

Abstract
Familial exudative vitreoretinopathy (FEVR) is a genetically heterogeneous retinal disorder characterized by abnormal vascularisation of the peripheral retina, often accompanied by retinal detachment. To date, mutations in three genes (FZD4, LRP5, and NDP) have been shown to be causative for FEVR. In two large Dutch pedigrees segregating autosomal-dominant FEVR, genome-wide SNP analysis identified an FEVR locus of ∼40 Mb on chromosome 7. Microsatellite marker analysis suggested similar at risk haplotypes in patients of both families. To identify the causative gene, we applied next-generation sequencing in the proband of one of the families, by analyzing all exons and intron-exon boundaries of 338 genes, in addition to microRNAs, noncoding RNAs, and other highly conserved genomic regions in the 40 Mb linkage interval. After detailed bioinformatic analysis of the sequence data, prioritization of all detected sequence variants led to three candidates to be considered as the causative genetic defect in this family. One of these variants was an alanine-to-proline substitution in the transmembrane 4 superfamily member 12 protein, encoded by TSPAN12. This protein has very recently been implicated in regulating the development of retinal vasculature, together with the proteins encoded by FZD4, LRP5, and NDP. Sequence analysis of TSPAN12 revealed two mutations segregating in five of 11 FEVR families, indicating that mutations in TSPAN12 are a relatively frequent cause of FEVR. Furthermore, we demonstrate the power of targeted next-generation sequencing technology to identify disease genes in linkage intervals.
Main Text
Familial exudative vitreoretinopathy (FEVR) is a well-characterized hereditary ocular disorder that was first described by Criswick and Schepens.1 The pathologic features of the disease initiate from the abnormal retinal development due to the incomplete vascularization of the peripheral retina and/or retinal blood vessel differentiation.2 The latter can lead to various complications, such as retinal neovascularization and exudates, vitreous haemorrhage, ectopia of the macula, retinal folds, and retinal detachment.3 FEVR is genetically heterogeneous and shows autosomal-dominant, autosomal-recessive, and X-linked recessive modes of inheritance.1,4–9 Mutations in the genes coding for frizzled 4 (FZD4, located on 11q14.2 [MIM ∗604579]) and low-density lipoprotein 5 (LRP5, located on 11q13.2 [MIM ∗603506]), which act as Wnt coreceptors, and Norrin (NDP, located on Xp11.3 [MIM ∗300658]), a ligand for these Wnt receptors, have been shown to result in FEVR. A consistent feature is the aberrant retinal vascular development caused by the impaired action of the Ndp/β-catenin signaling pathway, which, among others, orchestrates eye organogenesis and blood supply.7,10–13
To unravel the genetic causes underlying FEVR, we included patients and their relatives in our study. Informed consent was obtained from all patients and their participating relatives in adherence to the tenets of the Declaration of Helsinki. In 11 families with autosomal-dominant FEVR, we excluded mutations in FZD4 and LRP5 via sequence analysis of all coding exons and intron-exon boundaries (unpublished data). In those families in which X-linked inheritance could not be excluded on the basis of the pattern of inheritance, mutations in NDP were excluded via sequence analysis. In the two largest families (families A and B) that did not carry exonic mutations in FZD4 and LRP5, detailed analysis of microsatellite markers surrounding these two genes excluded them as causative for FEVR. Patients in both families invariably show a peripheral avascular area, the most typical feature in FEVR (Figure 1). The visual acuity varies considerably (normal to light perception) as a result of secondary defects such as retinal detachment and retinal exudates. Subsequently, eight individuals from family A and 15 individuals from family B (Figure 2A) were genotyped on the Infinium II HumanLinkage-12 Panel (Illumina, San Diego, CA, USA) that contains 6090 SNP markers. Genome-wide linkage analysis via the GeneHunter 2.1r5 program in the easyLinkage v5.052beta software package14 revealed a 40.5 Mb genomic region on chromosome 7 suggestive for linkage (LOD score 2.34) in family A (Figure 2B, left panel), whereas in family B, a significant LOD score of 3.31 was obtained for a 16.7 Mb genomic region on chromosome 7 (Figure 2B, right panel), completely contained within the 40.5 Mb region detected in family A (Figure 3A). Moreover, the affected individuals in both families shared the same SNP and microsatellite alleles for a region of at least 10 Mb. These results strongly suggested that the region on chromosome 7 is likely to contain a gene causative for FEVR. Two obvious candidate genes within the shared linkage intervals were WNT2 (MIM ∗147870) and WNT16 (MIM ∗606267), since the canonical Wnt signaling pathway has been implicated in regulating the formation of intraretinal capillaries.15 Sequence analysis of the exons and intron-exon boundaries of these two genes in probands III-7 (family A) and II-5 (family B) did not, however, reveal any sequence variant causative for FEVR.
Figure 1.
