is essential for outer hair cell function. J. Neurosci. 2007;27:2163–2175. doi: 10.1523/JNEUROSCI.4975-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grillet N., Schwander M., Hildebrand M.S., Sczaniecka A., Kolatkar A., Velasco J., Webster J.A., Kahrizi K., Najmabadi H., Kimberling W.J. Mutations in LOXHD1, an evolutionarily conserved stereociliary protein, disrupt hair cell function in mice and cause progressive hearing loss in humans. Am. J. Hum. Genet. 2009;85:328–337. doi: 10.1016/j.ajhg.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kurima K., Peters L.M., Yang Y., Riazuddin S., Ahmed Z.M., Naz S., Arnaud D., Drury S., Mo J., Makishima T. Dominant and recessive deafness caused by mutations of a novel gene, TMC1, required for cochlear hair-cell function. Nat. Genet. 2002;30:277–284. doi: 10.1038/ng842. [DOI] [PubMed] [Google Scholar]
- 39.Waterhouse A.M., Procter J.B., Martin D.M., Clamp M., Barton G.J. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Larkin M.A., Blackshields G., Brown N.P., Chenna R., McGettigan P.A., McWilliam H., Valentin F., Wallace I.M., Wilm A., Lopez R. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.