

Abstract
Osteochondritis dissecans is a disorder in which fragments of articular cartilage and subchondral bone dislodge from the joint surface. We analyzed a five-generation family in which affected members had autosomal-dominant familial osteochondritis dissecans. A genome-wide linkage analysis identified aggrecan (ACAN) as a prime candidate gene for the disorder. Sequence analysis of ACAN revealed heterozygosity for a missense mutation (c.6907G > A) in affected individuals, resulting in a p.V2303M amino acid substitution in the aggrecan G3 domain C-type lectin, which mediates interactions with other proteins in the cartilage extracellular matrix. Binding studies with recombinant mutated and wild-type G3 proteins showed loss of fibulin-1, fibulin-2, and tenascin-R interactions for the V2303M protein. Mass spectrometric analyses of aggrecan purified from patient cartilage verified that V2303M aggrecan is produced and present in the tissue. Our results provide a molecular mechanism for the etiology of familial osteochondritis dissecans and show the importance of the aggrecan C-type lectin interactions for cartilage function in vivo.
Introduction
The skeletal disorder osteochondritis dissecans is defined as a separation of cartilage and subchondral bone from the surrounding tissue and primarily affects the knee, ankle, and elbow joints.1 Most cases of osteochondritis dissecans are sporadic, with a prevalence of knee osteochondritis of 15–29 per 100,000,2 and its etiology remains unclear. Familial osteochondritis dissecans (MIM 165800) is a rare disorder involving disturbed chondroskeletal development, disproportionate growth, and deformation of the skeleton.3–7 The familial osteochondritis dissecans phenotype presents as multiple osteochondritic lesions in knees and/or hips and/or elbows (Figures 1A–1D), disproportionate short stature, and early osteoarthritis (OA).4
Figure 1.
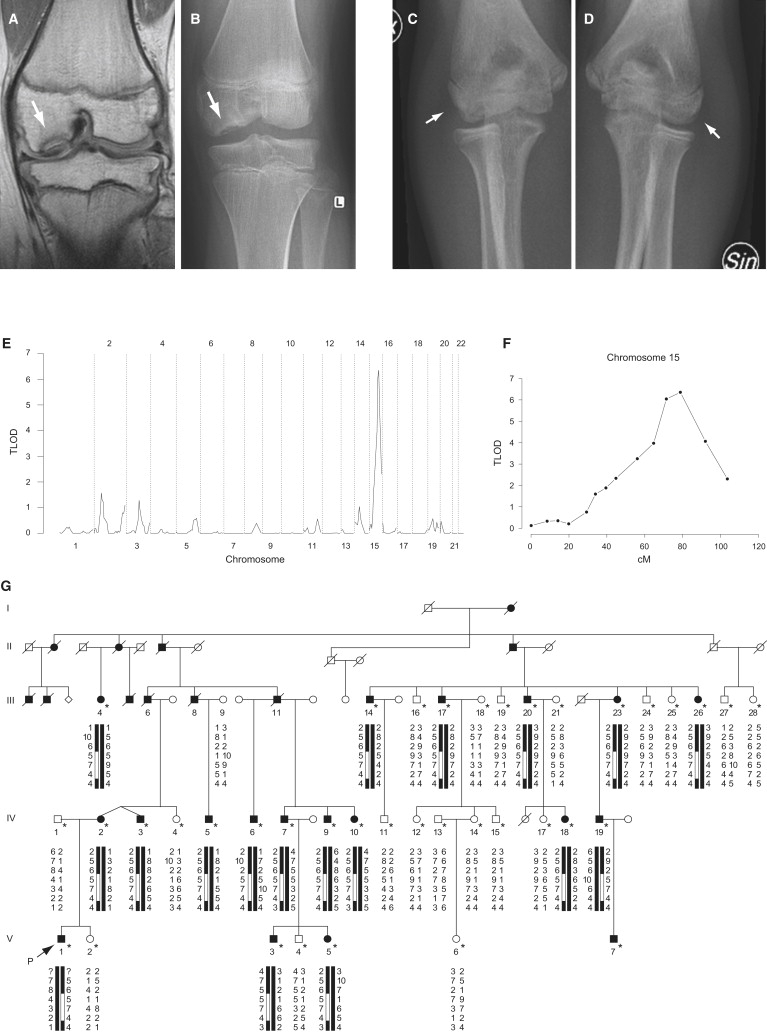
Radiographic Phenotype of Familial Osteochondritis Dissecans and Gene Linkage with Chromosome 15q
(A–D) MRI (A) and radiographs (B–D) from a 14-year-old male patient with familial osteochondritis dissecans. The image shows osteochondritic lesions (arrows) at the lateral aspect of the medial femoral chondyle in the knee (A and B) and at the humeral capitellum in the elbow (C and D).
(E) Genome-wide scan showing linkage to chromosome 15q in a family displaying autosomal-dominant familial osteochondritis dissecans over five generations.
(F) Multipoint analysis, with the polymorphic markers D15S128, D15S1002, D15S165, D15S1007, D15S1012, D15S994, D15S978, D15S117, D15S153, D15S131, D15S205, D15S127, D15S130, and D15S120, resulting in a maximal lod score of 6.36 at marker locus D15S127.
(G) Haplotype showing polymorphic marker alleles from chromosome 15q in familial osteochondritis dissecans. The markers shown are (from top to bottom): D15S117, D15S153, D15S131, D15S205, D15S127, D15S130, and D15S120. The disease haplotype is highlighted by the open boxes within the haplotypes below the symbols. Filled symbols represent affected individuals, the arrow indicates the proband, and asterisks denote individuals examined (by E.-L. S.). In addition, the diagnoses of individuals 6, 8, and 11 in generation III have been verified through archived medical journals and radiographs.
Aggrecan is a major cartilage component and confers resilience to the tissue. The extreme fixed charge density of the proteoglycan molecule creates a high osmotic activity and swelling pressure retained by the cartilage collagen network, making joint cartilage ideal for resisting compressive load with minimal deformation.8 This large proteoglycan consists of globular domains flanking an elongated domain substituted with over 100 chondroitin sulfate and additional keratan sulfate glycosaminoglycan chains and oligosaccharides (Figure 2A).9 The globular domains mediate interactions with other components of the extracellular matrix (ECM); the N-terminal G1 domain binds to hyaluronan,10 and the C-terminal G3 domain binds tenascins and fibulins through its C-type lectin repeat (CLD).11–15 Data from a mutation in the chick aggrecan gene causing nanomelia suggests that the CLD repeat may also be involved in the secretion of aggrecan from chondrocytes.16
Figure 2.
