

Abstract
Carbon monoxide dehydrogenase/acetyl-CoA synthase catalyzes acetyl-CoA synthesis from CO, CoA, and a methylated corrinoid iron-sulfur protein, which acts as a methyl donor. This reaction is the last step in the Wood-Ljungdahl pathway of anaerobic carbon fixation. The binding sequence for the three substrates has been debated for over a decade. Different binding orders imply different mechanisms (i.e. paramagnetic versus diamagnetic mechanisms). Ambiguity arises because CO and CoA can each undergo isotopic exchange with acetyl-CoA, suggesting that either of these two substrates could be the last to bind to the acetyl-CoA synthase active site. Furthermore, carbonylation, CoA binding, and methyl transfer can all occur in the absence of the other two substrates. Here, we report pulse-chase studies, which unambiguously establish the order in which the three substrates bind. Although a CoA pulse is substantially diluted by excess CoA in the chase, isotope recovery of a pulse of labeled CO or methyl group is unaffected by the presence of excess unlabeled CO or methyl group in the chase. These results demonstrate that CoA is the last substrate to bind and that CO and the methyl group bind randomly as the first substrate in acetyl-CoA synthesis. Up to 100% of the methyl groups and CoA and up to 60–70% of the CO employed in the pulse phase can be trapped in the product acetyl-CoA.
There are three types of CO dehydrogenase (CODH)2 (1, 2). These include a copper molybdopterin iron-sulfur protein that is present in aerobic bacteria and an NiFeS enzyme that is present in anaerobic microbes. These enzymes allow microbes to utilize CO at the low concentrations present in the atmosphere as a carbon and electron source. A third type of CODH is highly homologous to the monofunctional NiFeS enzyme and is found tightly associated with another NiFeS enzyme called acetyl-CoA synthase (ACS), forming the CODH·ACS complex.
CODH·ACS is a macromolecular machine that catalyzes the last step of the Wood-Ljungdahl pathway of anaerobic carbon dioxide fixation (Reaction 1). The 300-kDa α2β2 heterotetramer is composed of two types of subunits. The 72-kDa β-subunit is CODH, which catalyzes the reversible oxidation of CO to CO2 (Reaction 2) at the C-cluster, a novel [NiFe4S4–5] cluster (3, 4), whereas the 82-kDa α-subunit is ACS, which catalyzes the condensation of three substrates, CoA, CO, and a methyl group from the methylated corrinoid iron-sulfur protein (CH3-CFeSP), to produce acetyl-CoA. Methylation of the CFeSP is accomplished by a methyltransferase (MeTr), which catalyzes the transfer of a methyl group from CH3-H4folate to the Co(I) state of the CFeSP, according to Reaction 3 (5). Although MeTr contains no metals or other cofactors, the CFeSP contains a corrinoid cofactor and a [Fe4S4] cluster, which is involved in the reactivation of the cobalt center back to the 1+ state whenever it has undergone oxidation (approximately once in every 1000 catalytic cycles) (6, 7). The resulting CH3-CFeSP becomes the methyl donor to ACS (8, 9). Acetyl-CoA generated by the Wood-Ljungdahl pathway can either serve as a source of energy, by linking thioester bond cleavage to the formation of ATP, or as a building block for cell carbon synthesis by entry into the reductive (or incomplete) tricarboxylic acid cycle. A variant of CODH·ACS, which is found in methanogenic archaea, is called acetyl-CoA decarbonylase synthase and allows methanogens to generate cell carbon from CO2 and to use acetate as a source of carbon and energy. In this case, acetyl-CoA is disassembled by acetyl-CoA decarbonylase synthase by a reversal of Reaction 1 (10).
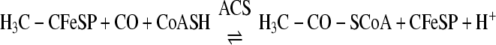 |
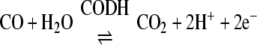 |
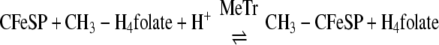 |
ACS carries out the condensation reaction shown by Reaction 1 at a novel [Fe4S4]-Nip-Nid cluster (11, 12) known as the A-cluster. The Nip and Nid designations correspond to the nickel atoms that are proximal and distal to the [Fe4S4] cubane, respectively. The A-cluster has unusual electronic and magnetic properties in that the [Fe4S4] and Nid components appear to be redox-inactive and to remain in the EPR-silent +2 state during the catalytic cycle (13–16). On the other hand, the Nip atom is stable in the +2 and +1 states (2, 11, 17), and it has been proposed that it can even cycle through the 0+ state during catalysis (18). Another unusual feature of the A-cluster is that the +1 state has been observed for both CODH·ACS (19) and ACSHT (20, 21) only in complexes of the reduced enzymes with CO. There is agreement that CO (17) and the methyl group (8) bind to the Nip site.
