

Abstract
Familial exudative vitreoretinopathy (FEVR) is an inherited blinding disorder of the retinal vascular system. Although mutations in three genes (LRP5, FZD4, and NDP) are known to cause FEVR, these account for only a fraction of FEVR cases. The proteins encoded by these FEVR genes form part of a signaling complex that activates the Norrin-β-catenin signaling pathway. Recently, through a large-scale reverse genetic screen in mice, Junge and colleagues identified an additional member of this signaling complex, Tspan12. Here, we report that mutations in TSPAN12 also cause autosomal-dominant FEVR. We describe seven mutations identified in a cohort of 70 FEVR patients in whom we had already excluded the known FEVR genes. This study provides further evidence for the importance of the Norrin-β-catenin signaling pathway in the development of the retinal vasculature and also indicates that more FEVR genes remain to be identified.
Main Text
Familial exudative vitreoretinopathy (FEVR) is an inherited disorder characterized by the incomplete development of the retinal vasculature (MIM 133780).1 Its clinical appearance varies considerably, even within families, with severely affected patients often registered as blind during infancy and mildly affected patients having few or no visual problems. These mildly affected patients may have only a small area of avascularity in their peripheral retina, visible only by fluorescein angiography, and patients usually undergo this invasive exam only when a severely affected family member is diagnosed. It is believed that this peripheral avascularity is the primary anomaly in FEVR and results from defective retinal angiogenesis. The sight-threatening features of the FEVR phenotype are considered secondary to retinal avascularity and develop because of the resulting retinal ischemia. They include the development of hyperpermeable blood vessels, neovascularisation, vitreo-retinal traction, retinal folds, and retinal detachments (Figure 1).2
Figure 1.
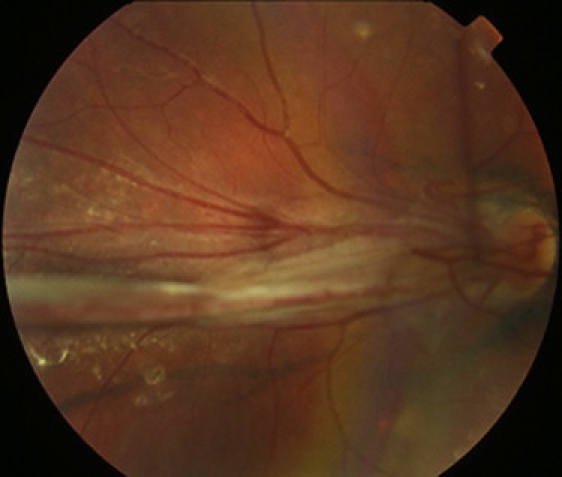
Clinical Appearance of FEVR
Fundus photograph of the right eye of the proband from family 1 (individual III:2 in Figure S1), showing the retinal vessels drawn up in a retinal fold that is obscuring the macula.