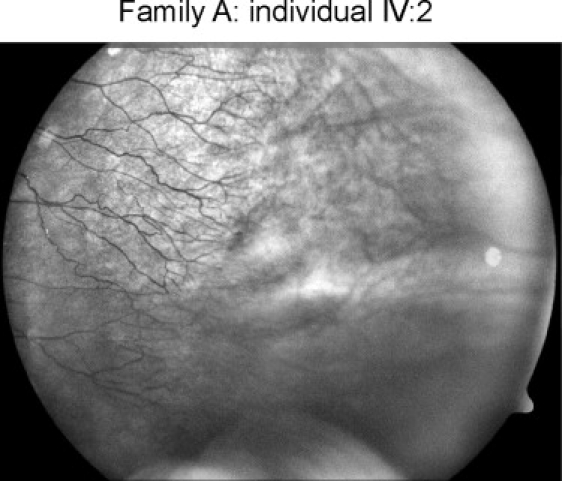
Fundus Photograph Showing Part of the Avascular Periphery of Patient IV:2 from Family A
Temporal periphery of the left fundus in patient IV:2 from family A (numbering according to the pedigree presented in Figure 2A). The retinal vasculature (at the left side) ends in small aberrant ramifications in the equatorial area of the fundus. The peripheral retina (at the right side) is avascular; only choroidal vessels are present in this area.
Figure 2.
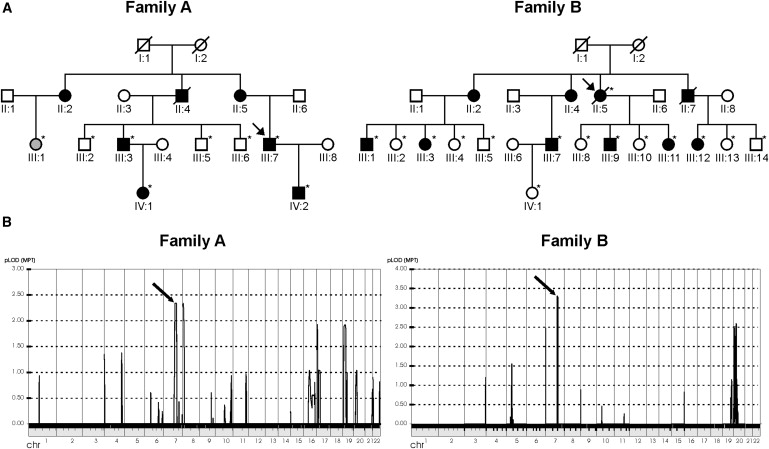
Pedigree Structure and Genome-wide Linkage Analysis for FEVR Families A and B
(A) Pedigree structure of those parts of families A and B that were selected for Illumina 6K SNP genotyping analysis. Individuals of whom the clinical status is uncertain are depicted in gray. Probands are indicated with an arrow. Eight members of family A and 15 individuals of family B, indicated with asterisks, were genotyped.
(B) Genome-wide LOD score calculations, with the use of the 6K SNP array genotyping data. In family A, two peaks are visible, on chromosomes 7 and 8, respectively. In family B, only one clear peak is visible, on chromosome 7, with a genome-wide-significant LOD score of 3.31. Interestingly, the two peaks on chromosome 7 in families A and B, indicated by arrows, represent the same genomic region.
Figure 3.
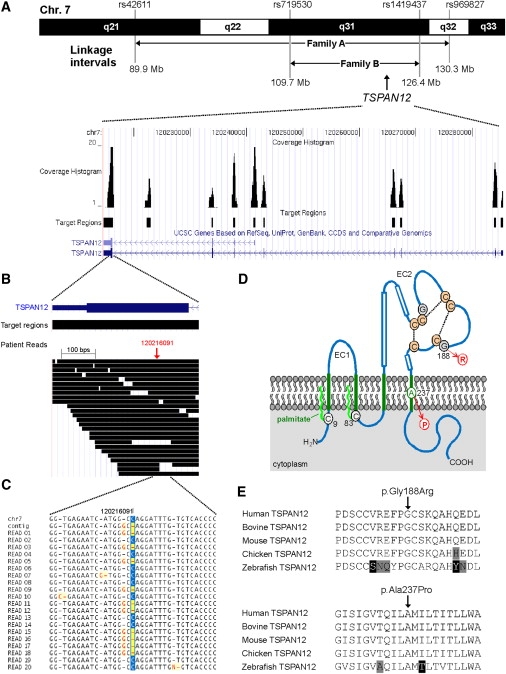
Genomic Structure, Next-Generation Sequencing, and Mutation Analysis of TSPAN12
(A) Upper panel, part of chromosome 7 showing the linkage intervals and the corresponding flanking SNPs for FEVR families A and B. The linkage interval from family A was enriched for targeted next-generation sequencing technology. The TSPAN12 gene is located in the shared interval between families A and B. Lower panel, overview of the genomic structure of the TSPAN12 gene, showing all introns and exons. For all exons, as well as a highly conserved region between exons 7 and 8, the sequence coverage histograms are presented.
(B) Overview of all patient reads for exon 8 of TSPAN12. In total, more than 30 reads were representing parts of this exon.
