

Abstract
Five components have thus far been identified that are necessary for the incorporation of selenocysteine (Sec) into ~25 mammalian proteins. Two of these are cis sequences, a SECIS element in the 3′-untranslated region and a Sec codon (UGA) in the coding region. The three known trans-acting factors are a Sec-specific translation elongation factor (eEFSec), the Sec-tRNASec, and a SECIS-binding protein, SBP2. Here we describe a system in which the efficiency of Sec incorporation was determined quantitatively both in vitro and in transfected cells, and in which the contribution of each of the known factors is examined. The efficiency of Sec incorporation into a luciferase reporter system in vitro is maximally 5–8%, which is 6–10 times higher than that in transfected rat hepatoma cells, McArdle 7777. In contrast, the efficiency of Sec incorporation into selenoprotein P in vitro is ~40%, suggesting that as yet unidentified cis-elements may regulate differential selenoprotein expression. In addition, we have found that SBP2 is the only limiting factor in rabbit reticulocyte lysate but not in transfected rat hepatoma cells where SBP2 is found to be mostly if not entirely cytoplasmic despite having a strong putative nuclear localization signal. The significance of these findings with regard to the function of known Sec incorporation factors is discussed.
The best studied function for selenium in the mammalian diet is its incorporation as selenocysteine (Sec)1 into a distinct set of proteins (selenoproteins) that carry out a diverse array of cellular functions (reviewed in Ref. 1). Through genomic analysis, it has been estimated that there are 25 human selenoproteins (2). The expression of selenoproteins is highly regulated by selenium levels and in a tissue-specific manner (reviewed in Ref. 3). While some of the known regulation occurs at the level of mRNA stability through the nonsense-mediated mRNA decay pathway (4–6), the role of regulated translation is an important component. Our understanding of the mechanism involved in selenoprotein synthesis has expanded rapidly in the past few years, but our understanding of the regulation of selenoprotein production remains preliminary.
Sec is incorporated into mammalian selenoproteins by a translational recoding event at specific UGA codons that are found upstream of stable stem-loop structures known as Sec insertion sequence (SECIS) elements that reside in the 3′-untranslated region (UTR). While the UGA codon and the SECIS element are the only known cis-acting elements required for Sec incorporation, at least three trans-acting factors are also required: 1) the Sec-specific elongation factor (eEFSec), 2) a SECIS-binding protein (SBP2), and 3) the Sec-tRNASec. Although there is a report that SBP2 and eEFSec interact in a tRNA-dependent manner (7), the minimal requirements and basic mechanism of Sec incorporation have not been elucidated.
SBP2 is a SECIS-specific RNA-binding protein that interacts with the kink-turn motif (8) found in all SECIS elements (1, 9). This ~94 kDa protein contains distinct SECIS element binding and Sec incorporation domains in the C-terminal 447 amino acids (10). The N-terminal 399 amino acids have no known function, and the sequence is not similar to any known protein, but it does contain a strong putative nuclear localization signal (11). Previous studies have relied on the C-terminal portion of SBP2 to provide Sec incorporation activity (10, 12), but a quantitative comparison of full-length and truncated protein has not been performed. SBP2 has also been reported to be a ribosome binding protein (10), but the contribution of this activity to Sec incorporation remains unknown.
One of the key open questions in this field is whether or not Sec incorporation is inherently inefficient. This is reported to be the case for bacterial Sec incorporation where maximum efficiency is reported to be 7–10% (13). In mammals, several selenoproteins are made at very high levels, particularly PHGPx in the mammalian testis (14), suggesting that efficiency may be much higher than that found in prokaryotes. Several reports have attempted to investigate efficiency in transfected cells (6, 15–17), but a quantitative study of the efficiency of Sec incorporation as measured by the ratio of translation termination products to Sec incorporation both in vitro and in transfected cells has not been performed. This question is vitally important to our understanding of Sec incorporation because the regulation of efficiency may be the primary determinant of variable selenoprotein expression. That is, the tissue-specific expression of any given selenoprotein may be primarily regulated by factors that may not be required for Sec incorporation per se, but may be yet unidentified factors that regulate the efficiency of incorporation by altering the interplay between Sec incorporation and translation termination.
To address the questions of efficiency and regulation quantitatively, we have designed an assay system that allows us to measure the ratio of termination to Sec incorporation using a monocistronic luciferase system both in vitro and in transfected cells. In addition, we have analyzed the contribution of SBP2 expression and subcellular localization, eEFSec, and Sec-tRNASec to the efficiency of Sec incorporation.
EXPERIMENTAL PROCEDURES
Plasmid Construction and mRNA Synthesis
The luciferase coding region and 3′-UTR were PCR-amplified and subcloned into pCDNA3.1 by TOPO-TA cloning (Invitrogen). Site-directed mutagenesis (QuikChange, Stratagene) was used to generate a PacI site in the 3′-UTR, and the wild-type 105-nt PHGPx SECIS element or a mutant element lacking the AUGA motif (12) was PCR-amplified with PacI/NotI linkers, digested, and inserted into the luciferase 3′-UTR. Site-directed mutagenesis was also used to alter each of the Cys codons to Sec within the luciferase coding region. Mutant and wild-type luciferase plasmids were linearized with XhoI then used as templates for in vitro transcription with T7 RNA polymerase in the presence of m7G(5′)ppp(5′)G (mMessage mMachine Ambion).