The order in which the three substrates bind to ACS (or in which these products dissociate from acetyl-CoA decarbonylase synthase) has not been established and has been a controversial topic. Some insight has been gleaned from isotope exchange studies. In the absence of CFeSP, CODH·ACS catalyzes the CO/acetyl-CoA exchange reaction between free CO in solution and [1-14C]acetyl-CoA (22–24) (Reaction 4). This reaction occurs at a rate similar to that of acetyl-CoA synthesis and involves cleavage of the C–C and C–S bonds of acetyl-CoA to form a quaternary complex in which 14CO, CH3, and CoA independently bind to ACS; then bound 14CO undergoes isotopic exchange with CO in solution (25–27). This reaction occurs with retention of stereochemical configuration at the methyl moiety of acetyl-CoA (9). CODH·ACS also catalyzes a CoA/acetyl-CoA exchange between CoA and acetyl-CoA (Equation 5). Ramer et al. (27) concluded that CoA binds as the final substrate and that acetyl-CoA synthesis occurs through an acetyl-enzyme intermediate, because the CoA/acetyl-CoA exchange occurs ∼6-fold faster than the CO/acetyl-CoA exchange reaction. The rate of this reaction increases as the redox potential is lowered, following a Nernst curve with a mid-point redox potential of -520 mV and reaching a value ∼40-fold faster than the CO/acetyl-CoA exchange (28). Bashkar et al. (29) observed similar properties for the CoA/acetyl-CoA exchange reaction with the acetyl-CoA decarbonylase synthase from Methanosarcina thermophila. Thus, many cycles of cleavage and resynthesis of the C–S bond of acetyl-CoA can occur for each cycle of C–C bond synthesis/cleavage, indicating that CoA is the last substrate to add and that it reacts with an acetyl-enzyme intermediate.
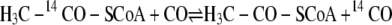 |
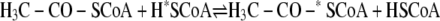 |
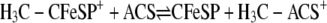 |
Although it is rather accepted that CoA is the last substrate to bind to ACS, it is controversial whether CO or the methyl group binds first. Lu et al. (30) first demonstrated that methylation of CODH·ACS with CH3-CFeSP (Reaction 6) could be carried out in the absence of CoA and CO. Since the carbonylated enzyme or the methylated enzyme could be converted to acetyl-CoA when the other two substrates were added, it was suggested that CO and the methyl group bind randomly (30). On the other hand, Barondeau and Lindahl (8) proposed that methylation of ACS is the first step in an ordered mechanism of acetyl-CoA synthesis because the methyl group of the CH3-CFeSP could be transferred to CODH·ACS (in the absence of CO), forming a methylated enzyme that, when treated with CO and CoA, forms acetyl-CoA. That methylation of CODH·ACS or recombinant (ACSHT) (31, 32) by CH3-CFeSP occurs faster than acetyl-CoA synthesis also suggests that methylation is the first step in the synthesis (33). Furthermore, CO acts as a substrate inhibitor of acetyl-CoA synthesis (34, 35). On the other hand, acetyl-CoA synthesis is linearly dependent on CO at concentrations likely to be physiologically relevant, with CO becoming inhibitory only at relatively high concentrations (>100 μm) (34). Furthermore, CODH·ACS can be carbonylated at catalytically relevant rates to form a paramagnetic adduct called the NiFeC species (17, 36), and the carbonylated enzyme can then undergo methylation and CoA binding to form acetyl-CoA at catalytically relevant rates (33). Thus, definitive experiments to establish the order of binding have not been previously described.