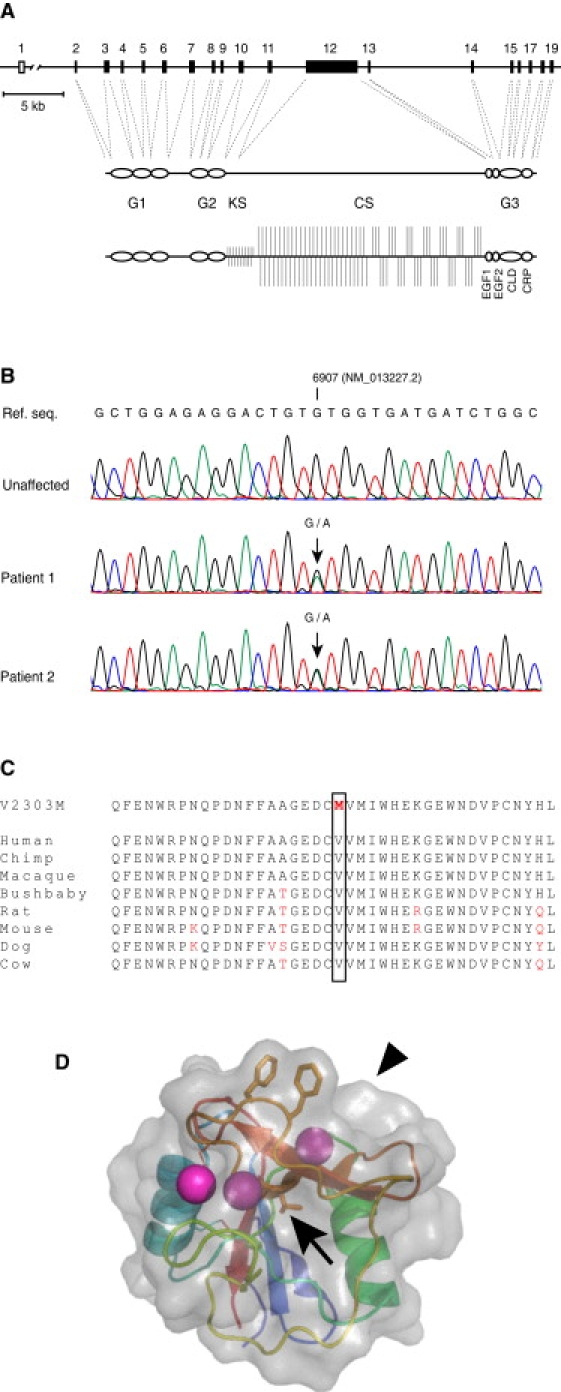
A Missense Mutation in the Aggrecan C-type Lectin Repeat Is Present in Patients with Familial Osteochondritis Dissecans
(A) Organization of the ACAN gene (top), domain structure of the aggrecan core protein (middle), and proteoglycan (bottom).
(B) Sequence analysis from individuals affected by familial osteochondritis dissecans (middle and bottom traces), but not from an unaffected family member (top trace), show heterozygosity for a G-to-A transition in the ACAN gene.
(C) The c.6907G > A mutation results in the replacement of the highly conserved Val2303 residue in the aggrecan C-type lectin (CLD) repeat with a Met residue.
(D) The Val2303 residue (arrow) is located in a beta strand underlying the CLD ligand-binding site, and its side chain is buried in the tightly packed hydrophobic core of the CLD repeat. The location of the CLD ligand-binding surface is indicated by an arrowhead.
In the present study, we performed genome-wide linkage analyses on a five-generation family with hereditary osteochondritis dissecans. We identified heterozygosity for a missense mutation, p.V2303M in the aggrecan CLD. Biochemical analysis using recombinant proteins showed that the mutation led to loss of interaction with known ECM protein ligands, and mass spectrometry analysis of patient cartilage extract verified that the mutated aggrecan proteoglycan was produced.
Methods
Clinical Information
A five-generation family from northern Sweden with autosomal-dominantly inherited osteochondritis dissecans was studied.4 In total, the study comprised 53 family members, including 15 affected individuals. Investigations included medical history, physical examination, MRI or radiography of knees, hips and hands in some individuals, and blood collection for DNA extraction. The case definition was osteochondritis dissecans in at least one joint. Fifteen family members were previously described in detail.4 In this study, four additional family members were included. Two of these (pedigree numbers V:5 and V:7) developed osteochondritis dissecans in the knees at the age of 13 years, whereas the remaining two family members (pedigree numbers IV:5 and IV:18) showed a milder phenotype but were found to carry the familial haplotype and mutation. The study was conducted after approval by the Research Ethics Committee of Umeå University Hospital, Sweden, and informed consent was obtained from each participating family member or that person's legal guardian.
Genome-wide Scan
Genomic DNA from 53 participants including healthy parents and siblings was prepared from white blood cells. In brief, buffy-coat blood cells were lysed, and nuclei were isolated through centrifugation. After denaturation, proteinase K digestion, and centrifugation, the DNA was precipitated from the supernatant with alcohol, washed, air-dried, and dissolved in TE buffer. Samples from 38 family members were analyzed with 400 microsatellite markers spanning all autosomes with an average spacing of 10 cM (Linkage Mapping Set 10 cM version 2.5, Applied Biosystems). Multiplex PCR reactions were performed according to the manufacturer's instructions. The PCR products were separated on 36 cm capillary arrays by the use of POP-4 polymer in an ABI PRISM 3170 DNA sequencer. Genotypes were analyzed manually, and linkage was calculated with MCLINK (Myriad Genetics),17 which utilizes the robust multipoint linkage statistic TLOD proposed by Goring and Terwilliger.18 An autosomal-dominant inheritance model was used in the analysis, and the disease allele frequency was set to 0.0001. Affected cases were assumed to be carriers of the disease allele, and the phenocopy rate was assumed to be 15%. All unaffected individuals were given an unknown phenotype. Marker allele frequencies were estimated empirically from all 38 individuals genotyped.
ACAN Gene Sequencing
The ACAN gene [MIM 155760] is located on chromosome 15q26 and consists of 19 exons ranging in size from 77–4224 bp.19 The 19 exons and intron-exon boundaries of the ACAN gene were amplified by PCR (see Table 1 for primers used). Samples from two affected and two unaffected family members were analyzed by bidirectional sequencing with an ABI PRISM 3170 DNA sequencer (Applied Biosystems).
Table 1.