In Vitro Translation
All in vitro translation reactions involving luciferase mRNAs used rabbit reticulocyte lysates (RRL; Promega). Recombinant SBP2 or eEFSec was diluted into 20 mM Tris acetate, pH 7.2, and added to the RRL reaction as indicated in the figure legends. Total reaction volumes were 12.5 μl and were incubated for 1 h at 30 °C. For luciferase labeling, [35S]Met was included in the translation reaction and 2 μl of labeled translation products were resolved by SDS-PAGE on a 12% gel and quantitated by PhosphorImager analysis. Relative protein amounts were calculated by adjusting PhosphorImager density values according to the number of methionine residues in the pre-Sec peptide (10) versus full-length luciferase (14). In order to determine luciferase activity, 2 μl of the translation reaction was added to 50 μl of 1× phosphate-buffered saline (PBS). Readings were conducted in a Dynex MLX plate luminometer using 50-μl luciferase assay substrate (Promega). For calculation of the efficiency for Sel P translation, density values were adjusted for the two methionine residues in the pre-Sec peptide. The third and final Met in this protein is located just beyond the third Sec residue, and for our calculations, we assumed that all upper molecular weight bands contained a total of three Met residues.
Northern Blot Analysis
RNA was extracted from cells grown in 6-well plates at 24 h post-transfection using TRIzol as outlined by the manufacturer (Invitrogen). Half of the total RNA extracted from each well (~25 μg) was resolved at 120 V for 2 h in 1% agarose, 7% formaldehyde gels that were transferred to Immobilon Ny+ membranes in 0.01 N NaOH/3 M NaCl buffer overnight. The membrane was probed with 10.2 ng (7.5 × 106 cpm) of [μ-32P]dCTP-labeled luciferase cDNA probe (7 × 108 cpm/μg) made by RadPrime DNA labeling system (Invitrogen) in Expresshyb (BD-Clontech) hybridization solution and conditions. The membrane was washed and exposed to a PhosphorImager screen.
Transfections and Western Blot Analysis
Transient transfections were executed in McArdle 7777 rat hepatoma cells grown in DMEM/F12 50/50 Mix with L-glutamine (Cellgro) supplemented with 10% fetal bovine serum (Cellgro) and 0.5 μM sodium selenite (Sigma). At ~70% confluence, cells were transfected (LipofectAMINE; Invitrogen) with 2 μg of each luciferase construct per 9.6 cm2 or as indicated in the figure legends. Cell extracts were taken 24 h post-transfection with 0.16 ml per 9.6 cm2 of Glo Lysis Buffer (Promega) according to the manufacturer’s protocol, and 20 μl of each sample was resolved on a 12% SDS-PAGE gel, blotted to nitrocellulose membrane (Amersham Biosciences), blocked overnight in 5% nonfat dried milk, and probed using a polyclonal, horseradish peroxidase-conjugated anti-luciferase antibody (Rockland Immunochemicals) at a 1:100,000 dilution, and was visualized using the SuperSignal West Femto kit (Pierce) according to the manufacturer’s protocol. This dilution of antibody resulted in excess probe based on the response of signal from the L/Cys/P construct (data not shown). For luciferase assays, 5 μl of each sample was diluted into 20 μl of PBS and analyzed in a Dynex MLX plate luminometer using 50 μl of luciferase assay substrate (Promega). Protein concentrations were determined by the Bradford protein assay kit (Bio-Rad).
Preparation of tRNAs
75Se-labeled Sec-tRNASec (D. L. Hatfield, NCI, National Institutes of Health) was prepared using HL60 cells (18, 19), and the two isoacceptors (mcmUm and mcmU) were resolved from each other by RPC-5 chromatography as described (20). For the experiments described here, the two isoacceptor fractions were pooled to a final specific activity of 1420 cpm/pmol. Phe-tRNAPhe (T. G. Kinzy, Robert Wood Johnson Medical School) was prepared as previously described (21). Yeast tRNAPhe (Sigma) was amino-acylated with 14C-Phe utilizing a partially purified preparation of rabbit reticulocyte amino-acyl-tRNA synthetases. Amino-acylated Phe-tRNAPhe was purified from unincorporated Phe by gel filtration chromatography. The yield of amino-acylated tRNA was 10%, as determined by the specific activity of the [14C]Phe and trichloroacetic acid-precipitable radioactivity.
Preparation of Recombinant Proteins
Full-length and C-terminal SBP2 constructs in pASK-IBA7 were transformed into Escherichia coli strain BL21 (Promega). Transformed bacteria were grown in LB medium to a density of ~1.0 OD600 then induced with anhydrotetracyclinen (0.2 mg/ml) for 1.5 h at 30 °C. The cells were pelleted, resuspended in Buffer W (100 mM Tris/HCl, pH 8.0, 1 mM EDTA) with protease inhibitors (Complete, Roche Applied Sciences) and lysed by freeze-thaw followed by sonication for 15 s per ml. The sonicate was then centrifuged at 10,000 × g for 15 min at 4 °C. The supernatant was applied directly to a 0.5 × 10-cm column containing Streptactin-Sepharose (Genosys) equilibrated in Buffer W with 25 mM KCl and tagged protein was eluted in Buffer W containing 2.5 mM desthiobiotin (Sigma). For eEFSec, the coding region was subcloned into pTrcHis (Invitrogen) so that the protein would contain N-terminal Xpress and His tags. Transformed E. coli (BL21) were grown in LB medium to a density of 1.0–1.1 OD600, and then induced with 1 mM isopropyl-1-thio-β-D-galactopyranoside (Fisher Biotech) for 1.5 h at 30 °C. The cells were pelleted, resuspended in Buffer E (20 mM Tris-HCl 7.6, 20 mM KCl, 0.1 mM EDTA, 50 mM imidazole, 25% glycerol, 1 mM dithiothreitol) with protease inhibitors (Complete) and lysed by freeze-thaw, followed by sonication for 12 s/ml. The sonicate was then centrifuged at 15,000 × g for 15 min at 4 °C. The supernatant was filtered through a 0.2-μm pore size Whatman syringe filter and applied directly to a tandem column system consisting of an HR 10/10 column containing DEAE Sepharose Fast Flow (Amersham Biosciences) followed by a 1-ml Hitrap chelating HP column (Amersham Biosciences) previously charged with 0.5 ml of 0.1 M NiSO4. The system was equilibrated in Buffer E and eEFSec protein was eluted in Buffer E using a 50–500 mM imidazole gradient. Further purification was achieved by loading the protein-containing fraction(s) onto an HR 5/5 column packed with Source 15S resin (Amersham Biosciences) and eluted with Buffer E in a linear 20–500 mM KCl gradient. Peak fractions were pooled and exchanged to Buffer E lacking imidazole using a Centricon YM-10 ultrafiltration device (Amicon).