Establishing the order of substrate addition is of paramount importance in terms of the mechanistic possibilities for ACS action. It was stated that a strictly ordered reaction sequence with the methyl group binding first would rule out the paramagnetic mechanism, which includes the intermediate NiFeC species (8). As described above, all possibilities have been proposed for the first step: (a) the methylation step (18), (b) methylation or CoA binding (37), (c) carbonylation (2), and (d) random binding of CO and the methyl group followed by CoA binding (30). The first two proposals include a two-electron activation of ACS followed by methylation in a diamagnetic mechanism, whereas the third and fourth alternatives assume a one-electron activation followed by methylation or carbonylation in a paramagnetic cycle (Fig. 1).
FIGURE 1.

A-cluster states according to the paramagnetic and diamagnetic cycles for acetyl-CoA synthesis by ACS.
Steady-state methods such as product inhibition experiments, which traditionally aid in establishing binding order and in ruling out random versus ordered mechanisms (38, 39) are difficult to apply to the ACS reaction. First, a large set of experiments would be needed, since acetyl-CoA synthesis has three substrates and two products, and patterns of inhibition are quite complex in a Tri-Bi mechanism. Second, steady-state experiments are complicated by the behavior of the substrates. Acetyl-CoA synthesis shows a linear dependence on the CH3-CFeSP concentration (40); moreover, CO is a potent uncompetitive substrate inhibitor (34). Another alternative for establishing the order of substrate binding is to use of uncompetitive (41) or bisubstrate (42) inhibitors, such as desulfo-CoA, but, again, the complexities associated with CO and CH3-CFeSP binding steps complicate the interpretation of the results.
The purpose of the present study is to establish the order of the addition of substrates by pulse-chase isotope-trapping experiments (43). Radiolabeled forms of the CH3-CFeSP and CO can be readily prepared. Since dephospho-CoA has been shown to have a Km value similar to that of CoA (29), it can be used in place of 32P-labeled CoA. Thus, in such experiments, the dephospho-CoA is equivalent in principle to the “isotopically labeled” substrate. Most importantly, since steady-state conditions are avoided, no substrate or product inhibition should occur.
Our results strongly indicate that acetyl-CoA synthesis occurs by a random mechanism, with either methyl transfer or CO binding occurring first, followed by the ordered addition of CoA as the last substrate in the reaction. That the only detectable carbonylated form of ACS is the paramagnetic NiFeC species (17) coupled with demonstration of a random mechanism of CO and methyl binding provides strong support for the paramagnetic mechanism of acetyl-CoA synthesis.
EXPERIMENTAL PROCEDURES
Materials—Moorella thermoacetica (DSM 521) was grown anaerobically at 55 °C as previously described (44, 45). CODH·ACS and CFeSP were purified as described (20). All the purification steps and sample preparations were carried out in an anaerobic chamber from Vacuum Atmospheres (Hawthorne, CA) under nitrogen gas containing <2 ppm of oxygen. Recombinant MeTr was overexpressed in Escherichia coli strain BL21(DE3) Star from Novagen (San Diego, CA) and purified as previously described (46, 47). Titanium citrate (TiCit) was prepared as described (48) from titanium chloride (Pfaltz and Bauer, Waterbury, CT). Radiolabeled sodium 14C-formate was obtained from MP Biochemicals (Solon, OH), and 14CH3-H4folate was purchased from Amersham Biosciences/GE Healthcare. CH3-H4folate monoglutamate (Ca2+ salt) was supplied by Schircks laboratories (Jena, Switzerland). All other chemicals were obtained either from Sigma or J. T. Baker Inc. and were used without further purification. EPR spectra were measured at 100 K, 1.0 milliwatt microwave power, 100 kHz modulation frequency, and 20,000 detector gain on a Bruker EMX instrument (Bruker, Billerica, MA) equipped with an Oxford ITC4 temperature controller, a Hewlett-Packard model 5340 automatic frequency counter, and Bruker gaussmeter.
Cloning, Expression, and Activation of the acsB Gene—The ACS (acsB) gene was amplified from M. thermoacetica genomic DNA by PCR using primers GGCGGCATATGACTGATTTTGATAAAATCTTCG and GGCGGCTCGAGCATAATGGGATCCATGGTCAAG and cloned into the pET29 vector (Novagen, San Diego, CA), which introduced a C-terminal His6 tag. After confirming the ACS DNA sequence, the resulting plasmid (pETACSMTHT) was transformed into BL21 (DE3) Star competent cells that had been previously transformed with pDB1282, which contains the iscS-iscU-iscA-hscB-hscA-fdx portion of the isc operon from Azotobacter vinelandii under the control of an arabinose promoter (a kind gift from Drs. Dennis Dean and Patricia DosSantos, Virginia Tech, Blacksburg, VA). Cells containing both pDB1282 and pETACSMTHT were isolated from agarose plates containing ampicillin and kanamycin.
