

Abstract
Purpose of review
Polycystic kidney disease (PKD) is the most common genetic cause of chronic renal failure. Mouse models of PKD, especially those with mutations in genes that are orthologous to human disease genes, have provided insights into the pathogenesis of cyst formation and advanced the preclinical testing of new drugs.
Recent findings
PKD is a ciliopathy that arises from abnormalities in the primary cilium, a sensory organelle present on the surface of most cells. The primary cilium is required for the maintenance of planar cell polarity, which regulates tubular diameter. Acute kidney injury stimulates cell proliferation and promotes cyst formation in a mouse model of PKD. Studies of signaling pathways that are perturbed in PKD have identified new potential therapeutic targets. Drugs that have shown beneficial effects in orthologous animal models of PKD include tolvaptan, octreotide, src inhibitors, CFTR inhibitors, pioglitazone, etanercept, and triptolide.
Summary
Abnormalities in the primary cilium perturb signaling pathways that regulate renal epithelial cell growth and differentiation and lead to the formation of kidney cysts. Acute kidney injury promotes cyst formation and may underlie the variability in disease progression that is observed in affected individuals. Several promising new therapeutic agents that have been validated in orthologous animal models have entered clinical trials in humans.
Keywords: Polycystic kidney disease, cilia, planar cell polarity, tolvaptan, rapamycin, acute kidney injury
Introduction
Polycystic kidney disease (PKD), the most common genetic cause of chronic kidney disease, is characterized by the accumulation of numerous fluid filled cysts in the renal parenchyma. The cysts originate from the renal tubules and are lined by a single layer of epithelial cells called the cyst epithelium. Over time, the cysts progressively increase in size due to increased rates of proliferation and active secretion of fluid by the cyst epithelium. The enlarging cysts compress surrounding normal nephrons resulting in a decline of renal function. In end-stage kidneys, the cysts are surrounded by areas of fibrosis containing atrophic tubules. PKD can be inherited as an autosomal dominant trait or an autosomal recessive trait. The autosomal dominant form of PKD (ADPKD) primarily affects adults and is caused by mutations in the PKD1 or PKD2 genes, which encode the proteins polycystin-1 and polycystin-2, respectively. Clinically, adults with ADPKD present with enlarged kidneys, abdominal pain, hematuria, and infected kidney cysts. Approximately half of the individuals affected with ADPKD will develop end-stage renal disease (ESRD) [1] The autosomal recessive form of PKD (ARPKD) primarily affects infants and children and is caused by mutations in the PKHD1 gene, which encodes the protein fibrocystin. ARPKD may present in neonates with massive kidney enlargement, intrauterine renal failure, oligohydramnios, and pulmonary hypoplasia or may present later in life with renal insufficiency accompanied by systemic and portal hypertension.
Primary cilia
Recent studies suggest that both the dominant and recessive forms of PKD arise from abnormalities in a cellular organelle called the primary cilium [2]. The primary cilium is a hairlike structure that can be found on the surface of most cells in the body. It consists of a bundle of microtubules, called the axoneme, surrounded by a membrane that is continuous with the cell membrane [3]. The primary cilium is anchored in the cell body by the basal body, which also functions as a centriole during mitosis. Cilia in the body can be classified into two major types based on the structure of their axonemes. Motile cilia, such as those in the respiratory tract, contain an axoneme that is composed of nine microtubule doublets surrounding two central microtubules (9+2 pattern). In contrast, most primary cilia are non-motile and contain nine peripheral microtubule doublets but lack the two central microtubules (9+0 pattern). In the kidney, a single, immotile primary (9+0) cilium is present on the apical surface of most epithelial cells composing the renal tubules. Renal cilia project into the tubular lumen and are believed to function as mechanosensors of urine flow. Fluid flows over the apical surface of the cells, bends the primary cilium, and produces an increase in intracellular calcium concentration, [Ca2+]i, which regulates cell signaling pathways [4].
Three lines of evidence suggest that PKD can be considered a ciliopathy or a disorder of primary cilia. First, polycystin-1, polycystin-2, and fibrocystin as well as proteins that are mutated in other cystic kidney diseases, such as nephronophthisis and Bardet-Biedl syndrome, are located in the primary cilium and/or basal body [5]. Second, Pkd1 mutant cells contain dysfunctional primary cilia as evidenced by a failure to increase [Ca2+]i in response to fluid flow. Treatment of wild-type cells with blocking antibodies against polycystin-2 or fibrocystin also inhibits the flow-dependent increase in [Ca2+]i [6, 7]. These findings suggest that polycystin-1, polycystin-2, and fibrocystin have a mechanosensory function in renal cilia that is coupled to [Ca2+]i. Third, inactivation of genes that are required for the synthesis of cilia, such as Kif3a and Ift88, results in the loss of primary cilia and produces PKD in mice [8, 9]. The molecular mechanism(s) by which abnormalities in the primary cilium result in PKD is not well understood. In addition to the regulation of [Ca2+]i, other signaling pathways that are regulated by the primary cilium include Wnt/β-catenin signaling, cAMP signaling[10], and planar cell polarity.