Subcellular Localization
McArdle 7777 cells growing on slides were transiently transfected at ~50% confluence with full-length V5/His-tagged SBP2 in pCDNA4 TO-E (Invitrogen). At 24 h post-transfection, cells were rinsed in PBS, fixed in 4% paraformaldehyde, and permeabilized with 0.2% Triton X-100 in PBS for 5 min. Cells were washed in three changes of PBS containing 1% bovine serum albumin and incubated for 1 h with a 1:200 dilution of anti V5 monoclonal antibody in PBS with 1% bovine serum albumin. Cells were washed as before prior to incubating with 5 μg/ml fluorescein-conjugated goat anti-mouse secondary antibody (Molecular Probes) for 1 h followed by counterstaining of DNA with 300 nM DAPI (Molecular Probes). Stained cells were viewed by standard epifluorescent microscopy.
RESULTS
Development of a Sec Incorporation Reporter System
In order to quantitatively examine the efficiency of Sec incorporation in vitro, we developed a reporter system utilizing a luciferase coding region containing a Sec codon followed by a functional SECIS element. It is common for stop codon suppression studies to make use of bicistronic messages containing two reporter genes separated by a stop codon. For Sec incorporation, we have chosen a monocistronic system in order to avoid two major artifacts: 1) internal translation initiation that may be regulated by the intercistronic sequence, and 2) modification of reporter enzyme activity in the presence of an intervening Sec residue. The problem of internal initiation persists for monocistronic reporters in RRL, so we set out to determine the best position for a Sec codon that would give the lowest background activity and have the least impact on luciferase function. Toward this goal we mutated each of the four codons encoding Cys to UGA codons. Each of these mutations were made in a luciferase construct containing the 105-nt SECIS element derived from rat PHGPx (12). Since the distance between the UGA (Sec) codon and the SECIS element has previously been shown to be unconstrained beyond 51 nucleotides (22), we do not expect that the relative positions of the two elements contribute to differences in incorporation. Rather, these constructs were made in order to test the effect of substituting Sec for Cys on luciferase enzyme activity as well as minimizing background activity as a result of internal initiation downstream of the Sec codon. 5′-Capped mRNA was synthesized for each construct and luciferase activity was determined in the presence and absence of added recombinant SBP2 protein after translation in rabbit reticulocyte lysate (Fig. 1A, RRL). The SBP2-dependent increase in luciferase activity ranges from 13- to 88-fold in this experiment, with the highest increase at codon 258. This experiment was performed under conditions of limiting mRNA (10 ng) in order to maximize detection of background readthrough activity in the absence of added SBP2. Further experiments that included higher levels of SBP2 showed that maximal Sec incorporation occurred in reactions containing 200 ng of mRNA for all constructs used in this study (data not shown). In order to verify that the activity observed corresponds to Sec incorporation, we verified that luciferase activity was both SECIS element and UGA-dependent. The luciferase constructs diagrammed in Fig. 1B are used throughout this study to confirm the specificity of Sec incorporation. Fig. 1C shows that luciferase activity is only observed when an in-frame UGA codon is found in the context of a wild-type SECIS element. These data provide a clear indication that the Cys at position 258 will best tolerate the insertion of Sec with minimal or no contribution to background and that this reporter system provides a reliable and quantitative measure of Sec incorporation.
Fig. 1. Design of luciferase-based Sec incorporation system.
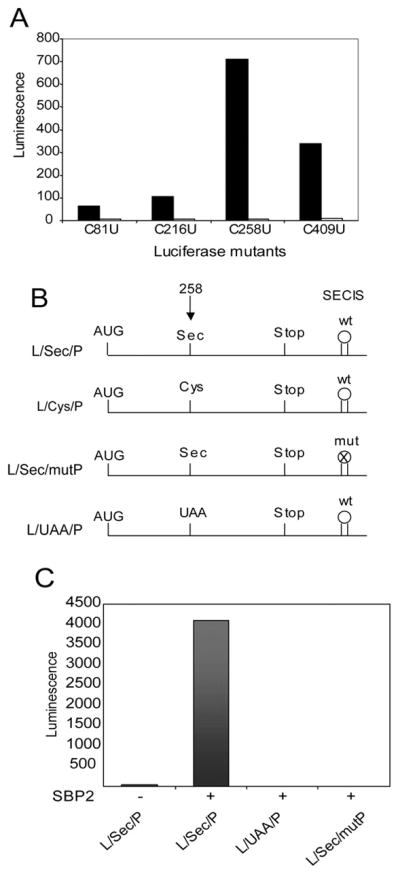
A, 10 ng of mRNA corresponding to Cys to Sec (U) mutants of luciferase mRNA were translated in a RRL Sec incorporation assay in the presence (black bars) or absence (white bars) of 200 ng of recombinant SBP2399–846. B, diagram of the constructs used in this study: L/Sec/p, luciferase coding region with UGASec at codon 258 and the wild-type PHGPx SECIS element in the 3′ UTR; L/Cys/p, same as L/Sec/P but with a Cys codon at position 258; L/Sec/mutP, same as L/Sec/P but with mutated PHPGx SECIS element lacking the conserved AUGA motif; L/UAA/p, same as L/Sec/P but with UAAterm at position 258. C, 200 ng of luciferase mRNA corresponding to three of the constructs diagrammed in B were translated in RRL in the presence of 160 ng of SBP2399–846.