Primers Used for ACAN Gene Sequencing
| NM_001135.2 Exon | NM_013227.2 Exon | Amplicon | Primera | Primer sequence (5′-3′) | TM | Amplicon Length |
|---|---|---|---|---|---|---|
| 1 UTR | 1 UTR | W1a | - | |||
| 1 UTR | 1 UTR | W1b | W1b F | GGAGTCCGCAACCCAGCAG | 58°C | 248 bp |
| W1b R | TGCAGTCTCAGTCCCGTTCC | |||||
| 2 | 2 | 2 | 2F | ATCTCACAAACCACGTGCAG | 58°C | 403 bp |
| 2R | GGGTGCTATCCCATCACAGT | |||||
| 3 | 3 | 3 | 3F | GAGTGCATTGCTGGAAGGAT | 58° | 536 bp |
| 3R | CGGAGACTGCTCTGGTTCTT | |||||
| 4 | 4 | 4 | 4F | TCGGTGATCAGAGACTGTGC | 58°C | 417 bp |
| 4R | GTTTGGGATGGGTCATTGAG | |||||
| 5 | 5 | 5 | 5F | GAGGAGGATTCAAAGGCAGA | 58°C | 405 bp |
| 5R | TGAGACCCTCTCCAAAGTGG | |||||
| 6 | 6 | 6 | W6F | AGGACTCCCAAGACCTCG | 55°C | 443 bp |
| W6R | CCCATTTTAGTGGCATGAAC | |||||
| 7 | 7 | 7 | 7F | GCTGAGTACCCCTGAAATGG | 55°C | 801 bp |
| 7R | TGTGCCTGCTTGATACAGAGA | |||||
| 8 | 8 | 8 | 8F | GGCTACCAAGTGGGATTTCA | 55°C | 525 bp |
| 8R | TCTGATGGGGAGAAATGACC | |||||
| 9 | 9 | 9 | 9F | CAGGAGCCACCAGATCCTT | 58°C | 520 bp |
| 9R | TTCAGTAGGAGAGCAGGCACTA | |||||
| 10 | 10 | 10 | 10F | TGGTGAGGAGGGTTAGAGGA | 55°C | 578 bp |
| 10R | AGAAGGATGACCTTGCCTTG | |||||
| 11 | 11 | 11 | 11F | GGTGCCGATGGCTCTTACTA | 58°C | 518 bp |
| 11R | CTGGGGAAACAGCACAAATC | |||||
| 12b | 12 | W12a | W12a F | AGTGGGGCAGAGTTAATAAAC | 55°C | 256 bp |
| W12a R | GGCTCCTCTGAGGGGAAC | |||||
| W12b | W12b F | ATTCCCCTCAGTGGAGCTGT | 65°C | 375 bp | ||
| W12b R | TCACCAACCGTAGGAGTGC | |||||
| 13b | 13 | W12d | W12d F | GATTACATTTGTGGACACCAG | 55°C | 382 bp |
| W12d R | TCCTGCCTCTTGGGCTG | |||||
| W12e | W12e F | TTTGGTGGAATCTGTAACCCAG | 58°C | 403 bp | ||
| W12e R | GGAGACTTTTCCACTGGAC | |||||
| W12f | W12f F | CCCTGTGACACACACACC | 58°C | 427 bp | ||
| W12f R | TCCGCTGATTTCAGTCCTG | |||||
| W12 g | W12 g F | CTCCCACGGCTTCTGGAG | 58°C | 309 bp | ||
| W12 g R | CTACCTAAGCACATTTAAAGGG | |||||
| 14 | W13 | W13 F | GAGGTCTTGGAGGCCATGGTA | 65°C | 262 bp | |
| W13 R | ACCTGGGGACCCTTTGGGGGA | |||||
| EGF2c | EGF2 | 14 | 14F | GCAGCAACAGTTCTCAGG | 58°C | 261bp |
| 14R | GCAGCAGTTAGTAACATTAGC | |||||
| 14 | 15 | 15 | 15F | GAGTGGAAGCATGTGCAGAG | 58°C | 433 bp |
| 15R | CTGCCTTCTGAAAGGTGAGG | |||||
| 15 | 16 | 16 | 16F | GAGAGGTGGCCCGAAGTG | 55°C | 412 bp |
| 16R | CCCTGCCCTCAAGGGTTC | |||||
| 16 | 17 | 17 | 17F | ATTCCCAGCAGTTTTTGTGC | 55°C | 340 bp |
| 17R | GATCCTATCCTCCCCTGACC | |||||
| 18 | W18 | 18F | CCTCGGGTCACAACCAGC | 55°C | 318 bp | |
| 18R | GGAAGGCTGTGCTGGCAG | |||||
| 17 | 19 | 19 | 19F | CCAAATCAGGAAAGCCGATA | 55°C | 542 bp |
| 19R | CTTAACCCCTCCCCTTCTCC |
The sequences for primers labeled “W” were kindly provided by Gillian Wallis (University of Manchester, UK).
Because of VNTR's, only parts of exon 12 and 13 were sequenced.
Primers for EGF2 modified from reference number 19.
PCR-dHPLC Analysis
To evaluate the presence of the c.6907G > A mutation in 115 Swedish army recruits, we used dHPLC20 (Transgenomic Wave 3500HT system). We realized optimal heteroduplex formations by denaturing PCR products at 95°C for 5 min and gradually cooling them to 24°C over a period of 60 min. Analysis of a 340 bp amplicon across the missense mutation was performed on a wild-type sample, a positive control, and 230 Swedish control chromosomes. Five microliters of PCR products of the heteroduplex/homoduplex mixture was loaded on a DNASep HT Cartridge (Transgenomics, Omaha, USA) with an automatic sample injector Model HI/L 7100. PCR products were eluted from the column with an acetonitrile gradient in 0.1 M triethylamine acetate buffer (pH 7) at a constant flow rate of 1.5 ml/min. Wavemaker 4.1 software (Transgenomic, Omaha, NE) was used for calculating the acetonitrile concentration during the gradient and the optimal partially denaturing temperature (Tm) for the amplicon, as well as for evaluating the results. The Tm of this amplicon was determined as 62.5°C.
Recombinant Proteins
The production and purification of recombinant domain II of fibulin-121 and fibulin-2,22 full-length tenascin-R,23 fibronectin type III repeats 3–5 of tenascin-R,24 alkaline phosphatase-tagged aggrecan C-type lectin,12 and the wild-type aggrecan G3 splice variants Lt, LCt, and E1E2LCt23 have been described. The corresponding aggrecan G3 variants carrying the familial osteochondritis dissecans V2303M mutation were produced by site-directed mutagenesis of the expression plasmids (QuikChange, Stratagene) with the primers 5′-CTCGTGCCAGATCATGACCATACAGTCCTCTCCAGC-3′ and 5′-GCTGGAGAGGACTGTATGGTCATGATCTGGCACGAG-3′, transfection into 293 c18 cells (ATCC CRL-10852), selection with hygromycin, and purification of the recombinant proteins from the cell culture medium via nickel chelation affinity chromatography followed by MonoQ ion exchange chromatography, as described for the wild-type constructs.23 For use in BIAcore experiments, proteins were further purified by gel filtration on a Superdex 75 PC3.2/30 column in a SMART chromatography system (Pharmacia) using BIAcore running buffer (10 mM HEPES-HCl, 150 mM NaCl, 2 mM CaCl2, and 0.005% P20 [pH 7.5]) as eluent. To verify the Val-to-Met mutation, we digested the resulting recombinant proteins with trypsin and analyzed peptide masses by MALDI-TOF mass spectrometry as described.25
Analysis of Protein Interactions
Solid-Phase Competition Binding Experiments
Microtiter wells (NUNC Maxisorp, Nalge, Denmark) were coated with 2.5 μg/ml tenascin-R in 50 mM sodium carbonate buffer (pH 9.5) at 4°C overnight. The wells were washed with TTBS-Ca (5 mM CaCl2, 150 mM NaCl, 0.05% Tween 20, and 50 mM Tris-HCl [pH 7.4]), incubated with blocking solution (3% BSA in TTBS) for 1 hr at room temperature, and washed with BSA/TTBS-Ca (0.3% BSA in TTBS-Ca). Wild-type or V2303M aggrecan G3 domain variant E1E2LCt protein was loaded into the wells at final concentrations of 0.3–1000 nM, together with 1 μg/ml (14 nM) alkaline-phosphatase-tagged rat aggrecan lectin domain, and incubated for 1 hr. After extensive washing with TTBS-Ca, 1 mg/ml p-nitrophenyl phosphate (in 5 mM MgCl2, 100 mM NaCl, and 100 mM Tris-HCl [pH 9.5]) was added, and the absorbance was measured at 405 nm after a 3 hr incubation. Sigmoidal dose-response curves were fitted to the data by nonlinear regression, and IC50 values were determined with Prism 3.02 for Windows (GraphPad software).