Planar cell polarity and PKD
Planar cell polarity (PCP) refers to the spatial organization of cells along a tissue plane that is perpendicular to the apical-basal axis [11]. PCP was first discovered in Drosophila, in which the hairs on the wing are always pointed distally towards the wing tip. Gene mutations that disrupt PCP produce swirls and other distortions of the wing hair pattern. Two major signaling pathways regulate PCP in Drosophila. One pathway, called the core PCP pathway, involves the membrane proteins Frizzled, Strabismus/Van Gogh, and Flamingo/Starry Night and the cytoplasmic proteins Dishevelled, Prickle, and Diego. PCP signaling leads to the formation of a protein complex containing Frizzled, Dishevelled, and Diego and a second complex containing Strabismus and Prickle. These protein complexes antagonize each other and are distributed asymmetrically within the cell. Core PCP proteins are evolutionarily conserved in mammals, and asymmetric intracellular localization of Vangl2 and Celsr1 (homologues of Strabismus and Flamingo, respectively) is believed to underlie the orderly arrangement of body hairs in the mouse [12]. A second PCP pathway involves two protocadherins, Fat and Dachsous, a transmembrane protein called Four-jointed, and the transcriptional repressor Atrophin.
PCP plays important roles in embryonic development by regulating cell migration, cell orientation, the orientation of cell division, and other morphogenetic processes. For example, PCP is involved in convergent extension movements that are required for elongation of the body axis and neural tube formation. Defects in these processes produce shortening of the body axis and failure of neural tube closure. One of the clearest examples of PCP in mammals is in the inner ear where the hair cells lining the cochlear duct contain V-shaped bundles of stereocilia that are always oriented with the apex positioned on the lateral side of the cell. Disruption of PCP in the inner ear produces misorientation of the stereociliary bundles and deafness.
Fischer et al were the first to demonstrate an association between abnormalities of planar cell polarity and PKD [13]. They studied the orientation of cell division, which is a manifestation of PCP, in the kidneys of the PCK rat (carrying a mutation of Pkhd1) and mice with kidney-specific inactivation of the transcription factor HNF-1β. Using lineage tracing and staining of mitotic cells, they found that cells in wild-type renal tubules divide along an axis that is approximately parallel to the longitudinal axis of the tubule. As a result, cell division produces tubular elongation without changing the diameter of the tubule. In contrast, in pre-cystic tubules from PCK rats and HNF-1β knockout mice, the orientation of cell division is randomized. The randomization of the orientation of cell division contributes to tubular dilatation and leads to cyst formation. These results suggest that abnormalities in PCP are present during early stages of cystogenesis.
Additional support for the link between abnormalities in PCP and the pathogenesis of PKD is provided by studies on mice lacking Fat4, a mammalian homologue of the Drosophila PCP protein Fat [14]. Knockout mice lacking Fat4 exhibit classic PCP phenotypes such as misoriented stereocilia in the cochlea and neural tube defects. Moreover, mutation of Fat4 produces randomization of the orientation of cell division in renal tubules and leads to the development of polycystic kidney disease.
Primary Cilia and PCP in the Kidney
The defects in PCP that are found in PKD may involve the primary cilium. Deletion of ciliogenic genes in the cochlea results in misorientation of the stereocilia, indicating that primary cilia are required for the maintenance of PCP in the inner ear [15]. To test whether primary cilia also regulate PCP in the kidney, we measured the orientation of cell division in the collecting ducts of mice in which the ciliogenic gene Kif3a had been inactivated [16]. First, we showed that inactivation of Kif3a results in the loss of primary cilia prior to the formation of kidney cysts. In pre-cystic tubules that lack primary cilia, the orientation of cell division is randomized, indicating aberrant PCP. Similar findings have been observed in mice with collecting duct-specific inactivation of another ciliogenic gene, Ift20 [17]. These results suggest that abnormalities in primary cilia produce disturbances in PCP that lead to PKD.
The mechanism by which the primary cilium regulates PCP is not known but may involve Wnt signaling. Wnts are secreted glycoproteins that play important roles in growth and development. Wnts bind to Frizzled receptors on the cell surface, recruit and activate Dishevelled, and signal via at least two pathways: a canonical pathway that is dependent on b-catenin and a non-canonical pathway that is β-catenin-independent. Non-canonical Wnt signaling has been shown to be necessary for the establishment of PCP in several organisms, including mammals. We showed that the loss of primary cilia in the kidney results in the activation of b-catenin-dependent signaling as evidenced by increased abundance of b-catenin in the nucleus and increased expression of downstream target genes such as c-Myc [8]. These results suggest that deletion of the primary cilium results in activation of canonical (β-catenin-dependent) Wnt signaling, a finding that has subsequently confirmed by Reiter et al [18]. The mechanism may involve Inversin, a ciliary protein that is mutated in the cystic kidney disease nephronophthisis type II [19,20]. Inversin interacts with Dishevelled (Dsh) and facilitates its degradation, and the depletion of Dsh inhibits canonical Wnt signaling. Conversely, Inversin is required for convergent extension in Xenopus laevis embryos [20]. These studies suggest that Inversin acts as a switch that down-regulates canonical Wnt signaling and stimulates PCP signaling. Recently, several other ciliary proteins that are mutated in various forms of cystic kidney diseases have been shown to constrain canonical Wnt signaling and/or promote PCP signaling [18,21-23]. Loss of primary cilia may also disrupt the regulation of PCP by the Fat/Dachsous pathway, since Fat4 has been localized in the cilia of MDCK renal epithelial cells [14].
Is cyst formation linked to tubular growth and repair?