Comparison of Full-length and Truncated SBP2
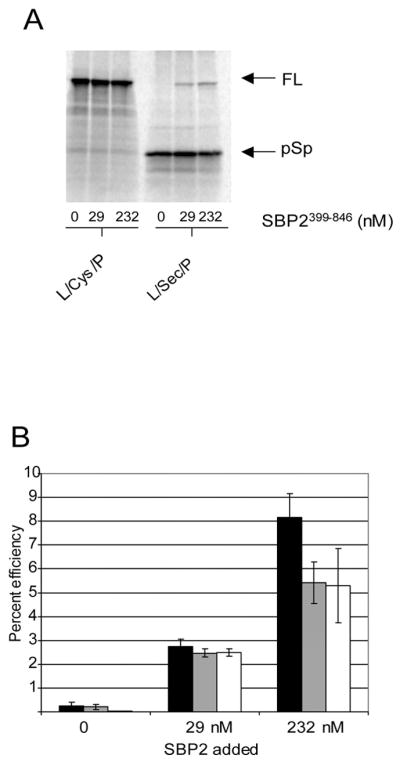
Our original report of SBP2 activity provided evidence that the C-terminal 447 amino acids of SBP2 (399–846) were sufficient for Sec incorporation activity in vitro (11). In order to directly compare the activity of full-length and C-terminal SBP2, we purified Strep-tagged full-length (SBP21–846) and C-terminal (SBP2399–846) and compared their function in the in vitro Sec incorporation assay. For this experiment, equimolar amounts of full-length and C-terminal SBP2 were added in increasing amounts to RRL reactions containing L/Sec/P mRNA. As shown in Fig. 2A, the dose-dependent increase in luciferase activity is nearly identical for both full-length and C-terminal SBP2 in the linear range of the reaction. In order to confirm the correlation between luciferase activity and the production of a full-length version of the protein, we analyzed the translation products from the L/Sec/P mRNA by [35S]Met labeling in RRL. To test the SBP2-dependent dose-response, varying amounts of purified, recombinant SBP2399–846 were added to an RRL reaction containing capped L/Sec/P mRNA (Fig. 2B). Two major translation products were observed, the larger of which (full-length luciferase) is only found in the presence of SBP2, and appears in an SBP2 concentration-dependent manner and corresponds exactly with the activity profile illustrated in Fig. 2A. These results suggest that the N-terminal half of SBP2 is not required for Sec incorporation in vitro and may therefore play a regulatory role in vivo under conditions not found in RRL. In addition, these results show that this assay system provides a clear distinction between the terminated “pre-Sec peptide” and full-length luciferase by [35S]Met labeling.
Fig. 2. Comparison of full-length and truncated SBP2.

A, purified strep-tagged full-length (gray bars) or C-terminal SBP2399–846 (white bars) were added to a Sec incorporation assay with the L/Sec/P mRNA. Average luciferase activity ± S.E. is plotted against SBP2 concentration (n = 3). B, purified Strep-tagged SBP2399–846 was added in increasing amounts (2-fold serial dilution ranging from 930 to 7 nM) to a Sec incorporation assay including [35S]Met and luciferase mRNAs as indicated. Radioactive proteins were resolved by SDS-PAGE and detected by PhosphorImage analysis. The positions of full-length (FL) luciferase versus the pre-Sec peptide (pSp) are indicated on the right.
Determination of the Efficiency of Sec Incorporation in Vitro
We have used three methods to calculate efficiency based on either quantitated amounts of [35S]Met-labeled protein after taking into consideration the number of Met residues or luciferase activity. First, efficiency was calculated as the amount of full-length luciferase divided by total translation product (pre-Sec peptide + full-length luciferase; Fig. 3B, black bar). Second, the amount of full-length luciferase derived from L/Sec/P was divided by that derived from L/Cys/P using 200 ng of each mRNA, which gives maximal protein expression for both constructs (Fig. 3B, gray bar). Third, the amount of activity derived from L/Sec/P was divided by that from L/Cys/P (Fig. 3B, white bar). In order to use this final method, a correction had to be made for the difference in specific activity between Sec- and Cys-containing luciferase. Based on a quantitation of luciferase activity per unit of quantified [35S]Met-labeled luciferase protein (Fig. 3A), we found that the L/Sec/P protein was 16% less active than that containing Cys at position 258. Together these results show an SBP2-dependent maximal efficiency ranging from ~5–8%. Thus, we are able to normalize our data between sets using the percent efficiency as a measure of SBP2 activity both in vitro and in vivo.
Fig. 3. Calculation of the efficiency of Sec incorporation.
A, 35S-labeled luciferase protein from a Sec incorporation reaction containing the indicated amount of SBP2399–846 was resolved by SDS-PAGE and detected by PhosphorImage analysis. B, percent efficiency of Sec incorporation. Black bars represent percent efficiency expressed as amount of full-length luciferase divided by total product (full-length plus pre-Sec peptide). Gray bars represent percent efficiency expressed as the amount of full-length Sec-containing luciferase protein divided by the amount of full-length Cys-containing luciferase protein as determined by PhosphorImage analysis. White bars represent percent efficiency expressed as the amount of luciferase activity derived from Sec-containing luciferase divided by the amount of activity derived from Cys-containing luciferase after correcting for differences in specific activity.