Surface Plasmon Resonance Measurements
Three different extracellular matrix ligands of the aggrecan G3 domain, i.e., tenascin-R, fibulin-1, and fibulin-2, were diluted with 10 mM sodium acetate (pH 4.0) and immobilized in different flow cells of CM5 sensor chips (BIAcore). Immobilization levels were between 400 and 750 resonance units. As a control for changes in the sample refraction index, a blank flow cell was subjected to the immobilization procedure without added protein. For affinity measurements, binding and dissociation were monitored in a BIAcore 2000 instrument (BIAcore). The purified wild-type or V2303M aggrecan G3 variants were injected in running buffer (10 mM HEPES-HCl, 150 mM NaCl, 2 mM CaCl2, and 0.005% P20 [pH 7.5] at 25°C) into the flow cells at concentrations ranging between 1.5 nM and 50 nM, and duplicate measurements were made at each concentration. The injections were at a flow rate of 50 μl/min, which we previously have shown prevents mass transfer limitations for these interactions.23 The ligand surfaces were regenerated by injection of a 300 μl pulse of 20 mM EDTA and 1.5 M NaCl in running buffer between each experiment. For confirmation of the results, control experiments were performed in the reverse orientation with wild-type or V2303M aggrecan G3 variants Lt, LCt, and E1E2LCt immobilized on a CM5 sensor chip (250–1700 RU immobilized). Fibulin-1A, fibulin-2, and a recombinant fragment comprising FnIII repeats 3–5 of tenascin-R were injected (in duplicate) at 50 μl/min over the surfaces at concentrations ranging from 1.5–50 nM. Surfaces were regenerated by four consecutive 300 μl injections of 20 mM EDTA and 1.5 M NaCl in running buffer. After X and Y normalization and subtraction of blank curves, the association (kass) and dissociation (kdiss) rate constants were determined simultaneously via Marquardt-Levenberg global curve fits to the equation for 1:1 Langmuir binding in the BIAevaluation 4.1 software (BIAcore). The equilibrium dissociation constant (KD) of each binding reaction was calculated from these values.
Extraction and Purification of Aggrecan from Cartilage
Articular cartilage was obtained from the tibial plateau and femoral condyles of a 44-year-old female patient who had familial osteochondritis dissecans and was undergoing arthroplasty because of severe OA. Cartilage was pulverized in liquid N2, and aggrecan was extracted with GuHCl and purified by dissociative CsCl gradient centrifugation as described.26
Mass Spectrometry
The aggrecan-containing D1 fraction was dialyzed into 25 mM ammonium bicarbonate buffer (pH 7.8) and digested with endoproteinase GluC (at 24°C overnight), and the resulting peptides were separated on a μRPC C2/C18 reverse-phase HPLC column in a SMART chromatography system (Pharmacia) with a gradient of 0%–100% acetonitrile in 0.1% TFA. The elution times of the peptides of interest (DCVVMIWHE and DCMVMIWHE) were determined with GluC-digested recombinant wild-type and V2303M mutant aggrecan G3 proteins. The fraction of the GluC-digested patient aggrecan containing the peptides of interest was analyzed by MALDI-TOF MS and ion trap MSMS. For MALDI-TOF MS, the sample was mixed with 2,5-dihydroxybenzoic acid (DHB) matrix, spotted onto an anchorchip target plate for MALDI-TOF MS (Bruker Daltonics, Bremen, Germany), and analyzed with a Bruker Reflex III apparatus. For corroboration of the mass peaks, the patient sample was spiked with wild-type and mutant peptides from recombinant aggrecan. For MSMS analysis, the fraction of interest was injected into an HCT ion trap mass spectrometer controlled by HyStar software, and spectra were processed with Data Analysis software (Bruker Daltonics). Selected Tandem MS spectra were checked with BioTools (Bruker). The mass spectrometer was programmed to perform tandem MS experiments only on peptide ions relating to aggrecan V2303M mutation. The following peptide ions were included on the basis of observed masses in MALDI MS data; mutant DCMVMIWHE, m/z = 610.75 Da (+2), wild-type DCVVMIWHE, m/z = 594.76 Da (+2), mutant NWRPNQPDNFFAAGEDCMVMIWHE, m/z = 988.76 Da (+3), and wild-type NWRPNQPDNFFAAGEDCVVMIWHE, m/z = 978.10 Da (+3).
Results
To identify the genetic defect causing familial osteochondritis dissecans, we performed a genome-wide multipoint linkage analysis on a Swedish family displaying the disease over five generations. The clinical details of the family have been described.4 We obtained evidence for linkage to the disease with chromosome 15q26 markers (Figure 1E) showing a peak lod score at D15S127 (TLOD 6.36, theta 0.00; Figure 1F). Refined mapping revealed that all affected individuals shared a 10.5 Mb haplotype restricted by the polymorphic markers D15S205 and D15S130 (Figure 1G). The candidate region spans approximately 100 genes (NCBI Map Viewer), including the ACAN gene, which encodes aggrecan.
Because of the fundamental importance of aggrecan for skeletal development and cartilage function, we considered ACAN to be a prime candidate gene for familial osteochondritis dissecans. We sequenced the complete coding region of the ACAN gene, including the alternatively spliced EGF2 repeat19 (absent from the two aggrecan isoforms in GenBank: NM_001135 and NM_013227) but not including parts of the VNTR-region of exon 12 and exon 13, in two affected and two unaffected family members. This revealed a G-to-A transition in exon 17 in the affected individuals (c.6907G > A in aggrecan isoform 2, GenBank NM_013227.2), predicting a Val-to-Met amino acid substitution of the coding sequence (p.V2303M) (Figure 2B). Exon 17 was sequenced in 49 additional family members, and the V2303M mutation was identified in all 19 affected individuals. The mutation was absent in all unaffected family members. Denaturing high-performance liquid chromatography (dHPLC, not shown) showed that the transition was also excluded on 230 control chromosomes derived from unrelated healthy Swedes.
The Val residue that is mutated in familial osteochondritis dissecans is located in the C-type lectin domain (CLD) of the aggrecan G3 domain, a protein region that is highly conserved between different species and that is involved in organizing the aggrecan network through interactions with other cartilage ECM molecules.11–15,23,27 The mutated Val residue is strictly conserved between species (Figure 2C) and is located in one of the beta strands at the known ligand-interaction site of the aggrecan CLD (labeled Val100 in Lundell et al.28); its side chain is buried within the tightly packed hydrophobic core of the protein fold (Figure 2D).28 Molecular structure modeling suggested that the V2303M substitution leads to a conformational change of the aggrecan CLD (not shown).