For overexpression of ACSMTHT, E. coli was grown aerobically at 37 °C in Terrific Broth. When the A600 reached ∼0.4, solid arabinose (0.5 g/liter of culture) was added to induce expression of the isc proteins. When the A600 reached ∼0.7, the gas phase was switched to N2, and 30 min later anaerobic sterile 0.1 mm Fe(NH4)(SO4)2, 2 mm cysteine, and 1.5 mm Na2S were added. When the optical density at 600 nm reached ∼0.8, expression of ACS was induced by the addition of 0.2 mm isopropyl thiogalactoside and 0.2 mm NiCl2. After 12 h, the cells were harvested and stored in liquid nitrogen. The cells were lysed by suspending in Buffer A (0.05 m sodium phosphate buffer pH 8.0, 0.3 m NaCl, 10 mm imidazole and 10 mm β-mercaptoethanol) supplemented with 0.25 mg/ml lysozyme, 0.2 mm phenylmethylsulfonyl fluoride, and 4 units/ml deoxyribonuclease I and sonicating for 10 min. The suspension was then ultracentrifuged at 4 °C at 30,000 rpm for 1 h. ACSMTHT was purified on an Ni2+-nitrilotriacetic acid Superflow column from Qiagen (Valencia, CA) and reconstituted by adding 4 eq of NiCl2 per ACS monomer. Reconstituted ACS was desalted by concentrating and diluting into 0.3 m MES, pH 6.1, using an Amicon concentrator equipped with a YM30 membrane (Millipore, Billerica, MA). After treating the reconstituted protein with 2 mm sodium dithionite and carbon monoxide for 10 min, the resulting protein showed 0.33 spins of the NiFeC signal at 100 K. The nickel-reconstituted protein contained ∼3.2 iron atoms and 2 nickel atoms per monomer, based on inductively coupled plasma emission spectrometric analysis, which was carried out at the Chemical Analysis Laboratory at the University of Georgia (Athens, GA). The specific activity of the reconstituted ACSMTHT in the exchange reaction between CO and 14C1-acetyl-CoA was 0.72 units/mg at 55 °C.
Methylation of the CFeSP—Methylation of the CFeSP was accomplished by incubating purified CFeSP (150 μm) with 30 μm MeTr, 3.0 mm CH3-H4folate (or 14CH3-H4folate for preparation of 14CH3-CFeSP), 1.0 mm TiCit in 10 mm Tris-HCl, pH 7.60, containing 100 mm NaCl at 55 °C. The level of CFeSP methylation was assessed by measuring the disappearance of Co1+ at 390 nm and the formation of CH3-Co3+ at 450 nm by UV-visible spectroscopy. The reaction mix was frozen in liquid nitrogen and stored until needed. For pulse-chase experiments in which 14CH3-CFeSP was used, TiCit and 14CH3-H4folate were removed by diafiltration into 10 mm Tris-HCl, pH 7.60, 0.1 m NaCl. Based on the incorporation of 14C, the protein contained ∼0.95 methyl groups per CFeSP dimer. The final specific activity of the methylated CFeSP was determined based on dilution of 14CH3-H4folate with unlabeled CH3-H4folate, using the measured dpm/μl of solution and the spectrophotometrically measured concentration with an extinction coefficient of 32,000 m-1 cm-1 at 298 nm.