(C) Sequence traces for the twenty unique reads covering the TSPAN12 heterozygous G-to-C mutation. For these 20 reads, ten were representing the reference allele and ten were showing the mutant allele. At the protein level, the G-to-C transition results in the substitution of a proline for an alanine residue (c.709G>C [p.Ala237Pro]).
(D) Topology of TSPAN12 and the positions of the missense mutions identified in the FEVR families. TSPAN12 shows the characteristic domain structure of tetraspanins, including four transmembrane (TM) domains (in green). A cysteine residue preceding TM domain 1 (C9) and another following TM domain 2 (C83) are probably palmitoylated (bright green wavy lines). The second extracellular loop (EC2) contains three α helices (boxes), the typical C-C-G motif characteristic for all tetraspanin proteins, followed by one pair and two single cysteine residues. These six cysteines are predicted to form disulphide bonds (stippled lines). The glycine residue that is mutated into an arginine (G188) precedes one of these cysteines. The alanine residue that is mutated into a proline (A237) resides within an α-helical structure within the fourth TM region.
(E) Sequence comparison of a number of vertebrate TSPAN12 proteins. Depicted are parts of TSPAN12 that harbor the amino acid residues that are mutated. Both the glycine residue at position 188 (upper panel) and the alanine residue at position 237 (lower panel) are completely conserved throughout vertebrate evolution. Residues identical in all sequences are black on a white background, whereas similar amino acids are black on a light gray background. Nonconservative changes are indicated in white on a black background. UniProt accession number of the protein sequences used for sequence comparison are as follows: human TSPAN12, O95859; bovine TSPAN12, Q29RH7; mouse TSPAN12, Q8BKT6; chicken TSPAN12, Q5ZIF5; zebrafish TSPAN12, Q7T2G0.
Given the high number of genes in the region, as well as the lack of other obvious candidate genes, we decided to apply array-based sequence capture followed by next-generation sequencing to simultaneously analyze all genes in the region. The complete 40.5 Mb region on chromosome 7, suggestive for linkage in family A (Figure 3A, upper panel), was used for sequence capture. The array design included all known exons, untranslated regions (UTRs), microRNAs, and highly conserved regions (PhastCons Conserved Elements, 28-way Mammal Multiz Alignment, LOD score ≥ 100), complemented with the complete genomic sequence of WNT2 and WNT16. For all exons, an additional 30 base pairs (bp) were added at each side for the detection of splice-site mutations. Targets smaller than 250 bp, which is, based upon our experience, the minimum size for the DNA capture protocol, were enlarged by extending both ends of the region. In total, 3048 exons from 338 RefSeq genes and 313 UCSC genes (see Web Resources), as well as 14 microRNAs and 23 noncoding RNAs, were targeted (Figure S1, available online). Together with the UTRs and regions of high conservation, 2,366,514 bp were selected. For reasons of efficiency, the array included targets for another linkage region representing 352 Kb on chromosome 17. After stringent probe selection by NimbleGen (Roche NimbleGen, Madison, WI, USA) (uniqueness tested by Sequence Search and Alignment by Hashing Algorithm-SSAHA),16 a total of 2,528,988 bases were represented on an array, with 385,000 oligonucleotide probes targeting the region of interest as well as the additional region on chromosome 17.
Sequence capture was performed in accordance with the manufacturer's instructions (Roche NimbleGen). In brief, 20 μg of genomic DNA of the FEVR proband from family A was used in the preparation of DNA for sequence-capture hybridization. A final amount of 5 μg DNA was hybridized to the customized array, subsequently eluted and amplified by ligation-mediated PCR. Three micrograms of amplified enriched DNA was used as input for generating an ssDNA library for massive parallel sequencing, with the use of a Roche 454 GS FLX sequencer with Titanium series reagents. The sample was sequenced by using one quarter lane of a Roche sequencing run, yielding 48.6 Mb of sequence data. Approximately 94.5% of the sequence data mapped back to unique regions of the human genome (hg18, NCBI Build 36.1), with the use of the Roche Newbler software (version 2.0.01.12). Of all mapped reads, 90.5% was located on or near the targeted regions (i.e., within 500 bp proximity). This was sufficient to reach an average of 13.2-fold coverage of all target regions and, more specifically, an average of 14.7-fold coverage of the targeted exons on chromsome 7. For the region of interest, only 5.2% (112,319 bp) of all targeted sequences were not covered, whereas 39.2% (1,099,873 bp) of the target sequence was covered less than ten times (Table S1).
The Roche software detected a total of 1915 variants on chromosome 7. We used a custom-made data analysis pipeline to annotate detected variants with various kinds of information, including known SNPs, amino acid consequences, genomic location, and evolutionary conservation. With this analysis, 1749 variants were found to correspond to known SNPs or to overlap with a known polymorphic region (dbSNP130). After we excluded nonexonic (145), synonymous (4), and putative homozygous variants (variation percentage ≥ 80%) (3), 14 variants remained (Table 1). From these, three promising candidates were selected (in the PTCD1, ZAN [MIM ∗602372], and TSPAN12 [MIM ∗613138] genes) because they were detected in at least four nonduplicate reads and had a high score for evolutionary conservation (vertebrate PhyloP).17–19
Table 1.