To determine whether the V2303M substitution has any effects on aggrecan G3 domain secretion and/or interactions, we produced wild-type and mutant human aggrecan G3 domains as recombinant proteins. Because the EGF and CRP repeats of the G3 domain are subject to alternative splicing,23 we produced three different splice variants of the G3 domain with and without the V2303M substitution (Figure 3A). All three mutated G3 variants were expressed and secreted by the human embryonic kidney 293 cell line, although the protein yields of the shorter mutant recombinant proteins appeared to be lower than those of the corresponding wild-type proteins (Figure 3B). The aggrecan CLD is known to interact with ECM proteins in the tenascin,13,14,23 fibrillin,27 and fibulin12,15 families. Interestingly, initial His-tag affinity precipitation of the recombinant proteins from conditioned medium via NiNTA beads resulted in copurification of the CLD ligand fibulin-1 with all three wild-type G3 variants but not with any of the V2303M mutated G3 domains (Figure 3B). The identity of fibulin-1 as well as that of the recombinant proteins was verified by MALDI-TOF mass spectrometry (not shown). The recombinant proteins were then purified to apparent homogeneity by His-tag metal-chelation-affinity, ion-exchange, and gel-filtration chromatography. Using a solid-phase inhibition assay, we found that the affinity for tenascin-R was decreased in the mutated G3 domain compared to the wild-type (Figure 3C). Analyses of interactions by surface plasmon resonance using a BIAcore verified that tenascin-R, fibulin-1, and fibulin-2 binding was affected for all V2303M mutated G3 splice variants (Figure 3D), which all showed weakened affinities or complete loss of interaction (Table 2). Taken together, this clearly demonstrates that the familial osteochondritis dissecans-associated V2303M substitution affects normal aggrecan G3 domain function.
Figure 3.
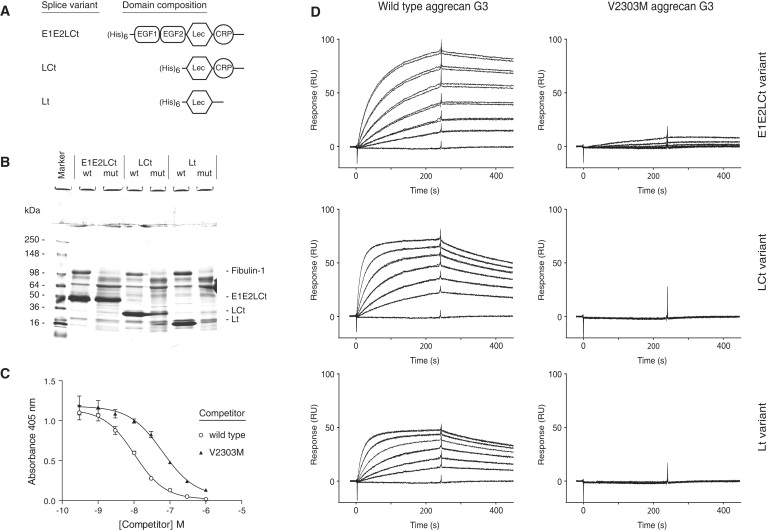
The V2303M Mutation Affects Aggrecan CLD Interactions
(A) Domain organization of three different splice variants of the wild-type aggrecan G3 domain, and the corresponding V2303M variants, produced as recombinant proteins in human embryonic kidney 293 cells.
(B) Ni2+-nitriloacetic acid (NiNTA)-agarose precipitation of His-tagged recombinant proteins from conditioned medium containing fetal calf serum. Bovine fibulin-1, a known aggrecan CLD ligand present in serum, copurified with the wild-type recombinant G3 variants, but not with the V2303M proteins. The identity of fibulin-1 and the recombinant proteins was verified by MALDI-TOF mass spectrometry (not shown).
(C) Solid-phase competition binding assay. Microtiter plate wells were coated with the FnIII 3-5 fragment of the CLD ligand tenascin-R, and alkaline phosphatase-tagged aggrecan CLD was allowed to bind in the presence of varying concentrations (0.3–1000 nM) of competitor proteins (open circles, wild-type E1E2LCt; filled triangles, V2303M E1E2LCt). Each data point represents the average of five measurements; error bars show standard deviation.
(D) Surface plasmon resonance interaction analysis. The sensorgrams show overlays of duplicate samples of the different recombinant aggrecan G3 variants. The lowest traces in each sensorgram show injections of running buffer alone. Samples (at 1.5 to 50 nM in HEPES-buffered saline containing 2 mM CaCl2) were injected for 4 min at 50 μl/min over a BIAcore sensor chip coupled with the CLD ligand fibulin-2, followed by injection of running buffer. The binding and dissociation of the G3 variants was registered in a BIAcore 2000 instrument. All wild-type G3 proteins showed strong binding and slow dissociation, whereas no or insignificant binding of the V2303M variants was observed.
Table 2.
Aggrecan G3 Domain Affinities for C-type Lectin Ligands
| G3 Variant | KD (nM) for Tenascin-Ra | KD (nM) for Fibulin-1b | KD (nM) for Fibulin-2c |
|---|---|---|---|
| E1E2LCt | 0.08 | 7.7 | 0.11 |
| V2303M E1E2LCt | 0.67 | -d | 43.6 |
| LCt | 0.26 | 38.1 | 0.93 |
| V2303M LCt | 2080 | -d | -d |
| Lt | 0.26 | 17.9 | 1.18 |
| V2303M Lt | 166 | -d | -d |
Aggrecan G3 variants were injected over ligand proteins immobilized on a BIAcore CM5 sensor chip by amine coupling.
Tenascin-R FnIII repeats 3–5.
Fibulin-1 domain II.
Fibulin-2 domain II.
No binding.
To determine whether aggrecan containing the V2303M mutation is actually produced and secreted in vivo, we studied proteoglycans from articular cartilage of an affected family member undergoing knee arthroplasty. After guanidine hydrochloride extraction, aggrecan was purified by CsCl density gradient centrifugation. After digestion with endoproteinase GluC, the resulting peptides were separated by reverse-phase HPLC, and the fractions were analyzed by mass spectrometry (Figure 4A). MALDI-TOF MS showed the presence of peptides with both wild-type sequence and the V2303M sequence, as we corroborated by spiking the sample with peptides from wild-type and mutated recombinant CLD (Figure 4B). To verify the peptide sequences, we analyzed the sample further by ion trap MSMS and confirmed the presence of both the wild-type (Figure 4C) and the V2303M (Figure 4D) aggrecan sequences.
Figure 4.
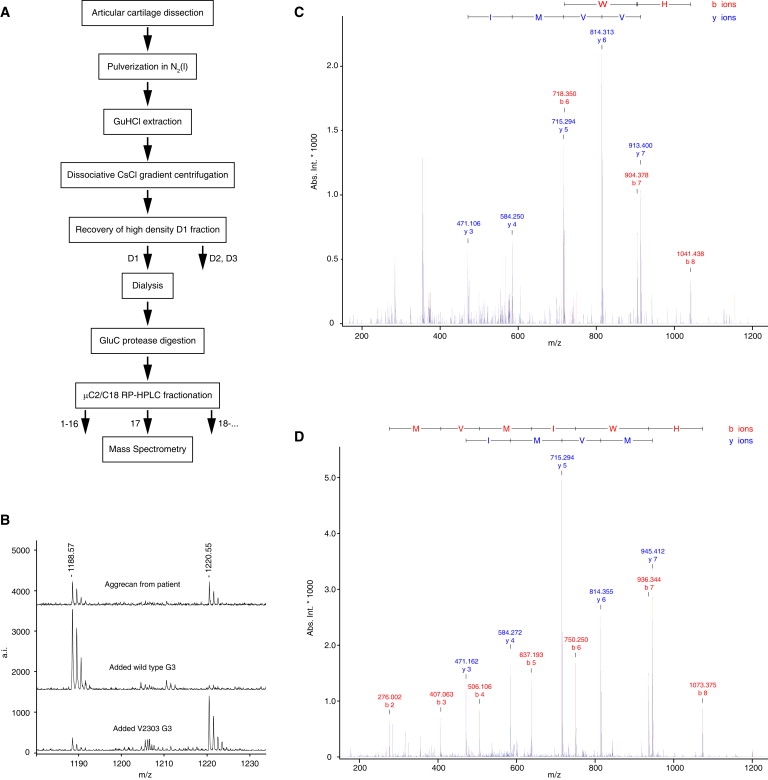
Aggrecan Containing the V2303M Mutation Is Produced and Present in Patient Cartilage
(A) Schematic representation of the extraction, purification, and analysis of aggrecan from articular cartilage obtained after knee-replacement surgery in a patient with familial osteochondritis dissecans.