Inactivation of ciliary genes, such as Kif3a and Pkd1, in the developing mouse kidney leads to the rapid development of renal cysts. To study the role of ciliary genes in the postnatal kidney, we and others have used inducible gene targeting to inactivate genes in mice at different ages. In this approach, transgenic mice in which Cre/loxP recombination can be induced by administration of drugs, such as tamoxifen, are mated to mice carrying a gene of interest flanked by loxP sites. In the absence of the drug the gene is expressed, whereas administration of the drug results in gene inactivation. Using this approach, we showed that inactivation of Kif3a in the early postnatal period results in the rapid formation of kidney cysts within two weeks. However, inactivation of Kif3a in adult mice does not produce a rapid onset of kidney cysts, despite a comparable loss of cilia [9,16,24]. The formation of kidney cysts after inactivation of Kif3a in adult mice requires up to six months. One difference between the developing kidney and the mature kidney is the rate of cell proliferation. The neonatal mouse kidney exhibits high rates of cell proliferation which decline at 10 days after birth. The decline in rates of proliferation coincides with the age-dependent effects of Kif3a gene inactivation. These findings suggest that cyst formation in mice lacking renal cilia requires high rates of cell proliferation. These results are also consistent with the role of aberrant PCP in cyst formation, since increased rates of cell proliferation would be predicted to promote the dilatation of tubules that have abnormalities in oriented cell division.
Piontek et al recently used inducible gene targeting to inactivate another ciliary protein, Pkd1, in postnatal mice [24]. Similar to the results of inactivation of Kif3a, they found that inactivation of Pkd1 in the early neonatal period resulted in the rapid formation of kidney cysts, whereas inactivation of Pkd1 in adult mice resulted in slow onset of kidney cysts. They attributed these findings to a developmental switch in gene expression that occurs in the kidney 14 days after birth [24]. Prior to this time point, the kidney exhibits an embryonic pattern of gene expression, whereas after this time point an adult pattern of gene expression appears. Taken together, these studies suggest that the rapid onset of kidney cysts following inactivation of ciliary genes requires high rates of cell proliferation and/or loss of cell differentiation
To test whether stimulation of cell proliferation promotes cyst formation, adult Kif3a knockout mice that lack renal cilia were subjected to acute kidney injury (AKI). Following AKI, surviving epithelial cells re-enter the cell cycle and proliferate to repair injured tubules. Therefore, if cyst formation in Kif3a knockout mice is dependent on increased cell proliferation, AKI would be predicted to stimulate cyst formation. When adult Kif3a knockout mice were subjected to renal ischemia-reperfusion injury, kidney cyst formation was accelerated compared to sham operated mice [16]. These findings are consistent with the hypothesis that cyst formation following disruption of ciliary genes requires high rates of cell proliferation. Since AKI is also associated with dedifferentiation of renal epithelial cells, these findings do not exclude a possible role of loss of cell differentiation in cyst formation.
Our studies are the first to show that acute kidney injury stimulates cyst formation in a mouse model of PKD. Along similar lines, administration of TNF-α, a potent cytokine induced by AKI or infection, stimulates cyst formation in adult Pkd2+/- heterozygous mice [25]. These findings suggest that cyst growth in adults with PKD may be affected by environmental factors such as sub-clinical kidney injury, kidney infection, or exposure to toxins. Individual variation in exposure to these environmental factors may contribute to the variation in severity of PKD that is observed between members of the same family who have inherited the identical gene mutation. Another conclusion of these studies is that one role of ciliary proteins in the mature kidney is to maintain tissue homeostasis by aiding repair at times of kidney injury. We speculate that similar to their role in the developing kidney, ciliary proteins maintain PCP and regulate tubular diameter during kidney repair.
Treatment of PKD
No specific treatment for PKD currently exists. Therefore, management of patients with PKD consists of supportive measures, such as analgesics for pain, antibiotics for cyst infection, blood pressure control, and avoidance of caffeine and estrogens. However, several promising investigational drugs are currently being studied in preclinical and clinical trials. These drugs target abnormal cell signaling pathways which lead to dysregulated cell proliferation, cell dedifferentiation, apoptosis, and fluid secretion. The development of rodent models that have mutations in genes that are orthologous to human PKD genes has been a major advance for validating the drugs prior to use in clinical studies. In this section, we will summarize the preclinical studies with emphasis on orthologous rodent models of PKD (also see Table 1).
Table 1. Novel treatments of PKD.
| Drug | Mechanism | Preclinical studies in animal models | Clinical studies | Results/Comments | |
|---|---|---|---|---|---|
| Non orthologous | Orthologous | ||||
| Triptolide | Stimulates polycystin 2-dependent calcium release | Kidney specific Pkd 1-/- mouse | Improved renal function | ||