The low efficiency of Sec incorporation in vitro could be due to the lack of either cis or trans effectors. That is, either the luciferase constructs we are using are missing critical unidentified elements or the system is lacking trans-acting efficiency factors. To test the former hypothesis, we wished to analyze the translation of a native selenoprotein mRNA. We were unable to use the native PHGPx mRNA for this assay because the pre-Sec peptide is not detectable in RRL,2 so we chose to translate rat selenoprotein P (Sel P) mRNA in RRL in the presence and absence of SBP2. Sel P is a unique selenoprotein in that it contains ten Sec codons and two SECIS elements in a large 3′-UTR thus allowing us to monitor Sec incorporation and translation termination at multiple positions as well as assess the processivity of Sec incorporation in vitro. The construct we chose to use contains the entire Sel P coding region plus 1 kb of 3′-UTR sequence containing both SECIS elements. If Sel P is efficiently translated, the full-length protein product containing 6–10 Sec residues is predicted to appear as a 40–43 kDa protein because the last four Sec residues are clustered within the last 12 amino acids. Fig. 4A shows the predicted molecular weights of Sel P protein products terminated successively at each of the ten UGA codons. Fig. 4B shows the translation of Sel P mRNA in RRL in the presence and absence of SBP2. There are three major [35S]Met-labeled translation products that appear in an SBP2-dependent manner. While the discrepancy between predicted molecular masses (Fig. 4A) and observed molecular masses (indicated to the left of Fig. 4B) makes it impossible to make definitive assignments to each product regarding the number of Sec residues incorporated, it is clear that the majority of Sec incorporation resulted in full-length or near-full-length Sel P, which under these conditions resolves as a 46.8 kDa protein. This provides clear evidence that Sec incorporation can proceed in a processive fashion in vitro. In addition, we found that 40% of the total translated product contained at least one Sec residue, suggesting a substantial increase in efficiency with this mRNA as compared with that found for L/Sec/P. These results suggest that at least part of the contribution to higher efficiency resides within the selenoprotein mRNA.
Fig. 4. Efficiency of Sel P translation in vitro.

A, all possible products of Sel P translation are listed with their predicted molecular masses. B, in vitro translation of Sel P mRNA in the presence (lane 1) or absence (lane 2) of recombinant SBP2399–846. Reactions lacking Sel P mRNA with and without added SBP2 are shown in lanes 3 and 4, respectively. Observed molecular masses are shown on the left.
SBP2 Is the Only Known Limiting Factor for Sec Incorporation in RRL
In an attempt to further increase Sec incorporation efficiency in RRL, we set out to determine if the other known Sec incorporation factors are limiting in this system, namely the elongation factor, eEFSec and the Sec-tRNASec. To address this, we purified 6× His-tagged mouse eEFSec expressed in E. coli and added it to a Sec incorporation assay in the presence of SBP2 and varying amounts of purified 75Se-labeled Sec-tRNASec. Fig. 5 shows that luciferase activity is not enhanced in the presence of added eEFSec alone or in combination with Sec-tRNASec. Even though we used the purification protocol designed to isolate highly active eEF1A,3 it is possible that our preparation of eEFSec is inactive. Since an in vitro functional test for eEFSec has not been developed, we chose to test another source of eEFSec in case the protein expressed in E. coli, which has previously been shown to bind Sec-tRNASec (23), is unable to function in Sec incorporation. To this end, we assessed the ability of in vitro translated eEFSec to stimulate Sec incorporation over background in RRL and it did not, even in combination with SBP2 (data not shown). Although we cannot definitively state that we have tested fully functional eE-FSec, our data strongly suggest that SBP2 is the only Sec incorporation factor limiting in reticulocyte lysate. As a control for supplemental tRNA, we added yeast Phe-tRNAPhe to the same set of reactions (Fig. 5B). The visualization of 75Se-labeled luciferase in these reactions allowed us to verify that the Sec-tRNASec was utilized in these reactions (data not shown). These results indicate that RRL is replete with Sec-tRNASec.
Fig. 5. SBP2 is the only limiting factor for Sec incorporation in RRL.

Purified Sec-tRNASec was added in the amounts indicated to a standard Sec incorporation assay. Reactions contained either 1 pmol each of SBP2399–846 and eEFSec (black bars), 1 pmol of SBP2399–846 alone (white bars), or no added protein (gray bars). B, same reactions as described for A but in the presence of Phe-tRNAPhe as a control for nonspecific effects.