Synthesis of 14CO—Radiolabeled CO was prepared from 14C-labeled sodium formate by acid-catalyzed dehydration. In an 8.0-ml vial containing a small stir bar, 2.0 ml of concentrated sulfuric acid was added, and the bottle was sealed with a butyl rubber stopper. The acid was extensively degassed by stirring under 100% N2 in a Schlenk line on which oxygen gas was removed with a heated copper catalyst (BASF, Florham Park, New Jersey). The degassed acid was incubated at 75 °C in the anaerobic chamber, and a mixture of 0.1 ml of 1 m unlabeled and 100 μCi of 14C-labeled sodium formate was slowly injected while stirring. The resulting 14CO was removed with a 5.0-ml disposable syringe containing two pellets of sodium hydroxide. Another disposable syringe was used to inject anaerobic water into the acid/formate mix to allow removal of more 14CO into the hydroxide-containing syringe, without creating back pressure. The gas was then injected into another sealed anaerobic bottle containing 3–4 ml of 10 mm Tris-HCl, pH 7.60, and 0.1 m NaCl. The concentration of CO was determined spectrophotometrically using 20 μm myoglobin, 2 mm sodium dithionite in 0.1 m Tris, pH 7.60, in a total volume of 1.0 ml. The injected sample contained between 50 and 100 nmol of 14CO. To quantify the amount of 14CO, a sample (50–100 μl) of buffer with a partial pressure of 14CO was injected into a scintillation vial, which was filled up to the top with scintillation fluid and sealed with a rubber septum. The specific radioactivity (2220 dpm/nmol of CO) was estimated from the 14C counts, and the concentration of CO was determined from a myoglobin assay at 425 nm, using an extinction coefficient of 80,000 m-1 cm-1 for the CO-myoglobin complex.
Pulse-Chase Experiments—Prior to the pulse-chase experiments, CODH·ACS or ACSHT was buffer exchanged into 10 mm Tris-HCl, pH 7.60, and 0.1 m NaCl using a 3.0-ml Amicon concentrator (Millipore), equipped with a YM 30 membrane. Dephospho-CoA was prepared at a concentration of 30 μm in the same buffer from a 1 mm stock solution. The concentrations of CoA and dephospho-CoA were determined from their absorption values at 260 nm, based on an extinction coefficient of 16,000 m-1 cm-1. TiCit was added to each solution used in the pulse-chase experiments to a final concentration of 1.0 mm. Experiments were carried out either in a sealed V-vial or in a rapid chemical quench apparatus. The pulse solution was composed of CODH·ACS (or ACSHT), which had been preincubated with one of the three “labeled” substrates (dephospho-CoA, 14CO, or 14CH3-CFeSP) for a given amount of time. Then the pulse solution was injected into the chase solution, which contained the two substrates missing in the pulse solution to complete the acetyl-CoA synthesis reaction plus an excess of the unlabeled substrate present in the pulse. Finally, 95 μl of 0.1 m hydrochloric acid was added to quench the reaction after the desired reaction time.
For the rapid mixing reactions, a rapid chemical quench apparatus from Update Instruments (Madison, WI) was used. The enzyme (ACS or CODH·ACS) solution, contained in the first syringe, was rapidly mixed with a solution containing a substoichiometric amount of one of the substrates (dephospho-CoA, 14CO, or 14CH3-CFeSP) in the second syringe. An empty 66-μl hose was coupled after the first mixer in order to incubate the pulse solution. The third syringe, containing the chase solution, was coupled to an empty 33-μl hose in order to add a delay time. These two hoses were attached to a second mixer in turn attached to an empty 100-μl final reaction hose, which was the reactor for the chase phase of the experiment. Three shots were collected in a single experiment with identical time delays for pulse and chase. The first shot filled the 66- and 33-μl hoses and started the pulse phase, the second initiated the chase phase and filled the 100-μl hose, and the third quenched the reaction into 1.5-ml Eppendorf tubes containing 95 μl of 0.1 m hydrochloric acid. In order to correct for any loss of acetyl-CoA during HPLC analysis, 5 μl of 3H-labeled acetyl-CoA (∼1.2 nmol) was added to the quenched reaction mixture (total volume ∼200 μl). The final concentration of acetyl-CoA was 225 μm in a total sample volume of 200 μl. The samples were then frozen in liquid nitrogen and stored at -20 °C until analyzed by HPLC and scintillation counting, as described below.
When dephospho-CoA was the substrate, the chase mix contained 1.0 mm CO, 0.1 m MES (pH 6.1), 0.33 mm TiCit, 10 μm MeTr, 50 μm 14CH3-CFeSP (3394 dpm/nmol), 0.35 mm 14CH3-H4folate, and either 0 (as the control) or 1.0 mm CoA (as the chase). 14CH3-H4folate was initially added to the chase mix at a concentration of 1.0 mm to ensure complete methylation of CFeSP but reduced to 0.35 mm by means of concentration and dilution of this mix in the Amicon concentrator, in order to reduce the amount of background radioactivity present in the final quenched samples.