| CFTR Inhibitors | Interferes with fluid secretion | Kidney specific Pkd 1-/- mouse | Improved renal function | ||
| Octreotide | Inhibits cAMP | PCK rat | Decreased the formation of cysts in the liver and kidney but no improvement in renal function. | ||
| Randomized crossover placebo-controlled | Smaller increase in kidney volume | ||||
| Vasopressin antagonists | Lowers cAMP | pcy mouse | PCK rat Pkd2 (ws25/-) mouse | Lowered kidney weights,Cr, BUN | |
| PCK and Brattleboro rat | PCK rats that lacked vasopressin show reduced cyst formation | ||||
| ongoing | TEMPO, a phase III study is underway | ||||
| Pioglitazone | Possibly inhibits β-catenin signaling | Pkd1-/- mouse | Improved phenotype and molecular defects. | ||
| Roscovitine | Inhibits cyclin dependent kinases | jck mouse cpk mouse | Intermittent administration of roscovitine showed a long lasting benefit | ||
| MAPK/ERK inhibitor | Inhibits MAPK/ERK signaling | pcy mouse | Kidney-specific Pkd1-/- mouse | Decreased cystic index in the pcy mouse but no beneficial effect in the kidney-specific Pkd1 mutant mouse | |
| Src Inhibition | Decreases ErbB1 and ErbB2 activity | bpk mouse | PCK rat | Src inhibition ameliorated renal and biliary lesions | |
| mTOR inhibitors | Prevents mTOR mediated cell growth | Han:SPRD rat oprk-rescue mutant mouse, bpk mouse | Retrospective studies | Decreased kidney volume in one study. Second study showed decrease in liver volume but not in kidney volume. | |
| Etanercept | Inhibits TNF-α | Pkd2+/- mouse | TNF-α stimulated and etanercept inhibited cyst formation. | ||
(A) Vasopressin receptor antagonists
The circulating levels of vasopressin are increased in mice with PKD. Many cysts in PKD are derived from collecting ducts, which express vasopressin V2 receptors. Vasopressin binds to the V2 receptors and activates adenylyl cyclase resulting in an increased level of cAMP. cAMP promotes cyst formation by stimulating fluid secretion and proliferative activity of cyst epithelial cells (Fig. 1). Preclinical studies with OPC-31260, a V2 receptor antagonist, in orthologous murine models of ARPKD (PCK rat), ADPKD (Pkd2WS25/- mouse), and nephronophthisis (pcy mouse) have shown significant inhibition of disease progression as measured by reductions in kidney volume, cyst volume, mitotic and apoptotic indices, and BUN levels [26]. Tolvaptan, another V2 receptor antagonist with higher affinity, has also been tested in orthologous animal models of ARPKD, ADPKD, and nephronophthisis and has shown similar efficacy [27]. To confirm that the protective effect of these drugs is due to antagonism of vasopressin, Wang et al crossed PCK rats (Pkhd1 mutant) with Brattleboro (AVP-/-) rats that are deficient in vasopressin [28]. PCK;AVP-/- rats had lower levels of renal cAMP and decreased cyst formation compared to PCK;AVP+/+ and PCK;AVP+/- rats. The beneficial effect was reversed when vasopressin-deficient rats were treated with a V2 receptor agonist. Based on these results, clinical trials to test the efficacy of Tolvaptan in humans with ADPKD are underway. The primary endpoint in these studies is the change in renal volume by MRI. The study participants consist of individuals age 18-50 years with preserved renal function and increased kidney volume >750 ml. The duration of treatment is 3 years. Of note, neither OPC-3120 nor tolvaptan has shown beneficial effects on the progression of fibrocystic liver disease. This finding could be explained by the lack of V2 receptor expression in the liver. Drinking large volume of water also suppresses vasopressin secretion and decreases cyst formation as demonstrated in an animal study [29]. However, a retrospective analysis of the MDRD study suggests that high urine volume and low urine osmolality were associated with faster progression of PKD [30]. More studies to determine the effects of water intake on PKD progression are needed to resolve this issue.
Figure 1. Mechanism of action of novel therapeutic agents in the treatment of PKD.
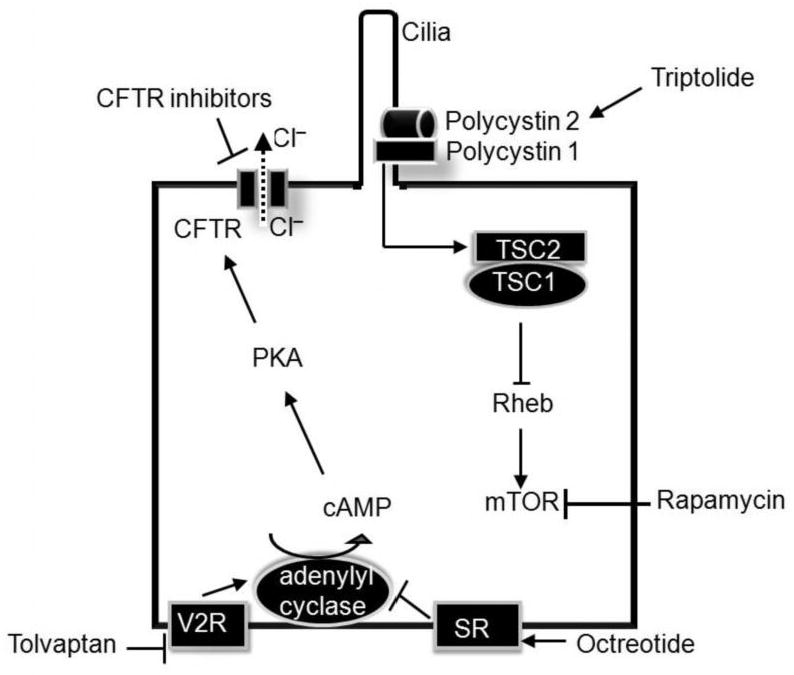
Vasopressin and octreotide decrease cAMP production by inhibiting V2 receptors (V2R) and activating stomatostatin receptors (SR), respectively. Decreased cAMP levels have been shown to retard cyst growth in several rodent models of PKD. CFTR inhibitors prevent the secretion of chloride (Cl-) into the cyst lumen by inhibiting the CFTR channel and thereby preventing cyst enlargement. The CFTR channels are activated by cAMP. Triptolide, a naturally occurring product, binds to polycystin-2 and stimulates calcium entry. Calcium entry is critical for the function of several signaling pathways. Polycystin-1 has been shown to interact with TSC2 (tuberin) which together with TSC1 inhibits Rheb. Rheb is an activator of mTOR. The net effect of interaction of polycystin-1 with TSC2 is mTOR inhibition. This mechanism may underlie the beneficial effects of the mTOR inhibitor rapamycin in rodents with PKD.
(B) Octreotide
Octreotide is a long-acting analogue of the hormone somatostatin. Octreotide binds to somatostatin receptors and inhibits cAMP production (Fig. 1). Octreotide has been shown to decrease cAMP levels and inhibit cyst formation in the livers of PCK rats; however, there was no significant improvement in renal function [31]. A small randomized, crossover, placebo-controlled study involving 12 patients followed over a 6-month period showed that octreotide decreased kidney volume but did not improve GFR [32]. Two other clinical trials on octreotide are being conducted at this time.