Determination of the Efficiency of Sec Incorporation in Transfected Cells
Because Sec incorporation into our luciferase constructs in vitro is maximally ~8% efficient, we set out to determine if the efficiency of Sec incorporation in cells is similar. Several reports have used transient transfection assays to assess Sec incorporation, but the question of efficiency has not been addressed. To study efficiency in cells, we transfected varying amounts of either L/Sec/P or L/Cys/P in 2-fold serial dilutions starting from 2 μg per well. Fig. 6A shows that maximal luciferase activity is found at 2 μg for both L/Sec/P and L/Cys/P but with significantly lower total activity for the Sec-containing construct. In order to determine efficiency we compared luciferase activity derived from L/Sec/P to that derived from L/Cys/P and corrected for the difference in specific activity based on our calculations from RRL reactions. As shown in Fig. 6B, the efficiency of Sec incorporation averages 0.8%, about 6–10 times lower in transfected cells than in RRL, even at lower DNA concentrations. This result predicts that the full-length luciferase to pre-Sec peptide ratio would be similarly affected and dramatically decreased. To test this hypothesis, we analyzed total cell lysates by Western blot analysis using polyclonal anti-luciferase antibody in order to detect both forms of the protein. Fig. 6C shows that the full-length to pre-Sec peptide ratio, while not strictly quantitative, does not correspond to the efficiency based on activity, suggesting that total luciferase translation is reduced in L/Sec/P versus L/Cys/P in transfected cells. This is confirmed by the Western blot as the amount of protein expressed from L/Cys/P vastly exceeds the total luciferase translation for L/Sec/P (pre-Sec peptide plus full-length; Fig. 6C, lane 2 versus lane 5) whereas these two quantities were very similar in RRL (Fig. 3A; note: although the full-length proteins appear to be migrating differently in this experiment, a side by side comparison of the product from L/Sec/P and L/Cys/P shows them to migrate identically during SDS-PAGE). There are two likely explanations for the difference in protein amounts: differential RNA stability or limiting and/or differential pre-Sec peptide versus full-length protein stability. In order to address the first point, Northern analysis was performed from RNA isolated from cells transfected with varying amounts of L/Sec/P and L/Cys/P as described above. Fig. 6D shows Northern blot analysis of RNA isolated from cells transfected with varying amounts of L/Sec/P and L/Cys/P DNA from a maximum of 2 μg as well as transfections of 2 μg each L/Sec/mutP and L/UAA/P. This analysis shows that the steady state levels of L/Sec/P and L/Cys/P mRNAs are not significantly different (~10% difference between lanes 2 and 8 when normalized to rRNA levels) indicating that RNA stability cannot account for the lack of efficiency in cells. The most likely explanations are thus either lower pre-Sec peptide stability or reduced overall translation of stop-codon containing luciferase constructs.
Fig. 6. Efficiency of Sec incorporation in transfected cells.

A, McArdle 7777 cells were transfected with varying amounts of L/Sec/P or L/Cys/P plasmids as indicated. Extracts were analyzed for luciferase activity (expressed as luminescence per mg of total protein). Activity for the L/Cys/P transfections was divided by a factor of 10 to allow visual comparison. B, percent efficiency of Sec incorporation was calculated by dividing activity derived from L/Sec/P and L/Cys/P at each of the DNA amounts used for transfection. The data was gathered from two luciferase assays for two independent experiments. C, Western blot analysis of 2% of the total protein (~85 μg) extracted from cells transfected with luciferase constructs as indicated. For L/Cys/P extracts, only 0.2% was loaded to enhance clarity. Full-length luciferase and the pre-Sec peptide are noted with arrows. D, Northern blot analysis of one half of the total RNA (~25 μg) extracted from cells transfected with 2, 0.5, 0.125, and 0.03 μg of L/Sec/P and L/Cys/P DNA (lanes 2–5 and 8–11) and 2 μg each of L/Sec/mutP and L/UAA/P DNA (lanes 6 and 12). RNA isolated from mock transfections was loaded in lanes 1 and 7. Bottom panel shows ethidium bromide-stained rRNA as a loading control.
Subcellular Localization of SBP2 in Rat Hepatoma Cells
To address a potential limitation of SBP2 in these cells, we transfected varying amounts of cDNA encoding SBP21–846 or SBP2399–846 but found no improvement in luciferase activity despite having confirmed SBP2 expression by Western blot (data not shown). Although the likely explanation for this observation is that SBP2 is not limiting in this cell type, another possibility would be nuclear localization of the overexpressed protein as is predicted by the presence of a strong putative nuclear localization signal between amino acids 368 and 382 (11). In order to address this question we used anti-V5 antibody and a fluorescein-conjugated secondary antibody to probe McArdle 7777 cells transfected with full-length V5/His-tagged SBP2. Fig. 7 (top) shows the resulting immunofluorescence as mostly if not entirely cytoplasmic. Fig. 7 (bottom) shows a merged image of DAPI-stained nuclei and the SBP2 immunofluorescence allowing direct comparison to adjacent non-transfected cells. These results allow us to conclude that at least in this cell type, SBP2 is predominantly cytoplasmic.
Fig. 7. Subcellular localization of SBP21–846.

McArdle 7777 cells were transfected with SBP21–846 cDNA, and the V5/His-tagged SBP2 protein was detected with anti-V5 mouse monoclonal antibody followed by fluorescein-conjugated secondary antibody. Top panel shows SBP2 fluorescence. Bottom panel shows merged image of DAPI-stained nuclei and SBP2 fluorescence.
DISCUSSION
In this report we have demonstrated that the efficiency of Sec incorporation into an in vitro luciferase reporter system from which we can calculate efficiency using both luciferase activity as well as the ratio of truncated to full-length translated product ranges from 5–8%. We have used the same set of constructs to assess efficiency in transfected cells and found up to a 10-fold decrease in efficiency. A ~5-fold improvement in the efficiency of Sec incorporation in vitro was obtained when we translated an authentic selenoprotein mRNA, suggesting that at least the Sel P mRNA possesses cis sequences that may be involved in regulating efficiency. In addition we have found that of the known Sec incorporation factors, SBP2 is the only one limiting in RRL. Together these results suggest that the known Sec incorporation factors are not sufficient to allow efficient and regulated selenoprotein synthesis.