FEVR is genetically heterogeneous and can be inherited as a dominant, recessive, or X-linked trait. Three FEVR genes have been identified to date: NDP (MIM 300658, X-linked), FZD4 (MIM 604579, dominant), and LRP5 (MIM 603506, dominant and recessive).3–6 Mutation screening of these genes suggests that they account for less than half of all FEVR cases, indicating that additional genes remain to be identified.7–12 However, only one additional locus has been mapped to chromosome 11p12-p13: autosomal-dominant EVR3.13
The proteins encoded by the three FEVR genes have all been shown to participate in the same molecular pathway, the Norrin-β-catenin signaling pathway.14,15 This pathway is similar to the Wnt-β-catenin signaling pathway with the exception that the Wnt ligand is replaced by Norrin, the protein encoded by NDP. FZD4 and LRP5 encode the receptors Frizzled-4 and low-density lipoprotein receptor-related protein-5, and these two proteins act as coreceptors for Wnts and Norrin. In the absence of the Wnt or Norrin ligand, signaling is not activated in a cell. This results in cytoplasmic β-catenin becoming phosphorylated and targeted for degradation through the ubiquitin-proteasome pathway. As a result, prospective target genes remain repressed. Signaling is activated by Wnt or Norrin binding at the cell surface to the Frizzled-4 and LRP5 receptor complex. These receptors transduce a signal that inhibits the destruction of β-catenin, allowing it to accumulate within the cytoplasm. Subsequently, β-catenin translocates to the nucleus, where it interacts with the T cell factor (TCF)/lymphoid enhancing factor (LEF) family of transcription factors to turn on the expression of Wnt and/or Norrin target genes.16
A number of mouse models of FEVR have been created by disrupting the sequence of the FEVR genes.17–21 Studies have shown that all of these mice have defects in the development of the superficial retinal vasculature and are missing the two intraretinal capillary beds.21–24 Recently, Junge and colleagues created Tspan12 mutant mice as part of a large-scale reverse genetic screen and showed that they had a phenotype similar to that of the FEVR mouse models.24 Prompted by this finding, they showed that Tspan12 is a component of the Norrin-LRP5-FZD4 signaling complex and enhances the level of Norrin-β-catenin signaling but not Wnt-β-catenin signaling.24
These results suggest that TSPAN12 (MIM 613138) on chromosome 7q31.31 is an excellent candidate for involvement in FEVR, and we therefore screened this gene in a panel of 70 FEVR patients. Informed consent was obtained from all subjects tested, and ethical approval was obtained from the Leeds Teaching Hospitals Trust Research Ethics Committee. We designed primers (see Supplemental Data, available online) and screened all seven coding exons and flanking intronic sequences by direct sequencing, using standard protocols. In total, we discovered seven heterozygous TSPAN12 mutations not present in controls (Figure 2).
Figure 2.
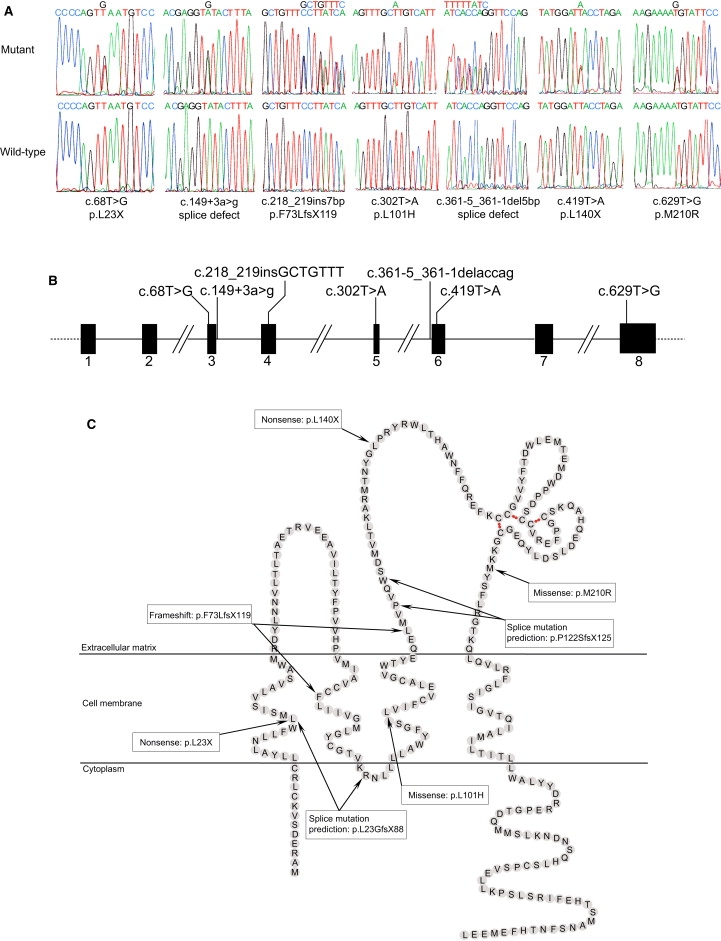
FEVR Is Caused by Mutations in TSPAN12
(A) Sequence traces of the seven mutations identified and the corresponding wild-type alleles.