When 14CO was the substrate, the chase mix contained the same components as for dephospho-CoA as substrate, with the following changes: 1) either no CO or 1 atm CO (the chase); 2) 50 μm unlabeled CH3-CFeSP (instead of 14C-labeled); 3) 1.0 mm unlabeled CH3-H4folate; and 4) 1 mm CoA and no dephospho-CoA.
When 14CH3-CFeSP was the substrate, the chase consisted of 0.1 m MES (pH 6.1), 0.33 mm TiCit, 1.0 mm CoA, 1 atm CO, and unlabeled CH3-CFeSP at the concentrations indicated under “Results.” The total amount of acetyl-CoA present in the sample cannot be accurately determined, since 3H-labeled and unlabeled acetyl-CoA were both present in order to identify and collect the product by HPLC. The 14C label is incorporated into the C1 of acetyl-CoA with 14CO as a substrate, whereas the same label is incorporated into the C2 with 14CH3-CFeSP and dephospho-CoA as substrates.
Analysis of the Pulse-Chase Reactions—The samples were divided into two portions. A total of 100 μl of the quenched sample was injected into a Waters high pressure liquid chromatograph (Milford, MA) with a μBondapak 125-Å C18-Reverse Phase column (5 × 300 mm) equilibrated with 0.1 m potassium phosphate buffer (pH 5.60) and 15% (v/v) methanol. The separation was developed by isocratic elution at 2.0 ml/min and was monitored at 260 nm. The elution times for the different substrates and products were as follows: 3 min for H4folate, 4–5 min for CoA, 6–8 min for CH3-H4folate and dephospho-CoA, 10–12 min for acetyl-CoA, and 20–22 min for dephospho-acetyl-CoA. The acetyl-CoA or dephospho-acetyl-CoA peaks were collected and mixed with scintillation fluid. The remainder of the sample was counted separately. The sum of the volumes of the HPLC-injected fraction and the remainder fraction volumes minus 100 μl (from the quench plus [3H]acetyl-CoA) was assumed to be the total volume of the reaction (Vr). The radioactivity for all samples was counted on a Packard Instruments scintillation counter (PerkinElmer Life Sciences). The protocol used measures the counts/min between 0 and 18 keV in one channel (3H, low energy) and the counts/min between 18 and 156 keV in the second channel (14C, high energy). The instrument then uses the correction for 14C-scintillation within the low energy channel to calculate the 3H and 14C dpm. The protocol was tested with known standards for both isotopes using the 12.2-year half-life decay curve for 3H. The percentage of 3H recovery (P.T.R.), which was between 70 and 100%, was calculated from the ratio of 3H dpm/μl of the acetyl-CoA peak and the 3H dpm/μl of the remainder fraction. The recovery of acetyl-CoA (in nmol) was calculated from the 14C dpm in the acetyl-CoA peak from the HPLC elution. The concentration of acetyl-CoA (in nmol/ml) was calculated according to Equation 1. The percentage recovery of substrate as acetyl-CoA product was calculated according to Equation 2, where [substrate] is the concentration of the limiting labeled substrate (CO, CH3-CFeSP, or dephospho-CoA).
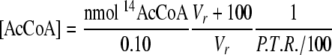 |
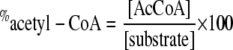 |
In Equation 1, the volume injected into the HPLC system was 0.1 ml. The concentration of substrate in Equation 8 refers to the substrate (14CO, 14CH3-CFeSP, or dephospho-CoA) being chased during a given experiment.
RESULTS
Isotope Trapping as Acetyl-CoA and Establishment of the Time Course—We used pulse-chase methodology to establish the order in which ACS binds its three substrates. These experiments involved reacting the enzyme with labeled CO or CH3-CFeSP or with dephospho-CoA (“labeled” CoA) in the pulse phase and reacting with excess unlabeled substrates in the chase. The samples of CODH·ACS and ACS exhibited ∼0.33 NiFeC species per monomer upon treatment with 2 mm sodium dithionite and 1 atm CO.