One of the confounding aspects regarding potential regulatory factors of Sec incorporation has been our inability to assign a function for the N-terminal half of SBP2. Our previous studies suggested that full-length and N-terminally truncated SBP2 may function similarly, and in this report we provide quantitative evidence in vitro that the N-terminal 399 amino acids of SBP2 are not required for maximal Sec incorporation in RRL. In addition, neither the full-length nor truncated form of SBP2 enhances Sec incorporation in transfected McArdle 7777 cells (data not shown). The role of this domain is further confounded by the lack of sequence similarity to any proteins of known function. The N-terminal domain does have one revealing segment of sequence and that is one predicted to be a strong nuclear localization signal. Here we have tested the subcellular localization of transfected full-length SBP2 and found that it is only detectable in the cytoplasm. This result suggests that nuclear localization may be a form of regulation for SBP2 either as a mode of sequestration from its cytoplasmic binding partners (ribosomes and SECIS elements) or as a means to pre-associate with SECIS elements prior to nuclear export. In either case, it is likely that SBP2 subcellular localization will be both cell-type and possibly condition-specific. That is, it is tempting to speculate that variable nuclear localization could regulate the amount of cytoplasmic SBP2 under conditions where selenoprotein expression is likely to be regulated such as during selenium deficiency, selenium excess, or oxidative stress.
Zavacki et al. (7) have reported that SBP2 and eEFSec directly interact in a Sec-tRNASec-dependent manner, but that this interaction cannot be observed in RRL because of the lack of tRNA in this system. Our own efforts to identify a direct interaction between SBP2 and eEFSec in RRL have been unsuccessful.2 However, here we have presented strong evidence that Sec-tRNASec is present in non-limiting amounts in RRL. This apparent discrepancy is further evidence that a factor found in some cell types is missing in RRL, and that one of the functions of this factor is the bridging of SBP2 and eEFSec. Interestingly, the original description of an SBP2/eEFSec interaction showed the complex to be RNase sensitive (23). If the Sec-tRNASec and SECIS element are not sufficient to bridge SBP2 and eEFSec in RRL, then the interaction observed in transfected cells could be the result of bridging by an RNA species distinct from Sec-tRNASec or the SECIS. However, this “bridging” factor must be nonessential since relatively robust Sec incorporation is achievable in RRL. It is precisely this type of factor, one that is regulatory but not essential, that will likely define the mechanisms of differential selenoprotein regulation.
To examine the importance of mRNA sequence context, we used an authentic selenoprotein mRNA encoding selenoprotein P, a plasma protein that contains up to 10 Sec residues. 75Se labeling in vivo clearly indicates that the vast majority of the selenium is incorporated into a ~57 kDa protein in rat plasma, and this corresponds to the molecular mass of full-length selenoprotein P as shown by immunopurification of 75Se-labeled proteins (24). Further work has demonstrated that some of the Sel P found in plasma corresponds to termination at the 2nd, 3rd, and 7th Sec codons, but the relative amount of each product has not been determined (25). These results indicate that although some termination does occur during Sel P synthesis in vivo, Sec incorporation at UGA codons 3–10 is relatively processive and efficient. This is in contrast to previous work with transfected cells where UGA readthrough of a bicistronic reporter construct with multiple UGA codons was reduced to near background levels (26). We have found that total Sec incorporation into Sel P protein in vitro is up to 5–8 times greater than that obtained for our luciferase constructs with a PHGPx SECIS element. From these results we must conclude that something in the Sel P mRNA allows for increased efficiency. Since the Sel P mRNA contains two SECIS elements in the context of over 1 kb of 3′-UTR, a detailed analysis of this sequence will be required to determine the functional components within this mRNA.
One of the surprising results obtained in this study is that in transfected cells the total pre-Sec peptide to full-length luciferase ratio does not reflect efficiency as measured by the difference in activity between L/Sec/P and L/Cys/P. As stated above the two most likely explanations are low pre-Sec peptide stability or reduced overall translation of UGA or UAA containing messages. As shown by Northern analysis, the steady-state levels of the mRNAs generated by these constructs are approximately equal indicating that transfection efficiency and mRNA stability are not likely candidates for the discrepancy. In addition, it seems unlikely that the cell would consider a fragment of luciferase (the pre-Sec peptide) any more “foreign” than the full-length protein, thus making differential protein stability an unlikely explanation as well. The argument can therefore be made that the combination of an in-frame stop codon and at least a partially functional SECIS element may regulate the overall translation rate. This finding may implicate the Sec incorporation machinery in regulating translation initiation. A detailed study of the contribution of regulated overall translation will be required to confirm this hypothesis.
These studies form the foundation for subsequent analysis of the regulation of selenoprotein synthesis. Our results indicate that SBP2 is a key regulatory component of the known Sec incorporation machinery, but that it is likely another factor exists, perhaps an “efficiency factor,” that is required to maximize selenoprotein synthesis. There are several theoretical candidates that stand out as possible “missing” factors. First, it has long been known that the terminal loop of the SECIS element is required for Sec incorporation. However, a binding partner has not yet been identified. Since SBP2 has been demonstrated to be ribosome associated and thus is likely to exert its effects during translation elongation, it is likely that a free cytoplasmic factor is involved in SECIS element recruitment and/or stabilization. The key to efficiency is likely to be at the elongation/termination “decision,” and it may be that SBP2 alone is insufficient to adequately prevent termination or enhance elongation. In addition, because we are able to observe increased efficiency when using an authentic selenoprotein mRNA (Sel P), it is likely that the luciferase constructs used in this study do not contain as of yet unidentified cis-elements that may be required for maximum efficiency. Such elements are likely to be found as Sec codon context or SECIS element context within the 3′-UTR. The methods described here will allow us to systematically and quantitatively test each of these hypotheses.