(C) Rapamycin
Activation of the mTOR (mammalian target of rapamycin) pathway has been detected in the cyst epithelium of mice and humans with PKD. Polycystin-1 has been shown to interact with tuberin (TSC2), which inhibits Rheb, an activator of mTOR (Fig. 1). In the absence of polycystin-1, disinhibition of Rheb results in activation of mTOR and increased cell growth. Rapamycin is an immunosuppressive drug that inhibits mTOR and is used to prevent transplant rejection. To test whether inhibition of mTOR prevents cyst growth, rapamycin and a related drug everolimus were evaluated in a non-orthologous animal model of PKD, the Han:SPRD rat [33,34]. These studies showed that inhibition of mTOR significantly slows cyst growth in male Han:SPRD rats. Other studies have tested the efficacy of rapamycin in three non-orthologous mouse models of PKD, Tg737orpk/orpk mice (mutation in the ciliary protein Polaris), bpk mice, and kidney-specific BHD (gene mutated in patients with Birt-Hogg-Dubé syndrome) mutant mice [35,36]. These studies also revealed significant improvement in cyst size and BUN levels compared to control animals. There are no published studies of rapamycin in orthologous animal models of PKD.
In a retrospective study, Shillingford et al examined a small group of patients who had undergone kidney transplantation for ESRD due to PKD and retained their native cystic kidneys [36]. In four patients who received rapamycin as a part of their post-transplant immunosuppression, the volume of the native cystic kidneys was reduced by 24% compared to 8.6% in three patients treated with other immunosuppressants. The change in kidney size was significantly different between the rapamycin and non-rapamycin groups. Another retrospective study by Qian et al demonstrated that rapamycin decreased the size and number of liver cysts but failed to show a benefit on kidney cysts [37].
(D) Roscovitine
The cyst epithelium in mice and humans with PKD exhibits high rates of proliferation suggesting abnormal cell cycle regulation. To test whether inhibition of cell cycle progression arrests cyst growth, roscovitine, an inhibitor of cyclin-dependent kinases was administered to two non-orthologous mouse models of PKD, jck mice and cpk mice [38]. Administration of roscovitine resulted in arrest of cyst growth in the jck and cpk mice. More significantly, intermittent administration of this drug produced a long-lasting anti-cystic effect. This result is promising considering the chronicity of PKD in which life-long therapy may be required.
(E) Others
Triptolide, a natural product isolated from the medicinal herb, Thunder God Vine, has been shown to retard cyst growth in kidney-specific Pkd1 mutant mice. Triptolide binds to polycystin-2 and induces calcium release in a polycystin 2-dependent manner. Intracellular calcium is thought to stimulate phosphodiesterase and inhibit adenylyl cyclase resulting in decreased levels of cAMP, thereby retarding cyst growth [39]. The cystic fibrosis transmembrane conductance regulator protein (CFTR), a cAMP-regulated chloride channel, is thought to be the primary route for chloride entry into cysts resulting in their expansion. CFTR inhibitors have been shown to slow cyst growth in some orthologous mouse models of PKD [40](Fig. 1). Pioglitazone has been shown to improve the renal as well as extra-renal abnormalities in Pkd1 knockout mice [41]. The mechanism of action is not well understood but may involve changes in Wnt/β-catenin signaling. Administration of a MAPK/ERK inhibitor decreased the cystic index in a non-orthologous animal model of PKD [42] but not in an orthologous mouse model [43]. Inhibition of Src activity ameliorates cyst formation in the non-orthologous bpk mouse model as well as the orthologous PCK rat model of ARPKD [44]. More recently, tumor necrosis factor-alpha (TNF-α) has been implicated in promoting PKD. Pkd2 mutant mice were treated with etanercept, a TNF-α inhibitor. The control animals developed cysts at the expected frequency, whereas none of the etanercept-treated animals developed kidney cysts at the end of ten weeks of treatment [25]. ACE inhibitors and ARBs have not been convincingly shown to increase renal survival or reduce mortality in humans with PKD. An ongoing NIH-funded study (HALT-PKD) is aimed at trying to clarify the role of ACE inhibitors and ARBs as well as aggressive blood pressure control in ADPKD [45].
Conclusions
Recent studies indicate that the pathogenesis of PKD is linked to abnormalities in the primary cilium that result in aberrant PCP signaling in the kidney. Inactivation of ciliary proteins in the postnatal kidney has uncovered novel roles of primary cilia in regulating tubular growth and repair after injury. Furthermore, these studies suggest that defective tubular repair after injury may contribute to the progression of PKD. Studies of signaling pathways that are perturbed in PKD have identified potential targets for pharmacological therapy. Drugs that have shown efficacy in preclinical studies utilizing orthologous animal models of PKD include tolvaptan, octreotide, src inhibitors, CFTR inhibitors, pioglitazone, etanercept, and triptolide. Several of these drugs are currently being evaluated in humans.
Acknowledgments
We thank Courtney Karner and Dr Thomas Carroll for helpful comments during the preparation of this manuscript. Work from the authors' laboratory is supported by grants from the National Institutes of Health (R01DK67565 and R01DK042921) and the UT Southwestern O'Brien Kidney Research Core Center (NIH P30DK079328). VP was supported by an NIH training grant (T32DK007257).