Overview of All Nonsynonymous Sequence Variants Not Present in dbSNP
| Position of the Reference Allele on Chr. 7 | Reference Allele | Variant Allele | No. of Reads | No. of Variant reads | Variation (%) | Reference Amino Acid | Variant Amino Acid | Gene | PhyloP Score |
|---|---|---|---|---|---|---|---|---|---|
| 92570705 | T | C | 7 | 3 | 43 | D | G | SAMD9 | 1.56 |
| 98870495 | G | A | 26 | 16 | 62 | R | C | PTCD1 | 3.06 |
| 99835402 | C | T | 13 | 6 | 46 | P | L | PILRA | 1.75 |
| 100122289 | - | CCT | 5 | 3 | 60 | EQ | ERQ | GIGYF1 | 3.98 |
| 100209410 | G | A | 15 | 8 | 53 | R | H | ZAN | 1.81 |
| 100473466 | A | G | 38 | 13 | 34 | T | A | MUC17 | 0.60 |
| 105066159 | TC | - | 19 | 3 | 16 | E | X | ATXN7L1 | 4.92 |
| 113306419 | C | T | 15 | 6 | 40 | S | N | PPP1R3A | 1.05 |
| 115411632 | C | T | 14 | 5 | 36 | D | N | TFEC | −0.45 |
| 119702880 | T | C | 29 | 3 | 10 | C | R | KCND2 | 5.00 |
| 120216091 | C | G | 20 | 10 | 50 | A | P | TSPAN12 | 5.32 |
| 120555712 | T | C | 10 | 3 | 30 | L | S | C7orf58 | 3.00 |
| 128099699 | C | G | 7 | 5 | 71 | I | M | FAM71F2 | 0.42 |
| 128221650 | T | C | 13 | 3 | 23 | L | P | CCDC136 | 1.26 |
After initial filtering of the 1915 variants on the basis of overlap with dbSNP (build 130) and amino acid consequences, only 14 candidate variants remained. Variants marked in bold were selected after further prioritization on the basis of the number of variant reads and evolutionary conservation. The position of the reference base pair is based on UCSC Genome Browser build hg18. The table lists the reference allele and the variant allele for that position, the total number of high-confidence sequence reads for this position, and the number of times that a variant was read. The percentage of variation indicates how often a variant was read in comparison to the total number of reads. The amino acid of the reference and mutant alleles are indicated, as well as the gene in which the variant was detected. In the right column, the vertebrate PhyloP score for evolutionary conservation is presented.17–19
The missense variant detected in PTCD1 changes an arginine to a cysteine, whereas the variant in ZAN alters an arginine to a histidine residue. Both arginine residues are not completely conserved throughout vertebrate evolution. In addition, the presumed physiological roles of the proteins encoded by PTCD1 and ZAN, namely controlling leucine tRNA levels in mitochondria20 and regulating the adhesion of sperm cells to the zona pellucida,21 respectively, do not support their involvement in causing FEVR. Finally, PTCD1 and ZAN are both located outside the smaller linkage interval in family B and will therefore definitely not be causative for FEVR in that family. The TSPAN12 gene, however, is located within the shared linkage interval, and the variant detected via next-generation sequencing technology substitutes a proline residue for a highly conserved alanine residue, with the highest PhyloP conservation score (Table 1). In addition, very recently, TSPAN12 was found to regulate retinal vascular development in cooperation with the proteins encoded by FZD4, LRP5, and NDP.22 Therefore, the variant detected in TSPAN12 was considered to be the mutation causative for FEVR in family A.
TSPAN12 contains eight exons, for which exons 2–8 are protein coding, plus an alternative noncoding exon for a different isoform (Figure 3A). All exons of TSPAN12 were covered by sequence reads from the targeted next-generation sequencing experiment (Figure 3A). The missense variant that was detected by this approach (c.709G>C [p.Ala237Pro]) is located in exon 8. This exon was covered by more than 30 sequence reads (Figure 3B). The mutated nucleotide (at position 120,216,091) was covered by 20 unique sequence reads. In ten of the reads, the reference allele was detected, whereas in the other ten reads, the mutant allele was observed (Figure 3C), indicating that the mutation is present in a heterozygous state.
Conventional Sanger sequencing confirmed the mutation to be present in the proband of family A, as well as in the proband of family B who shared the at-risk haplotype for the region containing TSPAN12. Furthermore, the mutation was present in all relatives that were clearly affected with FEVR in both families (Figure S2). In three other relatives of whom the clinical status was uncertain (III:1 in family A, III:15 and IV:2 in family B) and in one healthy individual (III:8 in family B), the mutation was also present, which points to nonpenetrance, a feature that is also often observed in FEVR families with mutations in FZD4, LRP5, and NDP.3
To determine the prevalence of TSPAN12 mutations as a cause of FEVR, we selected nine additional FEVR probands from unrelated families for sequence analysis of the complete gene (primer sequences are listed in Table S2). In two of these nine families, the same p.Ala237Pro change was detected as segregating with the phenotype (families C and D; Figure S2). Microsatellite marker analysis surrounding TSPAN12 showed that the affected individuals of all four families showed a shared at-risk haplotype of at least 5.8 Mb (data not shown). In addition, a second variant was detected in the proband of family E, substituting an arginine residue for a glycine residue (c.562G>C [p.Gly188Arg]). This change was also present in his affected brother (Figure S2). Both changes were not detected in 140 ethnically matched control individuals (data not shown), nor were they present in a database containing more than two million variants from various in-house and published next-generation sequencing projects.23–25 An overview of the mutations that were identified is presented in Table 2.