Acknowledgments
We thank Dolph Hatfield and Brad Carlson for providing Sec-tRNASec and Terri Kinzy for a critical reading of this manuscript as well as providing Phe-tRNAPhe. Thanks also to Ray Burk and Kristina Hill for providing the Sel P cDNA as well as to Marla Berry for providing the clone for eEFSec.
Footnotes
The abbreviations used are: Sec, selenocysteine; SECIS, Sec insertion sequence; UTR, untranslated region; PBS, phosphate-buffered saline; DAPI, 4′,6-diamidino-2-phenylindole; RRL, rabbit reticulocyte lysates; eEFSec, a Sec-specific translation elongation factor.
P. R. Copeland, unpublished data.
M. Anand and T. G. Kinzy, personal communication.
This work was supported by Public Health Service Grant GM068077 from the National Institutes of Health.
References
- 1.Driscoll DM, Copeland PR. Annu Rev Nutr. 2003;23:17–40. doi: 10.1146/annurev.nutr.23.011702.073318. [DOI] [PubMed] [Google Scholar]
- 2.Kryukov GV, Castellano S, Novoselov SV, Lobanov AV, Zehtab O, Guigo R, Gladyshev VN. Science. 2003;300:1439–1443. doi: 10.1126/science.1083516. [DOI] [PubMed] [Google Scholar]
- 3.Sunde RA. In: Selenium: Its Molecular Biology and Role in Human Health. Hatfield DL, editor. Kluwer Academic Publishers; Boston: 2001. pp. 81–98. [Google Scholar]
- 4.Moriarty PM, Reddy CC, Maquat LE. Mol Cell Biol. 1998;18:2932–2939. doi: 10.1128/mcb.18.5.2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun X, Li X, Moriarty PM, Henics T, LaDuca JP, Maquat LE. Mol Biol Cell. 2001;12:1009–1017. doi: 10.1091/mbc.12.4.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weiss Sachdev S, Sunde RA. Biochem J. 2001;357:851–858. doi: 10.1042/0264-6021:3570851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zavacki AM, Mansell JB, Chung M, Klimovitsky B, Harney JW, Berry MJ. Mol Cell. 2003;11:773–781. doi: 10.1016/s1097-2765(03)00064-9. [DOI] [PubMed] [Google Scholar]
- 8.Klein DJ, Schmeing TM, Moore PB, Steitz TA. EMBO J. 2001;20:4214–4221. doi: 10.1093/emboj/20.15.4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Allmang C, Carbon P, Krol A. RNA. 2002;8:1308–1318. doi: 10.1017/s1355838202020034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Copeland PR, Stepanik VA, Driscoll DM. Mol Cell Biol. 2001;21:1491–1498. doi: 10.1128/MCB.21.5.1491-1498.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Copeland PR, Fletcher JE, Carlson BA, Hatfield DL, Driscoll DM. EMBO J. 2000;19:306–314. doi: 10.1093/emboj/19.2.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fletcher JE, Copeland PR, Driscoll DM, Krol A. RNA. 2001;7:1442–1453. [PMC free article] [PubMed] [Google Scholar]
- 13.Suppmann S, Persson BC, Bock A. EMBO J. 1999;18:2284–2293. doi: 10.1093/emboj/18.8.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ursini F, Heim S, Kiess M, Maiorino M, Roveri A, Wissing J, Flohe L. Science. 1999;285:1393–1396. doi: 10.1126/science.285.5432.1393. [DOI] [PubMed] [Google Scholar]
- 15.Low SC, Grundner-Culemann E, Harney JW, Berry MJ. EMBO J. 2000;19:6882–6890. doi: 10.1093/emboj/19.24.6882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kollmus H, Flohe L, McCarthy JE. Nucleic Acids Res. 1996;24:1195–1201. doi: 10.1093/nar/24.7.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berry MJ, Harney JW, Ohama T, Hatfield DL. Nucleic Acids Res. 1994;22:3753–3759. doi: 10.1093/nar/22.18.3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Judd PA, Long A, Butcher M, Caygill CP, Diplock AT. Br Med J. 1997;314:1834–1834. doi: 10.1136/bmj.314.7097.1834a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee BJ, Kang SG, Hatfield D. J Biol Chem. 1989;264:9696–9702. [PubMed] [Google Scholar]
- 20.Hatfield D, Lee BJ, Hampton L, Diamond AM. Nucleic Acids Res. 1991;19:939–943. doi: 10.1093/nar/19.4.939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Merrick WC. Methods Enzymol. 1979;60:101–108. doi: 10.1016/s0076-6879(79)60010-1. [DOI] [PubMed] [Google Scholar]
- 22.Martin GW, Harney JW, Berry MJ. RNA. 1996;2:171–182. [PMC free article] [PubMed] [Google Scholar]
- 23.Tujebajeva RM, Copeland PR, Xu XM, Carlson BA, Harney JW, Driscoll DM, Hatfield DL, Berry MJ. EMBO Rep. 2000;1:1–6. doi: 10.1093/embo-reports/kvd033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Himeno S, Chittum HS, Burk RF. J Biol Chem. 1996;271:15769–15775. doi: 10.1074/jbc.271.26.15769. [DOI] [PubMed] [Google Scholar]
- 25.Ma S, Hill KE, Caprioli RM, Burk RF. J Biol Chem. 2002;30:30. doi: 10.1074/jbc.M111462200. [DOI] [PubMed] [Google Scholar]
- 26.Nasim MT, Jaenecke S, Belduz A, Kollmus H, Flohe L, McCarthy JE. J Biol Chem. 2000;275:14846–14852. doi: 10.1074/jbc.275.20.14846. [DOI] [PubMed] [Google Scholar]