(B) Schematic diagram of TSPAN12, showing the locations of the mutations.
(C) Schematic diagram of the TSPAN12 protein, showing the location of the mutations within the protein domains. The locations of the transmembrane domains were obtained from Kovalenko et al., 2005.32 The intramolecular disulfide bonds crucial for the correct folding of large extracellular loop are indicated by red dots. Because we were unable to assess the actual consequences of the two splicing mutations, only predicted protein outcomes are shown.
We first identified a 7 bp insertion in exon 4, c.218_219insGCTCTTT, in an Australian family of European descent (family 1). This mutation causes a frameshift resulting in 45 incorrect amino acids after codon 72, followed by premature termination at codon 118 (p.F73LfsX118). The female proband in this family shows macula ectopia, with a large retinal fold across the fovea of her right eye and fibrovascular changes in the temporal periphery of her left eye (Figure 1). Her asymptomatic father also carries the mutation and has bilateral peripheral retinal pigmentary disturbances that are reminiscent of the bone-spicule pattern more commonly associated with retinitis pigmentosa (Supplemental Data). This unusual finding was interpreted as being old exudative retinal detachments that have spontaneously resolved. The proband's asymptomatic brother was also found to carry the mutation, but fundus examination revealed no signs of disease. However, he was only 10 yrs old when examined, and fundus fluorescein angiography was not performed, so a mild phenotype could not be excluded.25
In a second family, originating from Japan (family 2), we identified a nonsense mutation in exon 6, c.419T>A, segregating with the disease. This mutation causes the TSPAN12 protein to be truncated from 305 amino acids to 139 (p.L140X). The female proband was diagnosed in infancy with bilateral retinal folds. Her mutation-carrying father is asymptomatic but has areas of avascularity and abnormal vessels in the peripheral retina.
The third mutation identified was a nonsense change in exon 3, c.68T>G, in an isolated FEVR patient from the USA. This mutation is predicted to encode a significantly truncated protein of only 22 amino acids (p.L23X). The patient is a 6-yr-old white female with bilateral retinal folds and unilateral, persistent hyperplastic primary vitreous (persistent fetal vasculature). No DNA was available from other family members, but none reported eye problems.
The fourth mutation identified was a 5 bp deletion at the end of intron 5, c.361-1_361-5delACCAG, in an isolated FEVR patient from the UK. This mutation removes the splice acceptor site including the consensus AG. The precise effect of this mutation has not been determined because RNA was unavailable. However, possible outcomes include the deletion of exon 6 (which would result in the in-frame deletion of the 36 amino acids encoded by exon 6), the retention of intron 5 (which would result in a frameshift and premature-termination codon, p.P122SfsX125), or the activation of an unknown cryptic splice site. The white female patient had no family history of the disease and was not diagnosed herself until the fifth decade of life. She has bilateral temporal retinal avascularity and associated areas of exudation visible with fundus fluorescein angiography. In addition, both eyes show traction of the retinal vasculature at the posterior pole.
We identified a second putative splice-site mutation at the start of intron 4, c.149+3A>G, in an American family originating from Europe (family 3). No RNA was available for testing of the pathogenic nature of this DNA change, but it was excluded from over 500 ethnically matched control chromosomes. This mutation is in the splice donor site at the end of exon 3, and the most common outcome for these types of mutations is the deletion of the preceding exon (which would result in a frameshift and a premature-termination codon, p.L23GfsX88). The proband of the family is an 8-yr-old female with bilateral retinal folds affecting the macula. Her mutation-carrying mother is asymptomatic but was noted to have bilateral peripheral retinal pigment pallor, possibly indicating avascularity. A maternal cousin, who also carries the mutation, was diagnosed with a unilateral retinal fold in infancy. Her mutation-carrying mother (proband's maternal aunt) showed no signs of FEVR, although she was not examined by fundus fluorescein angiography. A maternal uncle was reported to have undergone a spontaneous retinal detachment at age 27, but no DNA was available for checking whether he carried the mutation.