References
- 1.Igarashi P, Somlo S. Genetics and pathogenesis of polycystic kidney disease. J Am Soc Nephrol. 2002;13:2384–2398. doi: 10.1097/01.asn.0000028643.17901.42. [DOI] [PubMed] [Google Scholar]
- 2.Yoder BK. Role of primary cilia in the pathogenesis of polycystic kidney disease. J Am Soc Nephrol. 2007;18:1381–1388. doi: 10.1681/ASN.2006111215. [DOI] [PubMed] [Google Scholar]
- 3.Hiesberger T, Igarashi P. Elucidating the function of primary cilia by conditional gene inactivation. Curr Opin Nephrol Hypertens. 2005;14:373–377. doi: 10.1097/01.mnh.0000172725.37252.d8. [DOI] [PubMed] [Google Scholar]
- 4.Praetorius HA, Spring KR. Removal of the MDCK cell primary cilium abolishes flow sensing. J Membr Biol. 2003;191:69–76. doi: 10.1007/s00232-002-1042-4. [DOI] [PubMed] [Google Scholar]
- 5.Hildebrandt F, Otto E. Cilia and centrosomes: a unifying pathogenic concept for cystic kidney disease? Nat Rev Genet. 2005;6:928–940. doi: 10.1038/nrg1727. [DOI] [PubMed] [Google Scholar]
- 6.Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AE, Lu W, Brown EM, Quinn SJ, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003;33:129–137. doi: 10.1038/ng1076. [DOI] [PubMed] [Google Scholar]
- 7.Wang S, Zhang J, Nauli SM, Li X, Starremans PG, Luo Y, Roberts KA, Zhou J. Fibrocystin/polyductin, found in the same protein complex with polycystin-2, regulates calcium responses in kidney epithelia. Mol Cell Biol. 2007;27:3241–3252. doi: 10.1128/MCB.00072-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin F, Hiesberger T, Cordes K, Sinclair AM, Goldstein LS, Somlo S, Igarashi P. Kidney-specific inactivation of the KIF3A subunit of kinesin-II inhibits renal ciliogenesis and produces polycystic kidney disease. Proc Natl Acad Sci U S A. 2003;100:5286–5291. doi: 10.1073/pnas.0836980100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- * * 9.Davenport JR, Watts AJ, Roper VC, Croyle MJ, van Groen T, Wyss JM, Nagy TR, Kesterson RA, Yoder BK. Disruption of intraflagellar transport in adult mice leads to obesity and slow-onset cystic kidney disease. Curr Biol. 2007;17:1586–1594. doi: 10.1016/j.cub.2007.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study was the first to describe the age-dependent effects of ciliary gene mutations in the kidney. This study also uncovered an unexpected role of primary cilia in regulating food intake and obesity in mice.
- * 10.Masyuk AI, Gradilone SA, Banales JM, Huang BQ, Masyuk TV, Lee SO, Splinter PL, Stroope AJ, Larusso NF. Cholangiocyte primary cilia are chemosensory organelles that detect biliary nucleotides via P2Y12 purinergic receptors. Am J Physiol Gastrointest Liver Physiol. 2008;295:G725–734. doi: 10.1152/ajpgi.90265.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study revealed that several components of the cAMP signaling pathway, which is abnormally regulated in PKD, are expressed in the primary cilia of cholangiocytes.
- 11.Karner C, Wharton KA, Jr, Carroll TJ. Planar cell polarity and vertebrate organogenesis. Semin Cell Dev Biol. 2006;17:194–203. doi: 10.1016/j.semcdb.2006.05.003. [DOI] [PubMed] [Google Scholar]
- * 12.Devenport D, Fuchs E. Planar polarization in embryonic epidermis orchestrates global asymmetric morphogenesis of hair follicles. Nat Cell Biol. 2008;10:1257–1268. doi: 10.1038/ncb1784. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reported that, similar to Drosophila, asymmetric localization of core PCP proteins underlies planar polarization of hair follicles in the mouse skin.
- 13.Fischer E, Legue E, Doyen A, Nato F, Nicolas JF, Torres V, Yaniv M, Pontoglio M. Defective planar cell polarity in polycystic kidney disease. Nat Genet. 2006;38:21–23. doi: 10.1038/ng1701. [DOI] [PubMed] [Google Scholar]
- * * 14.Saburi S, Hester I, Fischer E, Pontoglio M, Eremina V, Gessler M, Quaggin SE, Harrison R, Mount R, McNeill H. Loss of Fat4 disrupts PCP signaling and oriented cell division and leads to cystic kidney disease. Nat Genet. 2008;40:1010–1015. doi: 10.1038/ng.179. [DOI] [PubMed] [Google Scholar]; This study showed that loss of the PCP protein, Fat4, produces PKD in mice thereby providing a direct link between PCP and PKD.
- * 15.Jones C, Roper VC, Foucher I, Qian D, Banizs B, Petit C, Yoder BK, Chen P. Ciliary proteins link basal body polarization to planar cell polarity regulation. Nat Genet. 2008;40:69–77. doi: 10.1038/ng.2007.54. [DOI] [PubMed] [Google Scholar]; This study showed that loss of primary cilia in the mouse cochlea results in aberrant PCP.
- * * 16.Patel V, Li L, Cobo-Stark P, Shao X, Somlo S, Lin F, Igarashi P. Acute kidney injury and aberrant planar cell polarity induce cyst formation in mice lacking renal cilia. Hum Mol Genet. 2008;17:1578–1590. doi: 10.1093/hmg/ddn045. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study was the first to show that acute kidney injury stimulates cyst formation in a mouse model of PKD. Furthermore, it was shown that primary cilia are required to maintain PCP in the kidney.
- * 17.Jonassen JA, San Agustin J, Follit JA, Pazour GJ. Deletion of IFT20 in the mouse kidney causes misorientation of the mitotic spindle and cystic kidney disease. J Cell Biol. 2008;183:377–384. doi: 10.1083/jcb.200808137. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study showed that in addition to maintaining oriented cell division, primary cilia are required for planar polarization of the basal bodies in the collecting duct epithelium.