(B) Top: MALDI-TOF mass spectrometry of trypsin digests identified peptides from both wild-type (DCVVMIWHE, 1188.51 Da) and Val > Met (DCMVMIWHE, 1220.48 Da) aggrecan in C2/C18 fraction 17 from the patient cartilage extract. Middle: Patient cartilage extract with added wild-type recombinant protein. Bottom: Patient cartilage sample with added recombinant mutant protein.
(C and D) Ion trap MSMS analysis of C2/C18 fraction 17 from the patient sample confirmed the presence and sequence of both wild-type (C) and V2303M (D) aggrecan in patient cartilage. Wild-type DCVVMIWHE, m/z = 594.76 Da (+2); mutant DCMVMIWHE, m/z = 610.75 Da (+2).
We quantified the total amount of proteoglycan in the extracts (in principle representing aggrecan) and in aggrecan purified via CsCl density-gradient centrifugation by using Alcian blue precipitation.29 In this procedure the acid conditions and the elevated ionic strength provides a focus on the sulfate groups. The proteoglycan contents of a control extract from osteoarthritic knee cartilage (pooled from ten individuals) and of the patient extract, as well as of purified aggrecan (D1 fraction) from the OA cartilage pool and the patient, were found to be similar (26.8 versus 24.8 mg/g and 13.1 versus 14.0 mg/g, respectively), indicating that there is no major alteration in aggrecan secretion or glycosaminoglycan sulfation in the patient tissue.
Discussion
These results provide a molecular explanation for autosomal-dominant familial osteochondritis dissecans. In addition to osteochondritis dissecans, the clinical features of this disease include premature OA and mild, disproportionate short stature.4 All of these symptoms are consistent with an impaired matrix assembly and function due to the aggrecan G3 dysfunction.
The etiology of sporadic osteochondritis dissecans is likely to be multifactorial.30 Suggested contributing factors include trauma and microfractures resulting from the cumulative damage of repetitive stress or injury and/or fragility of the cartilage and bone. Disturbance of the endochondral ossification might cause areas of local ischemic necrosis, predisposing the joint to osteochondritis dissecans after microtrauma.31 Finally, the formation of an abnormal cartilage might predispose individuals to osteochondritis dissecans. This might result from an altered networking of molecules in the cartilage. Our results show that perturbed function in a domain of aggrecan interacting with other extracellular matrix molecules is a key factor in the etiology of familial osteochondritis dissecans.
Both the sporadic and familial forms of osteochondritis dissecans are associated with secondary OA when lesions occur after closure of the epihyseal growth plate.2 In contrast, lesions occurring before closure of the growth plate normally heal spontaneously without secondary OA development in patients with sporadic osteochondritis dissecans.2 In patients with familial osteochondritis dissecans, however, lesions before growth plate closure are associated with secondary OA.4 Interestingly, early-onset OA is part of the phenotype in the family under study. Premature OA is part of the pathology associated with many mutations affecting cartilage ECM assembly or stability, e.g., in Stickler syndrome type I [MIM 108300] (COL2A1 [MIM 120140]), type II [MIM 604841] (COL11A1 [MIM 120280]), and type III [MIM 184840] (COL11A2 [MIM 120290]) and in multiple epiphyseal dysplasia ([MIM 132400] (COL9A1 [MIM 120210], COL9A2 [MIM 120260], COL9A3 [MIM 120270], COMP [MIM 600310], and MATN3 [MIM 602109]).32 Because sporadic osteochondritis dissecans before growth arrest does not lead to OA, the premature OA associated with familial osteochondritis dissecans is consistent with a disorganized and weakened cartilage ECM caused by loss of G3 interactions in the V2303M aggrecan.
A characteristic clinical feature of familial osteochondritis dissecans is disproportionate short stature.4 The aggrecan gene has been identified by genome-wide association analysis as a locus influencing adult height.33 Mutations in the aggrecan gene that result in skeletal phenotypes have been identified in several species.34–38 Most described aggrecan mutations are either null (cmd-Bc and Dexter Bulldog cattle BD2) or functional null (cmd, nanomelia, Dexter Bulldog cattle BD1, and spondyloepiphyseal dysplasia type Kimberley [SEDK, (MIM 608361)]). The latter mutations result in premature stop codons with several downstream introns and show lowered aggrecan mRNA levels.39,40 In the case of ACANBD1, it has been verified that this is due to nonsense mediated decay,34 which is likely the case for all the functional null mutations. The remaining mutant aggrecan mRNA might be translated, but secretion of truncated aggrecan is prevented by proteosomal degradation.16 When homozygous, the aggrecan null and functional null mutations result in lethal skeletal dysplasias. In the heterozygous state, these mutations lead to aggrecan haploinsufficiency and dwarfism phenotypes, and in the case of SEDK, this is associated with early-onset OA in weight-bearing joints. 35,41 Aggrecan provides swelling pressure to the cartilage tissue ECM through osmotic attraction of water by counter ions bound to the thousands of sulfate and carboxyl groups carried on the glycosaminoglycan chains of each proteoglycan molecule. Thus, skeletal phenotypes similar to aggrecan deficiency also result from mutations affecting glycosaminoglycan sulfation.42 Our data are consistent with a normal V2303M aggrecan secretion, and we have no reason to believe that sulfation is affected. We suggest that the loss of G3 interactions in the mutant might contribute to a disorganized ECM in the growth plate and thus lead to decreased long bone growth in the affected individuals.
The importance of the G3 interactions for skeletal development is supported by the recent identification of an additional missense mutation (D2267N) in the human aggrecan CLD.43 The D2267N mutation results in recessive spondyloepimetaphyseal dysplasia (MIM 612813), and homozygous individuals display extreme short stature, brachydactyly, and craniofacial abnormalities. Unlike patients with familial osteochondritis dissecans, these patients showed no orthopedic complications or signs of OA. Mild proportionate short stature was reported for two individuals heterozygous for the D2267N mutation.
The D2267N mutation obliterates one of the two calcium coordinations provided by this residue and also generates a novel site for N-linked glycosylation, which at least in recombinant G3 proteins is glycosylated. This might affect aggrecan CLD interaction with ECM ligands such as tenascins or fibulins, and experiments showed lowered total binding and altered kinetics in D2267N variant CLD interactions with tenascin-C. The retained tenascin-C binding of the D2267N variant proteins suggests that this mutation is less disruptive to CLD function than the V2303M mutation, which showed strong effects on CLD ligand binding. Nonetheless, the severe phenotype observed in the D2267N homozygous individuals confirms the importance of CLD interactions for aggrecan function.