Table 2.
Overview of the Missense Mutations Identified in TSPAN12
| Family | Proband | No. of Affected Relatives with the Mutation | No. of Unaffected Relatives with the Mutation | Mutation (cDNA Level) | Mutation (Protein Level) | Frequency in Control Alleles |
|---|---|---|---|---|---|---|
| Family A | III-7 | 3 | 0 | c.709G>C | p.Ala237Pro | 0/280 |
| Family B | II-5 | 7 | 1 | c.709G>C | p.Ala237Pro | 0/280 |
| Family C | II-2 | 0 | 0 | c.709G>C | p.Ala237Pro | 0/280 |
| Family D | III-2 | 3 | 0 | c.709G>C | p.Ala237Pro | 0/280 |
| Family E | II-1 | 1 | 0 | c.562G>C | p.Gly188Arg | 0/280 |
Five families have been identified as carrying a dominant missense mutation in TSPAN12. The proband corresponds to the pedigrees depicted in Figure S2. For each family, the number of affected relatives that carry the mutation is listed. In family B, one healthy individual also carried the mutation, suggesting nonpenetrance. None of the two variants was detected in over 280 ethnically matched control alleles.
The TSPAN12 protein is a member of the tetraspanin family, whose members share certain specific structural features that distinguish them from other proteins that pass the membrane four times.26 The glycine residue at position 188 is located in the extracellular loop between the third and the fourth transmembrane region and is completely conserved throughout vertebrate evolution (Figures 3D and 3E). The arginine residue substituting the glycine has a bulky side chain that is positively charged. In addition, this small and flexible glycine residue precedes a cysteine residue that is involved in the formation of disulphide bridges (Figure 3D) and is highly conserved in the complete tetraspanin protein family.27 Therefore, the substitution of an arginine for this glycine residue is likely to affect TSPAN12 function. The alanine at position 237 resides within the fourth transmembrane region and is also completely conserved throughout vertebrate evolution (Figures 3D and 3E). A proline residue has the intrinsic property to induce bends in the three-dimensional protein structure. Therefore, a proline residue at position 237 is predicted to disrupt the helical structure of the transmembrane region, which would result in a TSPAN12 protein with a defective confirmation that might even be subject to proteolytic degradation. The p.Ala237Pro mutation was detected in four unrelated Dutch families. Genealogical analysis revealed, however, that the ancestors of families A, B, and C were born in the same region in the eastern part of The Netherlands, with those of families B and C even being born in the same village. These data, combined with the microsatellite marker analysis that showed a shared haplotype among all families, suggest that the p.Ala237Pro mutation is a regional founder mutation.
The TSPAN12 protein has recently been identified as playing a crucial role in the regulation of the development of the retinal vasculature.22 Mice lacking Tspan12 show a primary vascular defect in the developing retina between postnatal days 5 and 11, similar to the phenotypes described for Fzd4, Lrp5, and Ndph mutant mice, genes for which mutations in the human orthologs also cause FEVR.13,28,29 In addition, whereas heterozygous Tspan12, Lrp5, and Ndph single-mutant mice did not show any abnormalities, Tspan12/Lrp5 and Tspan12/Ndph double-heterozygous mice demonstrated a retinal vascular phenotype, indicating that these proteins act in a similar pathway. A series of dedicated experiments revealed that TSPAN12 is required for Norrin-induced β-catenin signaling, by promoting FZD4 multimerization and clustering of FZD4 via binding to Norrin multimers.22
A question that remains is how the heterozygous FEVR-mutations described in this study affect the function of TSPAN12 and the complex that it is associated with. The glycine residue at position 188 precedes a highly conserved cysteine among tetraspanin family members involved in the formation of disulfide bridges and is located in the large extracellular loop (EC2) between the third and fourth transmembrane region (Figure 3D). This extracellular loop is known to harbor most of the protein-protein interaction sites described for tetraspanins.30 As such, the substitution of an arginine residue for the glycine at position 188 might disrupt one of these interactions; for instance, the interaction with the Fzd4/Lrp5/Norrin protein complex. The alanine-to-proline change at position 237 is likely to disrupt the three-dimensional structure of TSPAN12. Both haploinsufficiency and a dominant-negative effect of the mutant TSPAN12 on the wild-type protein should be considered as the underlying disease mechanism.