- * 18.Corbit KC, Shyer AE, Dowdle WE, Gaulden J, Singla V, Chen MH, Chuang PT, Reiter JF. Kif3a constrains beta-catenin-dependent Wnt signalling through dual ciliary and non-ciliary mechanisms. Nat Cell Biol. 2008;10:70–76. doi: 10.1038/ncb1670. [DOI] [PubMed] [Google Scholar]
- 19.Otto EA, Schermer B, Obara T, O'Toole JF, Hiller KS, Mueller AM, Ruf RG, Hoefele J, Beekmann F, Landau D, et al. Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left-right axis determination. Nat Genet. 2003;34:413–420. doi: 10.1038/ng1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simons M, Gloy J, Ganner A, Bullerkotte A, Bashkurov M, Kronig C, Schermer B, Benzing T, Cabello OA, Jenny A, et al. Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat Genet. 2005;37:537–543. doi: 10.1038/ng1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- * 21.Gerdes JM, Liu Y, Zaghloul NA, Leitch CC, Lawson SS, Kato M, Beachy PA, Beales PL, DeMartino GN, Fisher S, et al. Disruption of the basal body compromises proteasomal function and perturbs intracellular Wnt response. Nat Genet. 2007;39:1350–1360. doi: 10.1038/ng.2007.12. [DOI] [PubMed] [Google Scholar]
- * 22.Lal M, Song X, Pluznick JL, Di Giovanni V, Merrick DM, Rosenblum ND, Chauvet V, Gottardi CJ, Pei Y, Caplan MJ. Polycystin-1 C-terminal tail associates with beta-catenin and inhibits canonical Wnt signaling. Hum Mol Genet. 2008;17:3105–3117. doi: 10.1093/hmg/ddn208. [DOI] [PMC free article] [PubMed] [Google Scholar]; Studies in references 18 and 21-22 showed that proteins located in the primary cilia/basal body complex inhibit Wnt/β-catenin signaling and/or promote PCP signaling.
- 23.Ross AJ, May-Simera H, Eichers ER, Kai M, Hill J, Jagger DJ, Leitch CC, Chapple JP, Munro PM, Fisher S, et al. Disruption of Bardet-Biedl syndrome ciliary proteins perturbs planar cell polarity in vertebrates. Nat Genet. 2005;37:1135–1140. doi: 10.1038/ng1644. [DOI] [PubMed] [Google Scholar]
- * * 24.Piontek K, Menezes LF, Garcia-Gonzalez MA, Huso DL, Germino GG. A critical developmental switch defines the kinetics of kidney cyst formation after loss of Pkd1. Nat Med. 2007;13:1490–1495. doi: 10.1038/nm1675. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study revealed that the age-dependent effects of Pkd1 gene inactivation correlate with a change in gene expression in the kidney at postnatal day 14.
- * * 25.Li X, Magenheimer BS, Xia S, Johnson T, Wallace DP, Calvet JP, Li R. A tumor necrosis factor-alpha-mediated pathway promoting autosomal dominant polycystic kidney disease. Nat Med. 2008;14:863–868. doi: 10.1038/nm1783. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study showed that administration of TNF-α to Pkd2+/- mice promoted cyst formation, whereas administration of a TNF-α inhibitor, etanercept, prevented cyst formation.
- 26.Gattone VH, 2nd, Wang X, Harris PC, Torres VE. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med. 2003;9:1323–1326. doi: 10.1038/nm935. [DOI] [PubMed] [Google Scholar]
- 27.Wang X, Gattone V, 2nd, Harris PC, Torres VE. Effectiveness of vasopressin V2 receptor antagonists OPC-31260 and OPC-41061 on polycystic kidney disease development in the PCK rat. J Am Soc Nephrol. 2005;16:846–851. doi: 10.1681/ASN.2004121090. [DOI] [PubMed] [Google Scholar]
- * 28.Wang X, Wu Y, Ward CJ, Harris PC, Torres VE. Vasopressin directly regulates cyst growth in polycystic kidney disease. J Am Soc Nephrol. 2008;19:102–108. doi: 10.1681/ASN.2007060688. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study described the effects of genetic and pharmacological manipulation of cAMP levels in the PCK rat. Decreased levels of cAMP prevented cyst formation, whereas increased levels of cAMP stimulated cyst formation.