The dominant phenotype seen in the heterozygous V2303M individuals is consistent with a critical role of the aggrecan G3-domain interactions in regulating matrix assembly. There was no significant difference in the levels of mutated and wild-type aggrecan in joint cartilage from a V2303M patient, as evidenced by the similar signals upon mass spectrometry (Figure 4B). There was, moreover, no apparent perturbation in sulfation of aggrecan from the patient given that the reactivity in the Alcian blue assay was normal. The pronounced phenotype of the heterozygous individuals therefore indicates a dominant negative role of the mutated molecules, which might compromise the organization and interaction of matrix molecules that are crucial for normal function in a load-bearing tissue such as cartilage.
Web Resources
The URL for data presented herein is as follows:
Online Mendelian Inheritance in Man (http://www.ncbi.nlm.nih.gov/Omim).
Acknowledgments
We sincerely thank the family members who participated in this study. We also thank Susann Haraldsson, Department of Medical and Clinical Genetics, Umeå University Hospital, Umeå, for expert technical assistance, Gillian Wallis, The Wellcome Trust Centre for Cell-Matrix Research, Manchester, United Kingdom, for sharing primer sequences with us, and Lisa Cannon-Albright, University of Utah, Salt Lake City, USA, for providing access to the MCLINK software used for linkage analysis. This work was supported by grants from the Northern County Councils Cooperation Committee “Visare Norr”; the Capio Research Foundation; Uppsala and Lund Universities; the Kock's, Crafoord, and Lundbeck foundations; and the Swedish Research Council. The mass spectrometers were funded by the Lundberg foundation, Gothenburg.
References
- 1.Smillie I.S. E. & S. Livingstone Ltd.; Edinburgh and London: 1960. Osteochondritis Dissecans. Loose Bodies in Joints, Etiology, Pathology, Treatment. [Google Scholar]
- 2.Linden B. The incidence of osteochondritis dissecans in the condyles of the femur. Acta Orthop. Scand. 1976;47:664–667. doi: 10.3109/17453677608988756. [DOI] [PubMed] [Google Scholar]
- 3.Stougaard J. Familial occurrence of osteochondritis dissecans. J. Bone Joint Surg. Br. 1964;46:542–543. [PubMed] [Google Scholar]
- 4.Stattin E.L., Tegner Y., Domellof M., Dahl N. Familial osteochondritis dissecans associated with early osteoarthritis and disproportionate short stature. Osteoarthritis Cartilage. 2008;16:890–896. doi: 10.1016/j.joca.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 5.Pick M.P. Familial osteochondritis dissecans. J. Bone Joint Surg. Br. 1955;37-B:142–145. doi: 10.1302/0301-620X.37B1.142. [DOI] [PubMed] [Google Scholar]
- 6.Phillips H.O., Grubb S.A. Familial multiple osteochondritis dissecans. Report of a kindred. J. Bone Joint Surg. Am. 1985;67:155–156. [PubMed] [Google Scholar]
- 7.Andrew T.A., Spivey J., Lindebaum R.H. Familial osteochondritis dissecans and dwarfism. Acta Orthop. Scand. 1981;52:519–523. doi: 10.3109/17453678108992141. [DOI] [PubMed] [Google Scholar]
- 8.Heinegard D., Oldberg A. Structure and biology of cartilage and bone matrix noncollagenous macromolecules. FASEB J. 1989;3:2042–2051. doi: 10.1096/fasebj.3.9.2663581. [DOI] [PubMed] [Google Scholar]
- 9.Doege K., Sasaki M., Horigan E., Hassell J.R., Yamada Y. Complete primary structure of the rat cartilage proteoglycan core protein deduced from cDNA clones. J. Biol. Chem. 1987;262:17757–17767. [PubMed] [Google Scholar]
- 10.Heinegård D., Hascall V.C. Aggregation of cartilage proteoglycans. 3. Characteristics of the proteins isolated from trypsin digests of aggregates. J. Biol. Chem. 1974;249:4250–4256. [PubMed] [Google Scholar]
- 11.Rauch U., Clement A., Retzler C., Frohlich L., Fässler R., Göhring W., Faissner A. Mapping of a defined neurocan binding site to distinct domains of tenascin-C. J. Biol. Chem. 1997;272:26905–26912. doi: 10.1074/jbc.272.43.26905. [DOI] [PubMed] [Google Scholar]
- 12.Olin A.I., Morgelin M., Sasaki T., Timpl R., Heinegard D., Aspberg A. The proteoglycans aggrecan and Versican form networks with fibulin-2 through their lectin domain binding. J. Biol. Chem. 2001;276:1253–1261. doi: 10.1074/jbc.M006783200. [DOI] [PubMed] [Google Scholar]
- 13.Aspberg A., Binkert C., Ruoslahti E. The versican C-type lectin domain recognizes the adhesion protein tenascin-R. Proc. Natl. Acad. Sci. USA. 1995;92:10590–10594. doi: 10.1073/pnas.92.23.10590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aspberg A., Miura R., Bourdoulous S., Shimonaka M., Heinegård D., Schachner M., Ruoslahti E., Yamaguchi Y. The C-type lectin domains of lecticans, a family of aggregating chondroitin sulfate proteoglycans, bind tenascin-R by protein-protein interactions independent of carbohydrate moiety. Proc. Natl. Acad. Sci. USA. 1997;94:10116–10121. doi: 10.1073/pnas.94.19.10116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aspberg A., Adam S., Kostka G., Timpl R., Heinegard D. Fibulin-1 is a ligand for the C-type lectin domains of aggrecan and versican. J. Biol. Chem. 1999;274:20444–20449. doi: 10.1074/jbc.274.29.20444. [DOI] [PubMed] [Google Scholar]
- 16.Vertel B.M., Walters L.M., Grier B., Maine N., Goetinck P.F. Nanomelic chondrocytes synthesize, but fail to translocate, a truncated aggrecan precursor. J. Cell Sci. 1993;104:939–948. doi: 10.1242/jcs.104.3.939. [DOI] [PubMed] [Google Scholar]
- 17.Abkevich V., Camp N.J., Gutin A., Farnham J.M., Cannon-Albright L., Thomas A. A robust multipoint linkage statistic (tlod) for mapping complex trait loci. Genet. Epidemiol. 2001;21(Suppl 1):S492–S497. doi: 10.1002/gepi.2001.21.s1.s492. [DOI] [PubMed] [Google Scholar]
- 18.Goring H.H., Terwilliger J.D. Linkage analysis in the presence of errors I: Complex-valued recombination fractions and complex phenotypes. Am. J. Hum. Genet. 2000;66:1095–1106. doi: 10.1086/302797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Valhmu W.B., Palmer G.D., Rivers P.A., Ebara S., Cheng J.F., Fischer S., Ratcliffe A. Structure of the human aggrecan gene: exon-intron organization and association with the protein domains. Biochem. J. 1995;309:535–542. doi: 10.1042/bj3090535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xiao W., Oefner P.J. Denaturing high-performance liquid chromatography: A review. Hum. Mutat. 2001;17:439–474. doi: 10.1002/humu.1130. [DOI] [PubMed] [Google Scholar]