Tetraspanins are suggested to form so-called tetraspanin-enriched microdomains that act as specific signaling compartments within the membrane and might form homomultimers, as well as protein complexes, with nontetraspanin membrane proteins.30,31 In the case that TSPAN12 indeed forms homomultimers, the two missense mutations that cause FEVR might impair multimerization of the protein and, as such, act in a dominant-negative fashion. This would be in line with the absence of any abnormalities in the heterozygous Tspan12 mutant mice.22 Recessive loss-of-function mutations in genes encoding other tetraspanins, such as TM4SF2 (MIM ∗300096) and CD151 (MIM ∗602243), causing X-linked mental retardation and kidney malfunctions, respectively, would favor a dominant-negative mechanism instead of haploinsufficiency.32,33 On the other hand, heterozygous loss-of-function mutations in the PRPH2 gene [MIM ∗179605], encoding the tetraspanin peripherin, cause autosomal-dominant retinal photoreceptor degeneration, favoring a model for haploinsufficiency.34,35 Additional functional studies are needed to determine the effect of the two missense mutations described here, in order to unravel the pathological mechanism in which dominant mutations in TSPAN12 are causative for FEVR.
This study shows the power of next-generation sequencing after initial identification of a disease-gene locus. So far, the only other example of this approach has been reported on by Brkanac et al., which led to the identification of IFRD1 (MIM ∗603502) as a candidate gene for autosomal-dominant sensory and motor neuropathy with ataxia (MIM %607458).36 This targeted resequencing approach may be an attractive alternative to whole-genome or -exome resequencing in those families for which disease loci have been defined. Recently, unbiased genome-wide resequencing of the whole human exome led to the first disease-gene identification in patients with Miller syndrome.37 The number of potential protein-truncating variants in their study was, however, approximately three orders of magnitude higher than those identified by our study. Such a genome-wide approach requires multiple affected individuals to limit the amount of potential candidate genes, especially when operating under an autosomal-dominant disease model.
In conclusion, we have identified TSPAN12 as a gene causative for autosomal-dominant FEVR. TSPAN12 is only the third autosomal gene described to be causative for FEVR and is mutated in five of the 11 Dutch families studied. On the basis of the function of TSPAN12 in regulating the development of the retinal vascular system, it is expected that mutations in TSPAN12 will also be causative in FEVR patients from different populations and, as such, will facilitate genetic diagnostics for FEVR. Moreover, we have proven the enormous power of next-generation sequencing technology in combination with array-based sequence capture, by identifying a single causative base-pair substitution within a 40 Mb region containing more than 300 genes. Given that for many inherited disorders, several loci have been published for which the causative gene still awaits discovery, applying this technology will be instrumental in identifying many disease genes in the near future.
Acknowledgments
The authors thank Christel Beumer, Diana T. Cremers, Michael Kwint, Kornelia Neveling, Saskia D. van der Velde-Visser, and Ilse de Wijs for excellent technical assistance. We also thank all of the participating FEVR patients and their relatives. This study was financially supported by (1) the European Union Research Training Network grant RETNET (MRTN-CT-2003-5040032 to F.P.M.C.); (2) the Netherlands Organization for Health Research and Development (ZonMW grants 917-66-36 and 911-08-025 to J.A.V.); (3) the EU-funded TECHGENE project (Health-F5-2009-223143 to P.A., H.S., and J.A.V.); (4) the AnEUploidy project (LSHG-CT-2006-37627 to A.H. and J.A.V.); (5) the following Dutch blindness foundations: the Algemene Nederlandse Vereniging ter Voorkoming van Blindheid, the F.P. Fischer Stichting, the Gelderse Blinden Stichting, the Landelijke Stichting voor Blinden en Slechtzienden, the Rotterdamse Vereniging Blindenbelangen, the Stichting Blindenhulp, the Stichting Blinden-Penning, the Stichting Nederlands Oogheelkundig Onderzoek, the Stichting OOG, the Stichting voor Ooglijders, the Stichting tot Verbetering van het Lot der Blinden (to F.P.M.C., F.N.B., M.A.D.T., and C.E.v.N.); and (6) the Vereniging Bartiméus (to F.N.B.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
dbSNP, build 130, http://www.ncbi.nlm.nih.gov/projects/SNP/snp_summary.cgi?build_id=130