- 29.Nagao S, Nishii K, Katsuyama M, Kurahashi H, Marunouchi T, Takahashi H, Wallace DP. Increased water intake decreases progression of polycystic kidney disease in the PCK rat. J Am Soc Nephrol. 2006;17:2220–2227. doi: 10.1681/ASN.2006030251. [DOI] [PubMed] [Google Scholar]
- 30.Hebert LA, Greene T, Levey A, Falkenhain ME, Klahr S. High urine volume and low urine osmolality are risk factors for faster progression of renal disease. Am J Kidney Dis. 2003;41:962–971. doi: 10.1016/s0272-6386(03)00193-8. [DOI] [PubMed] [Google Scholar]
- 31.Masyuk TV, Masyuk AI, Torres VE, Harris PC, Larusso NF. Octreotide inhibits hepatic cystogenesis in a rodent model of polycystic liver disease by reducing cholangiocyte adenosine 3′,5′-cyclic monophosphate. Gastroenterology. 2007;132:1104–1116. doi: 10.1053/j.gastro.2006.12.039. [DOI] [PubMed] [Google Scholar]
- 32.Ruggenenti P, Remuzzi A, Ondei P, Fasolini G, Antiga L, Ene-Iordache B, Remuzzi G, Epstein FH. Safety and efficacy of long-acting somatostatin treatment in autosomal-dominant polycystic kidney disease. Kidney Int. 2005;68:206–216. doi: 10.1111/j.1523-1755.2005.00395.x. [DOI] [PubMed] [Google Scholar]
- 33.Wahl PR, Serra AL, Le Hir M, Molle KD, Hall MN, Wuthrich RP. Inhibition of mTOR with sirolimus slows disease progression in Han:SPRD rats with autosomal dominant polycystic kidney disease (ADPKD) Nephrol Dial Transplant. 2006;21:598–604. doi: 10.1093/ndt/gfi181. [DOI] [PubMed] [Google Scholar]
- 34.Wu M, Wahl PR, Le Hir M, Wackerle-Men Y, Wuthrich RP, Serra AL. Everolimus retards cyst growth and preserves kidney function in a rodent model for polycystic kidney disease. Kidney Blood Press Res. 2007;30:253–259. doi: 10.1159/000104818. [DOI] [PubMed] [Google Scholar]
- * 35.Baba M, Furihata M, Hong SB, Tessarollo L, Haines DC, Southon E, Patel V, Igarashi P, Alvord WG, Leighty R, et al. Kidney-targeted Birt-Hogg-Dube gene inactivation in a mouse model: Erk1/2 and Akt-mTOR activation, cell hyperproliferation, and polycystic kidneys. J Natl Cancer Inst. 2008;100:140–154. doi: 10.1093/jnci/djm288. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study showed that administration of Rapamycin, an inhibitor of mTOR, in a non-orthologous mouse model of PKD slows the progression of cystic disease.
- 36.Shillingford JM, Murcia NS, Larson CH, Low SH, Hedgepeth R, Brown N, Flask CA, Novick AC, Goldfarb DA, Kramer-Zucker A, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci U S A. 2006;103:5466–5471. doi: 10.1073/pnas.0509694103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- * 37.Qian Q, Du H, King BF, Kumar S, Dean PG, Cosio FG, Torres VE. Sirolimus reduces polycystic liver volume in ADPKD patients. J Am Soc Nephrol. 2008;19:631–638. doi: 10.1681/ASN.2007050626. [DOI] [PMC free article] [PubMed] [Google Scholar]; In a retrospective study, the authors analyzed the effects of Sirolimus, an inhibitor of the mTOR pathway, in patients with ADPKD. Sirolimus decreased liver volume compared to tacrolimus.
- 38.Bukanov NO, Smith LA, Klinger KW, Ledbetter SR, Ibraghimov-Beskrovnaya O. Long-lasting arrest of murine polycystic kidney disease with CDK inhibitor roscovitine. Nature. 2006;444:949–952. doi: 10.1038/nature05348. [DOI] [PubMed] [Google Scholar]
- * 39.Leuenroth SJ, Okuhara D, Shotwell JD, Markowitz GS, Yu Z, Somlo S, Crews CM. Triptolide is a traditional Chinese medicine-derived inhibitor of polycystic kidney disease. Proc Natl Acad Sci U S A. 2007;104:4389–4394. doi: 10.1073/pnas.0700499104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- * * 40.Yang B, Sonawane ND, Zhao D, Somlo S, Verkman AS. Small-molecule CFTR inhibitors slow cyst growth in polycystic kidney disease. J Am Soc Nephrol. 2008;19:1300–1310. doi: 10.1681/ASN.2007070828. [DOI] [PMC free article] [PubMed] [Google Scholar]; Using a small molecule screen, these authors identified inhibitors of CFTR. Administration of the inhibitor slowed cyst formation in an orthologous mouse model of PKD.
- 41.Muto S, Aiba A, Saito Y, Nakao K, Nakamura K, Tomita K, Kitamura T, Kurabayashi M, Nagai R, Higashihara E, et al. Pioglitazone improves the phenotype and molecular defects of a targeted Pkd1 mutant. Hum Mol Genet. 2002;11:1731–1742. doi: 10.1093/hmg/11.15.1731. [DOI] [PubMed] [Google Scholar]
- * 42.Omori S, Hida M, Fujita H, Takahashi H, Tanimura S, Kohno M, Awazu M. Extracellular signal-regulated kinase inhibition slows disease progression in mice with polycystic kidney disease. J Am Soc Nephrol. 2006;17:1604–1614. doi: 10.1681/ASN.2004090800. [DOI] [PubMed] [Google Scholar]; This study showed that inhibition of ERK 1/2 signaling retards the progression of cyst formation in a non-orthologous mouse model of PKD.
- * * 43.Shibazaki S, Yu Z, Nishio S, Tian X, Thomson RB, Mitobe M, Louvi A, Velazquez H, Ishibe S, Cantley LG, et al. Cyst formation and activation of the extracellular regulated kinase pathway after kidney specific inactivation of Pkd1. Hum Mol Genet. 2008;17:1505–1516. doi: 10.1093/hmg/ddn039. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study showed focal activation of the MAPK/ERK pathway in cyst epithelia of Pkd1 and Pkd2 mutant mice. However, inhibition of Erk1/2 did not retard cyst formation.
- * * 44.Sweeney WE, Jr, von Vigier RO, Frost P, Avner ED. Src inhibition ameliorates polycystic kidney disease. J Am Soc Nephrol. 2008;19:1331–1341. doi: 10.1681/ASN.2007060665. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study revealed that Src inhibition slows the progression of cyst formation in orthologous as well as non-orthologous mouse models of ARPKD.
- 45.Chapman AB. Approaches to testing new treatments in autosomal dominant polycystic kidney disease: insights from the CRISP and HALT-PKD studies. Clin J Am Soc Nephrol. 2008;3:1197–1204. doi: 10.2215/CJN.00060108. [DOI] [PubMed] [Google Scholar]