The remaining two mutations were missense changes. We identified p.L101H (c.302T>A) in exon 5 in a white British family (family 4). The male proband and his father both carried the mutation and showed classic signs of FEVR, with the proband being severe and the father mild. We identified p.M210R (c.629T > G) in exon 8 in a white Australian male patient with bilateral retinal detachments. This patient had no history of prematurity and presented at 5 yrs of age with a divergent squint. Upon examination he was found to have bilateral macular traction and exudates in the temporal retina. DNA was not available for family members, but there is no reported family history of disease. To exclude the possibility that these changes were common polymorphisms, we screened 200 ethnically matched control individuals (400 chromosomes) for each of these changes.26 We also checked each of the mutated amino acids for conservation within orthologs of TSPAN12 (Figure 3). Both p.L101H and p.M210R are changes to highly conserved residues, and both mutations have negative Blosum62 scores (−3 and −1, respectively).27 Although these results suggest that these missense changes are indeed pathogenic, without further examination by means of a functional assay, we are unable to categorically prove that they are disease-causing mutations.
Figure 3.

Protein Sequence Alignment of Human TSPAN12 with Its Orthologs
Alignments were calculated with ClustalW.33 Accession numbers: Human, O95859; Orangutan, Q5R8B5; Mouse, AAH68240; Rat, Q569A2; Cattle, Q29RH7; Horse, XP_001502093; Dog, XP_855095; Opossum, XP_001364876; Chicken, XP_416007; Zebrafinch, XP_002192381; Platypus, XP_001516347; Zebrafish, NP_957446; Seasquirt Tspan,12 XP_002123238. Only 30 amino acid residues surrounding each mutation are shown. Conserved amino acid residues are highlighted. The positions of the missense mutations p.L101H and p.M210R are indicated.
All types of mutations were identified (insertion, deletion, nonsense, missense, and splicing), and at least four of these are predicted to lead to transcripts with premature-termination codons that are likely to be targeted by nonsense-mediated decay. Furthermore, there was no difference in phenotype between the patients with truncating mutations and those with missense changes. We therefore propose that haploinsufficency of TSPAN12 is the cause of FEVR. From the clinical phenotypes observed in the patients with TSPAN12 mutations, it is clear that there is no correlation between particular mutations or mutation types and phenotype for this form of FEVR. The eye phenotypes vary in a manner similar to those reported in FZD4, NDP, and LRP5 mutation carriers. LRP5 mutation carriers therefore remain the only subset of FEVR patients that can be clinically distinguished by the presence of low bone-mass density.5,9,28
TSPAN12 is a member of the tetraspanin superfamily, of which there are 33 members in humans.29 Tetraspanins contain four transmembrane domains with both the N and C termini located in the cytoplasm. The transmembrane domains are linked by three loops: a small extracellular loop, a large extracellular loop, and a tiny inner loop (Figure 2C). Members of this protein family are characterized by their ability to interact laterally with other tetraspanins and interacting proteins, thereby facilitating the production of large multimolecular membrane complexes, also called tetraspanin webs.30 Through a number of cell-based assays and genetic interaction screens, Junge and colleagues showed that TSPAN12 associates with the Norrin-FZD4-LRP5 signaling complex and that this association results in an increased level of Norrin-β-catenin signaling, assayed with the Topflash reporter assay. Using the same assay, they also showed that overexpression of TSPAN12 could rescue the reduction in β-catenin signaling observed with either mutant Norrin or mutant FZD4 proteins (p.C95R and p.M147V, respectively). On the basis of these results, the authors hypothesize that Norrin and TSPAN12 cooperatively induce the multimerization of the FZD4-LRP5 complex to induce β-catenin signaling.24 It is interesting to note that both of the missense mutations identified in this study are located in regions that are predicted to play a role in tetraspanin homo- and heterodimerization, indicating that they may disrupt the multimerization of TSPAN12.31 The identification of additional TSPAN12 mutations in the future will enable a more detailed analysis of the domains of TSPAN12 that are functionally important in Norrin-β-catenin signaling.