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
UniProt, http://www.uniprot.org
University of California-Santa Cruz (UCSC) Genome Bioinformatics, http://www.genome.ucsc.edu
References
- 1.Criswick V.G., Schepens C.L. Familial exudative vitreoretinopathy. Am. J. Ophthalmol. 1969;68:578–594. doi: 10.1016/0002-9394(69)91237-9. [DOI] [PubMed] [Google Scholar]
- 2.Canny C.L.B., Oliver G.L. Fluorescein angiographic findings in familial exudative vitreoretinopathy. Arch. Ophthalmol. 1976;94:1114–1120. doi: 10.1001/archopht.1976.03910040034006. [DOI] [PubMed] [Google Scholar]
- 3.Boonstra F.N., van Nouhuys C.E., Schuil J., de Wijs I.J., van der Donk K.P., Nikopoulos K., Mukhopadhyay A., Scheffer H., Tilanus M.A.D., Cremers F.P.M. Clinical and molecular evaluation of probands and family members with familial exudative vitreoretinopathy. Invest. Ophthalmol. Vis. Sci. 2009;50:4379–4385. doi: 10.1167/iovs.08-3320. [DOI] [PubMed] [Google Scholar]
- 4.Chen Z.Y., Battinelli E.M., Fielder A., Bundey S., Sims K., Breakefield X.O., Craig I.W. A mutation in the Norrie disease gene (NDP) associated with X-linked familial exudative vitreoretinopathy. Nat. Genet. 1993;5:180–183. doi: 10.1038/ng1093-180. [DOI] [PubMed] [Google Scholar]
- 5.Feldman E.L., Norris J.L., Cleasby G.W. Autosomal dominant exudative vitreoretinopathy. Arch. Ophthalmol. 1983;101:1532–1535. doi: 10.1001/archopht.1983.01040020534004. [DOI] [PubMed] [Google Scholar]
- 6.Gow J., Oliver G.L. Familial exudative vitreoretinopathy. An expanded view. Arch. Ophthalmol. 1971;86:150–155. doi: 10.1001/archopht.1971.01000010152007. [DOI] [PubMed] [Google Scholar]
- 7.Jiao X., Ventruto V., Trese M.T., Shastry B.S., Hejtmancik J.F. Autosomal recessive familial exudative vitreoretinopathy is associated with mutations in LRP5. Am. J. Hum. Genet. 2004;75:878–884. doi: 10.1086/425080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Nouhuys C.E. Dominant exudative vitreoretinopathy and other vascular developmental disorders of the peripheral retina. Doc. Ophthalmol. 1982;54 doi: 10.1007/BF00183127. 1–415. [DOI] [PubMed] [Google Scholar]
- 9.van Nouhuys C.E. Dominant exudative vitreoretinopathy. Ophthalmic Paediatr. Genet. 1985;5:31–38. doi: 10.3109/13816818509007853. [DOI] [PubMed] [Google Scholar]
- 10.Chen Z.Y., Battinelli E.M., Woodruff G., Young I., Breakefield X.O., Craig I.W. Characterization of a mutation within the NDP gene in a family with a manifesting female carrier. Hum. Mol. Genet. 1993;2:1727–1729. doi: 10.1093/hmg/2.10.1727. [DOI] [PubMed] [Google Scholar]
- 11.Robitaille J.M., Wallace K., Zheng B.Y., Beis M.J., Samuels M., Hoskin-Mott A., Guernsey D.L. Phenotypic overlap of familial exudative vitreoretinopathy (FEVR) with persistent fetal vasculature (PFV) caused by FZD4 mutations in two distinct pedigrees. Ophthalmic Genet. 2009;30:23–30. doi: 10.1080/13816810802464312. [DOI] [PubMed] [Google Scholar]
- 12.Toomes C., Bottomley H.M., Scott S., Mackey D.A., Craig J.E., Appukuttan B., Stout J.T., Flaxel C.J., Zhang K., Black G.C. Spectrum and frequency of FZD4 mutations in familial exudative vitreoretinopathy. Invest. Ophthalmol. Vis. Sci. 2004;45:2083–2090. doi: 10.1167/iovs.03-1044. [DOI] [PubMed] [Google Scholar]
- 13.Xu Q., Wang Y., Dabdoub A., Smallwood P.M., Williams J., Woods C., Kelley M.W., Jiang L., Tasman W., Zhang K. Vascular development in the retina and inner ear: control by Norrin and Frizzled-4, a high-affinity ligand-receptor pair. Cell. 2004;116:883–895. doi: 10.1016/s0092-8674(04)00216-8. [DOI] [PubMed] [Google Scholar]
- 14.Hoffmann K., Lindner T.H. easyLINKAGE-Plus–automated linkage analyses using large-scale SNP data. Bioinformatics. 2005;21:3565–3567. doi: 10.1093/bioinformatics/bti571. [DOI] [PubMed] [Google Scholar]
- 15.Fruttiger M. Development of the retinal vasculature. Angiogenesis. 2007;10:77–88. doi: 10.1007/s10456-007-9065-1. [DOI] [PubMed] [Google Scholar]
- 16.Ning Z., Cox A.J., Mullikin J.C. SSAHA: a fast search method for large DNA databases. Genome Res. 2001;11:1725–1729. doi: 10.1101/gr.194201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murphy W.J., Eizirik E., O'Brien S.J., Madsen O., Scally M., Douady C.J., Teeling E., Ryder O.A., Stanhope M.J., de Jong W.W. Resolution of the early placental mammal radiation using Bayesian phylogenetics. Science. 2001;294:2348–2351. doi: 10.1126/science.1067179. [DOI] [PubMed] [Google Scholar]
- 18.Siepel A., Bejerano G., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Siepel A., Pollard K.S., Haussler D. Research in Computational Molecular Biology. Springer Berlin / Heidelberg; 2006. New methods for detecting lineage-specific selection. pp. 190–205. [Google Scholar]