- 21.Adam S., Göhring W., Wiedemann H., Chu M.L., Timpl R., Kostka G. Binding of fibulin-1 to nidogen depends on its C-terminal globular domain and a specific array of calcium-binding epidermal growth factor-like (EG) modules. J. Mol. Biol. 1997;272:226–236. doi: 10.1006/jmbi.1997.1244. [DOI] [PubMed] [Google Scholar]
- 22.Sasaki T., Mann K., Wiedemann H., Göhring W., Lustig A., Engel J., Chu M.L., Timpl R. Dimer model for the microfibrillar protein fibulin-2 and identification of the connecting disulfide bridge. EMBO J. 1997;16:3035–3043. doi: 10.1093/emboj/16.11.3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Day J.M., Olin A.I., Murdoch A.D., Canfield A., Sasaki T., Timpl R., Hardingham T.E., Aspberg A. Alternative splicing in the aggrecan G3 domain influences binding interactions with tenascin-C and other extracellular matrix proteins. J. Biol. Chem. 2004;279:12511–12518. doi: 10.1074/jbc.M400242200. [DOI] [PubMed] [Google Scholar]
- 24.Xiao Z.C., Taylor J., Montag D., Rougon G., Schachner M. Distinct effects of recombinant tenascin-R domains in neuronal cell functions and identification of the domain interacting with the neuronal recognition molecule F3/11. Eur. J. Neurosci. 1996;8:766–782. doi: 10.1111/j.1460-9568.1996.tb01262.x. [DOI] [PubMed] [Google Scholar]
- 25.Lorenzo P., Aspberg A., Önnerfjord P., Bayliss M.T., Neame P.J., Heinegård D. Identification and characterization of asporin. a novel member of the leucine-rich repeat protein family closely related to decorin and biglycan. J. Biol. Chem. 2001;276:12201–12211. doi: 10.1074/jbc.M010932200. [DOI] [PubMed] [Google Scholar]
- 26.Heinegård D., Sommarin Y. Isolation and characterization of proteoglycans. Methods Enzymol. 1987;144:319–372. doi: 10.1016/0076-6879(87)44186-4. [DOI] [PubMed] [Google Scholar]
- 27.Isogai Z., Aspberg A., Keene D.R., Ono R.N., Reinhardt D.P., Sakai L.Y. Versican interacts with fibrillin-1 and links extracellular microfibrils to other connective tissue networks. J. Biol. Chem. 2002;277:4565–4572. doi: 10.1074/jbc.M110583200. [DOI] [PubMed] [Google Scholar]
- 28.Lundell A., Olin A.I., Morgelin M., Al-Karadaghi S., Aspberg A., Logan D.T. Structural basis for interactions between tenascins and Lectican C-type lectin domains: Evidence for a crosslinking role for tenascins. Structure. 2004;12:1495–1506. doi: 10.1016/j.str.2004.05.021. [DOI] [PubMed] [Google Scholar]
- 29.Bjornsson S. Simultaneous preparation and quantitation of proteoglycans by precipitation with alcian blue. Anal. Biochem. 1993;210:282–291. doi: 10.1006/abio.1993.1197. [DOI] [PubMed] [Google Scholar]
- 30.Schenck R.C., Goodnight J.M. Osteochondritis dissecans. J. Bone Joint Surg. Am. 1996;78:439–456. [PubMed] [Google Scholar]
- 31.Ytrehus B., Carlson C.S., Ekman S. Etiology and pathogenesis of osteochondrosis. Vet. Pathol. 2007;44:429–448. doi: 10.1354/vp.44-4-429. [DOI] [PubMed] [Google Scholar]
- 32.Li Y., Xu L., Olsen B.R. Lessons from genetic forms of osteoarthritis for the pathogenesis of the disease. Osteoarthritis Cartilage. 2007;15:1101–1105. doi: 10.1016/j.joca.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weedon M.N., Lango H., Lindgren C.M., Wallace C., Evans D.M., Mangino M., Freathy R.M., Perry J.R., Stevens S., Hall A.S. Genome-wide association analysis identifies 20 loci that influence adult height. Nat. Genet. 2008;40:575–583. doi: 10.1038/ng.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cavanagh J.A., Tammen I., Windsor P.A., Bateman J.F., Savarirayan R., Nicholas F.W., Raadsma H.W. Bulldog dwarfism in Dexter cattle is caused by mutations in ACAN. Mamm. Genome. 2007;18:808–814. doi: 10.1007/s00335-007-9066-9. [DOI] [PubMed] [Google Scholar]
- 35.Gleghorn L., Ramesar R., Beighton P., Wallis G. A mutation in the variable repeat region of the aggrecan gene (AGC1) causes a form of spondyloepiphyseal dysplasia associated with severe, premature osteoarthritis. Am. J. Hum. Genet. 2005;77:484–490. doi: 10.1086/444401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krueger R.C., Kurima K., Schwartz N.B. Completion of the mouse aggrecan gene structure and identification of the defect in the cmd-Bc mouse as a near complete deletion of the murine aggrecan gene. Mamm. Genome. 1999;10:1119–1125. doi: 10.1007/s003359901176. [DOI] [PubMed] [Google Scholar]
- 37.Li H., Schwartz N.B., Vertel B.M. cDNA cloning of chick cartilage chondroitin sulfate (aggrecan) core protein and identification of a stop codon in the aggrecan gene associated with the chondrodystrophy, nanomelia. J. Biol. Chem. 1993;268:23504–23511. [PubMed] [Google Scholar]
- 38.Watanabe H., Kimata K., Line S., Strong D., Gao L.Y., Kozak C.A., Yamada Y. Mouse cartilage matrix deficiency (cmd) caused by a 7 bp deletion in the aggrecan gene. Nat. Genet. 1994;7:154–157. doi: 10.1038/ng0694-154. [DOI] [PubMed] [Google Scholar]
- 39.Stirpe N.S., Argraves W.S., Goetinck P.F. Chondrocytes from the cartilage proteoglycan-deficient mutant, nanomelia, synthesize greatly reduced levels of the proteoglycan core protein transcript. Dev. Biol. 1987;124:77–81. doi: 10.1016/0012-1606(87)90461-1. [DOI] [PubMed] [Google Scholar]
- 40.Watanabe H., Nakata K., Kimata K., Nakanishi I., Yamada Y. Dwarfism and age-associated spinal degeneration of heterozygote cmd mice defective in aggrecan. Proc. Natl. Acad. Sci. USA. 1997;94:6943–6947. doi: 10.1073/pnas.94.13.6943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eyre S., Roby P., Wolstencroft K., Spreckley K., Aspinwall R., Bayoumi R., Al-Gazali L., Ramesar R., Beighton P., Wallis G. Identification of a locus for a form of spondyloepiphyseal dysplasia on chromosome 15q26.1: exclusion of aggrecan as a candidate gene. J. Med. Genet. 2002;39:634–638. doi: 10.1136/jmg.39.9.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schwartz N.B., Domowicz M. Chondrodysplasias due to proteoglycan defects. Glycobiology. 2002;12:57R–68R. doi: 10.1093/glycob/12.4.57r. [DOI] [PubMed] [Google Scholar]
- 43.Tompson S.W., Merriman B., Funari V.A., Fresquet M., Lachman R.S., Rimoin D.L., Nelson S.F., Briggs M.D., Cohn D.H., Krakow D. A recessive skeletal dysplasia, SEMD aggrecan type, results from a missense mutation affecting the C-type lectin domain of aggrecan. Am. J. Hum. Genet. 2009;84:72–79. doi: 10.1016/j.ajhg.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]