In summary, we have shown that heterozygous mutations in TSPAN12 can cause autosomal-dominant FEVR. Mutations in this gene accounted for 10% of FEVR cases in our patient series, although this figure is inflated because the majority of these patients had already been excluded as carrying mutations in FZD4, LRP5, and NDP. This result provides further evidence of significant genetic heterogeneity in FEVR and indicates that other FEVR genes remain to be found. In addition, this result confirms the importance of the Norrin-β-catenin signaling pathway in the development of the retinal vasculature and suggests that the remaining FEVR genes will also be part of this new signaling pathway.
Acknowledgments
We thank the FEVR families for their help in this study. The financial support of The Royal Society (C.T. is a Royal Society University Research Fellow) and the Wellcome Trust (080313/Z/06) is gratefully acknowledged. J.A.P. is supported by an Emma and Leslie Reid Scholarship from the University of Leeds.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Clustal W, http://www.ebi.ac.uk/clustalw2/
NCBI protein database, http://www.ncbi.nlm.nih.gov/protein
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
References
- 1.Criswick V.G., Schepens C.L. Familial exudative vitreoretinopathy. Am. J. Ophthalmol. 1969;68:578–594. doi: 10.1016/0002-9394(69)91237-9. [DOI] [PubMed] [Google Scholar]
- 2.Benson W.E. Familial exudative vitreoretinopathy. Trans. Am. Ophthalmol. Soc. 1995;93:473–521. [PMC free article] [PubMed] [Google Scholar]
- 3.Chen Z.Y., Battinelli E.M., Fielder A., Bundey S., Sims K., Breakefield X.O., Craig I.W. A mutation in the Norrie disease gene (NDP) associated with X-linked familial exudative vitreoretinopathy. Nat. Genet. 1993;5:180–183. doi: 10.1038/ng1093-180. [DOI] [PubMed] [Google Scholar]
- 4.Robitaille J., MacDonald M.L.E., Kaykas A., Sheldahi L.C., Zeisler J., Dube M.P., Zhang L.H., Singaraja R.R., Guernsey D.L., Zheng B. Mutant Frizzled-4 disrupts retinal angiogenesis in familial exudative vitreoretinopathy. Nat. Genet. 2002;32:326–330. doi: 10.1038/ng957. [DOI] [PubMed] [Google Scholar]
- 5.Toomes C., Bottomley H.M., Jackson R.M., Towns K.V., Scott S., Mackey D.A., Craig J.E., Jiang L., Yang Z., Trembath R. Mutations in LRP5 or FZD4 underlie the common familial exudative vitreoretinopathy locus on chromosome 11q. Am. J. Hum. Genet. 2004;74:721–730. doi: 10.1086/383202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiao X., Ventruto V., Trese M.T., Shastry B.S., Hejtmancik J.F. Autosomal recessive familial exudative vitreoretinopathy is associated with mutations in LRP5. Am. J. Hum. Genet. 2004;75:878–884. doi: 10.1086/425080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kondo H., Hayashi H., Oshima K., Tahira T., Hayashi K. Frizzled 4 Gene (FZD4) mutations in patients with familial exudative vitreoretinopathy with variable expressivity. Br. J. Ophthalmol. 2003;87:1291–1295. doi: 10.1136/bjo.87.10.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Toomes C., Bottomley H.M., Scott S., Mackey D.A., Craig J.E., Appukuttan B., Stout J.T., Flaxel C.J., Zhang K., Black G.C. Spectrum and frequency of FZD4 mutations in familial exudative vitreoretinopathy. Invest. Ophthalmol. Vis. Sci. 2004;45:2083–2090. doi: 10.1167/iovs.03-1044. [DOI] [PubMed] [Google Scholar]