- 20.Rackham O., Davies S.M., Shearwood A.M., Hamilton K.L., Whelan J., Filipovska A. Pentatricopeptide repeat domain protein 1 lowers the levels of mitochondrial leucine tRNAs in cells. Nucleic Acids Res. 2009;37:5859–5867. doi: 10.1093/nar/gkp627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bi M., Hickox J.R., Winfrey V.P., Olson G.E., Hardy D.M. Processing, localization and binding activity of zonadhesin suggest a function in sperm adhesion to the zona pellucida during exocytosis of the acrosome. Biochem. J. 2003;375:477–488. doi: 10.1042/BJ20030753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Junge H.J., Yang S., Burton J.B., Paes K., Shu X., French D.M., Costa M., Rice D.S., Ye W. TSPAN12 regulates retinal vascular development by promoting Norrin- but not Wnt-induced FZD4/beta-catenin signaling. Cell. 2009;139:299–311. doi: 10.1016/j.cell.2009.07.048. [DOI] [PubMed] [Google Scholar]
- 23.Ng S.B., Turner E.H., Robertson P.D., Flygare S.D., Bigham A.W., Lee C., Shaffer T., Wong M., Bhattacharjee A., Eichler E.E. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–276. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pushkarev D., Neff N.F., Quake S.R. Single-molecule sequencing of an individual human genome. Nat. Biotechnol. 2009;27:847–852. doi: 10.1038/nbt.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang J., Wang W., Li R., Li Y., Tian G., Goodman L., Fan W., Zhang J., Li J., Zhang J. The diploid genome sequence of an Asian individual. Nature. 2008;456:60–65. doi: 10.1038/nature07484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garcia-Espana A., Chung P.J., Sarkar I.N., Stiner E., Sun T.T., Desalle R. Appearance of new tetraspanin genes during vertebrate evolution. Genomics. 2008;91:326–334. doi: 10.1016/j.ygeno.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 27.Hemler M.E. Specific tetraspanin functions. J. Cell Biol. 2001;155:1103–1107. doi: 10.1083/jcb.200108061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luhmann U.F.O., Neidhardt J., Kloeckener-Gruissem B., Schafer N.F., Glaus E., Feil S., Berger W. Vascular changes in the cerebellum of Norrin/Ndph knockout mice correlate with high expression of Norrin and Frizzled-4. Eur. J. Neurosci. 2008;27:2619–2628. doi: 10.1111/j.1460-9568.2008.06237.x. [DOI] [PubMed] [Google Scholar]
- 29.Xia C.H., Liu H.Q., Cheung D., Wang M., Cheng C., Du X., Chang B., Beutler B., Gong X. A model for familial exudative vitreoretinopathy caused by LPR5 mutations. Hum. Mol. Genet. 2008;17:1605–1612. doi: 10.1093/hmg/ddn047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hemler M.E. Tetraspanin functions and associated microdomains. Nat. Rev. Mol. Cell Biol. 2005;6:801–811. doi: 10.1038/nrm1736. [DOI] [PubMed] [Google Scholar]
- 31.Boucheix C., Rubinstein E. Tetraspanins. Cell. Mol. Life Sci. 2001;58:1189–1205. doi: 10.1007/PL00000933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karamatic Crew V., Burton N., Kagan A., Green C.A., Levene C., Flinter F., Brady R.L., Daniels G., Anstee D.J. CD151, the first member of the tetraspanin (TM4) superfamily detected on erythrocytes, is essential for the correct assembly of human basement membranes in kidney and skin. Blood. 2004;104:2217–2223. doi: 10.1182/blood-2004-04-1512. [DOI] [PubMed] [Google Scholar]
- 33.Zemni R., Bienvenu T., Vinet M.C., Sefiani A., Carrie A., Billuart P., McDonell N., Couvert P., Francis F., Chafey P. A new gene involved in X-linked mental retardation identified by analysis of an X;2 balanced translocation. Nat. Genet. 2000;24:167–170. doi: 10.1038/72829. [DOI] [PubMed] [Google Scholar]
- 34.Keen T.J., Inglehearn C.F. Mutations and polymorphisms in the human peripherin-RDS gene and their involvement in inherited retinal degeneration. Hum. Mutat. 1996;8:297–303. doi: 10.1002/(SICI)1098-1004(1996)8:4<297::AID-HUMU1>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 35.Kohl S., Christ-Adler M., Apfelstedt-Sylla E., Kellner U., Eckstein A., Zrenner E., Wissinger B. RDS/peripherin gene mutations are frequent causes of central retinal dystrophies. J. Med. Genet. 1997;34:620–626. doi: 10.1136/jmg.34.8.620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brkanac Z., Spencer D., Shendure J., Robertson P.D., Matsushita M., Vu T., Bird T.D., Olson M.V., Raskind W.H. IFRD1 is a candidate gene for SMNA on chromosome 7q22-q23. Am. J. Hum. Genet. 2009;84:692–697. doi: 10.1016/j.ajhg.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ng S.B., Buckingham K.J., Lee C., Bigham A.W., Tabor H.K., Dent K.M., Huff C.D., Shannon P.T., Jabs E.W., Nickerson D.A. Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet. 2010;42:30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.