- 9.Qin M., Hayashi H., Oshima K., Tahira T., Hayashi K., Kondo H. Complexity of the genotype-phenotype correlation in familial exudative vitreoretinopathy with mutations in the LRP5 and/or FZD4 genes. Hum. Mutat. 2005;26:104–112. doi: 10.1002/humu.20191. [DOI] [PubMed] [Google Scholar]
- 10.Nallathambi J., Shukla D., Rajendran A., Namperumalsamy P., Muthulakshmi R., Sundaresan P. Identification of novel FZD4 mutations in Indian patients with familial exudative vitreoretinopathy. Mol. Vis. 2006;12:1086–1092. [PubMed] [Google Scholar]
- 11.Kondo H., Qin M., Kusaka S., Tahira T., Hasebe H., Hayashi H., Uchio E., Hayashi K. Novel mutations in Norrie disease gene in Japanese patients with Norrie disease and familial exudative vitreoretinopathy. Invest. Ophthalmol. Vis. Sci. 2007;48:1276–1282. doi: 10.1167/iovs.06-1042. [DOI] [PubMed] [Google Scholar]
- 12.Boonstra F.N., van Nouhuys C.E., Schuil J., de Wijs I.J., van der Donk K.P., Nikopoulos K., Mukhopadhyay A., Scheffer H., Tilanus M.A., Cremers F.P., Hoefsloot L.H. Clinical and molecular evaluation of probands and family members with familial exudative vitreoretinopathy. Invest. Ophthalmol. Vis. Sci. 2009;50:4379–4385. doi: 10.1167/iovs.08-3320. [DOI] [PubMed] [Google Scholar]
- 13.Downey L.M., Keen T.J., Roberts E., Mansfield D.C., Bamashmus M., Inglehearn C.F. A new locus for autosomal dominant familial exudative vitreoretinopathy maps to chromosome 11p12-13. Am. J. Hum. Genet. 2001;68:778–781. doi: 10.1086/318790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ye X., Wang Y., Cahill H., Yu M., Badea T.C., Smallwood P.M., Peachey N.S., Nathans J. Norrin, frizzled-4, and Lrp5 signaling in endothelial cells controls a genetic program for retinal vascularization. Cell. 2009;139:285–298. doi: 10.1016/j.cell.2009.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clevers H. Eyeing up new Wnt pathway players. Cell. 2009;139:227–229. doi: 10.1016/j.cell.2009.09.027. [DOI] [PubMed] [Google Scholar]
- 16.van Amerongen R., Nusse R. Towards an integrated view of Wnt signaling in development. Development. 2009;136:3205–3214. doi: 10.1242/dev.033910. [DOI] [PubMed] [Google Scholar]
- 17.Berger W., van de Pol D., Bächner D., Oerlemans F., Winkens H., Hameister H., Wieringa B., Hendriks W., Ropers H.H. An animal model for Norrie disease (ND): gene targeting of the mouse ND gene. Hum. Mol. Genet. 1996;5:51–59. doi: 10.1093/hmg/5.1.51. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y., Huso D., Cahill H., Ryugo D., Nathans J. Progressive cerebellar, auditory, and esophageal dysfunction caused by targeted disruption of the frizzled-4 gene. J. Neurosci. 2001;21:4761–4771. doi: 10.1523/JNEUROSCI.21-13-04761.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kato M., Patel M.S., Levasseur R., Lobov I., Chang B.H., Glass D.A., 2nd, Hartmann C., Li L., Hwang T.H., Brayton C.F. Cbfa1-independent decrease in osteoblast proliferation, osteopenia, and persistent embryonic eye vascularization in mice deficient in Lrp5, a Wnt coreceptor. J. Cell Biol. 2002;157:303–314. doi: 10.1083/jcb.200201089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holmen S.L., Giambernardi T.A., Zylstra C.R., Buckner-Berghuis B.D., Resau J.H., Hess J.F., Glatt V., Bouxsein M.L., Ai M., Warman M.L., Williams B.O. Decreased BMD and limb deformities in mice carrying mutations in both Lrp5 and Lrp6. J. Bone Miner. Res. 2004;19:2033–2040. doi: 10.1359/JBMR.040907. [DOI] [PubMed] [Google Scholar]
- 21.Xia C.H., Liu H., Cheung D., Wang M., Cheng C., Du X., Chang B., Beutler B., Gong X. A model for familial exudative vitreoretinopathy caused by LPR5 mutations. Hum. Mol. Genet. 2008;17:1605–1612. doi: 10.1093/hmg/ddn047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu Q., Wang Y., Dabdoub A., Smallwood P.M., Williams J., Woods C., Kelley M.W., Jiang L., Tasman W., Zhang K., Nathans J. Vascular development in the retina and inner ear: control by Norrin and Frizzled-4, a high-affinity ligand-receptor pair. Cell. 2004;116:883–895. doi: 10.1016/s0092-8674(04)00216-8. [DOI] [PubMed] [Google Scholar]
- 23.Luhmann U.F., Lin J., Acar N., Lammel S., Feil S., Grimm C., Seeliger M.W., Hammes H.P., Berger W. Role of the Norrie disease pseudoglioma gene in sprouting angiogenesis during development of the retinal vasculature. Invest. Ophthalmol. Vis. Sci. 2005;46:3372–3382. doi: 10.1167/iovs.05-0174. [DOI] [PubMed] [Google Scholar]
- 24.Junge H.J., Yang S., Burton J.B., Paes K., Shu X., French D.M., Costa M., Rice D.S., Ye W. TSPAN12 regulates retinal vascular development by promoting Norrin- but not Wnt-induced FZD4/beta-catenin signaling. Cell. 2009;139:299–311. doi: 10.1016/j.cell.2009.07.048. [DOI] [PubMed] [Google Scholar]
- 25.Ober R.R., Bird A.C., Hamilton A.M., Sehmi K. Autosomal dominant exudative vitreoretinopathy. Br. J. Ophthalmol. 1980;64:112–120. doi: 10.1136/bjo.64.2.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Collins J.S., Schwartz C.E. Detecting polymorphisms and mutations in candidate genes. Am. J. Hum. Genet. 2002;71:1251–1252. doi: 10.1086/344344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Henikoff S., Henikoff J.G. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Downey L.M., Bottomley H.M., Sheridan E., Ahmed M., Gilmour D.F., Inglehearn C.F., Reddy A., Agrawal A., Bradbury J., Toomes C. Reduced bone mineral density and hyaloid vasculature remnants in a consanguineous recessive FEVR family with a mutation in LRP5. Br. J. Ophthalmol. 2006;90:1163–1167. doi: 10.1136/bjo.2006.092114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garcia-España A., Chung P.J., Sarkar I.N., Stiner E., Sun T.T., Desalle R. Appearance of new tetraspanin genes during vertebrate evolution. Genomics. 2008;91:326–334. doi: 10.1016/j.ygeno.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 30.Hemler M.E. Tetraspanin functions and associated microdomains. Nat. Rev. Mol. Cell Biol. 2005;6:801–811. doi: 10.1038/nrm1736. [DOI] [PubMed] [Google Scholar]
- 31.Stipp C.S., Kolesnikova T.V., Hemler M.E. Functional domains in tetraspanin proteins. Trends Biochem. Sci. 2003;28:106–112. doi: 10.1016/S0968-0004(02)00014-2. [DOI] [PubMed] [Google Scholar]
- 32.Kovalenko O.V., Metcalf D.G., DeGrado W.F., Hemler M.E. Structural organization and interactions of transmembrane domains in tetraspanin proteins. BMC Struct. Biol. 2005;28 doi: 10.1186/1472-6807-5-11. 5:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chenna R., Sugawara H., Koike T., Lopez R., Gibson T.J., Higgins D.G., Thompson J.D. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003;31:3497–3500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.