The goal of the pulse-chase experiments is to measure the amount of isotope dilution that occurs for each of the labeled substrates, not rates. To ensure that the incubation times, for the pulse and the chase phases of the reaction, are sufficiently long for binding and product formation, we used a chemical quench apparatus to mix 50 μm CODH·ACS with 7 μm 14CH3-CFeSP (final concentrations), incubated for 30 s, and then mixed this solution with another solution containing 0.1 mm CO and 1.0 mm CoA (also final concentrations) for variable periods of time. We included 1 mm TiCit to accomplish reductive activation of CODH·ACS without consuming CO, which is present in approximately stoichiometric amounts. As shown in Fig. 2, acetyl-CoA formation follows a biphasic exponential increase with up to 14% of the methyls trapped as acetyl-CoA, and, after 30 s, over 90% of the reaction is complete. Thus, 100 s was chosen as the mixing time for both the pulse and the chase phases of the experiments. For CO, we have previously determined the rates of carbonylation of CODH·ACS (33) and of reduced ACSMTHT (17) to be 1.0 s-1. The rate of CoA binding has not been measured, but the kcat for the acetyl-CoA/dephospho-CoA exchange reaction is 1.87 units/mg or 4.8 s-1 at 20 °C (20). It is likely that binding of dephospho-CoA and CoA occurs much more rapidly than 4.8 s-1; regardless, the binding of these two substrates would be fully equilibrated after 10 half-lives, or 70 s.
FIGURE 2.
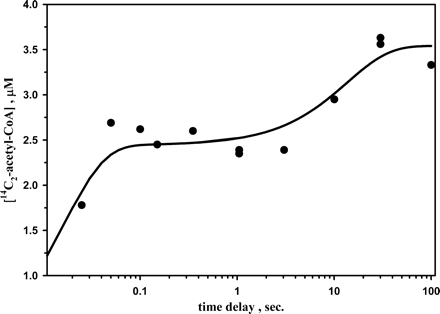
Trapping of 14CH3-CFeSP as 14C2-acetyl-CoA by rapid chemical quench. The final concentrations of the reactants were 50 μm CODH·ACS, 14 μm 14CH3-CFeSP, 1 mm CoA, 1 mm TiCit, and 0.1 mm CO, all in 0.1 m MES, pH 6.10. The data were fitted to a double exponential increase function with rate constants equal to 50 and 0.1 s-1 and corresponding amplitudes of 2.5 and 1.0 μm.
Methyl Group Pulse-Chase Reactions—When 50 μm CODH·ACS was rapidly mixed with 6.2 μm 14CH3-CFeSP and this solution was mixed with 0.5 atm (490 μm) CO, and 1 mm CoA (all are final concentrations), about 20–50% of the methyl groups were converted to product, acetyl-CoA (Table 1). The pulse phase lasted ∼100 s; thus, the equilibrium between 14CH3-CFeSP and CODH·ACS was clearly established, since methyl transfer from CH3-CFeSP to ACS occurs with rate constants between 3.0 and 7.0 s-1 (31).
TABLE 1.
Pulse-chase experiments for the trapping of 14CH3 groups with CODH/ACS or with ACS with 1 mm TiCit, 1 mm CoA, and 1.0 atm CO
The background of radioactivity detection was measured using a control without CODH/ACS or ACS. It was estimated as 1.2 μm. The concentrations given are final concentrations.
|
Time delay |
[CODH/ACS] |
[14CH3-CFeSP] pulse |
[CH3-CFeSP] chase |
[14C2-acetyl-CoA] formeda |
Percentage recovery in pulse/chaseb |
Expected percentage recoveryc |
|---|---|---|---|---|---|---|
| s | μm | μm | μm | μm | % | % |
| 100 | 50 | 6.2 | 0 | 3.80 ± 0.5 | ||
| 80 | 1.80 ± 0.5 | 47 ± 18 | 7 | |||
| 15 | 0 | 3.1 ± 0.5 | ||||
| 80 | 3.5 ± 0.5 | 110 ± 24 | 16 | |||
| 10 | 0 | 5.0 ± 0.5 | ||||
| 80 |
6.8 ± 0.5 |
135 ± 17 |
11 |
|||
|
[ACS] |
||||||
| s | μm | |||||
| 120 | 24 | 24 | 0 | 4.8 ± 0.5 | ||
| 70 |
5.7 ± 0.5 |
120 ± 16 |
26 |
Recovery was based on 14C recovered in acetyl-CoA as calculated by Equation 1
Percentage recovery was calculated by Equation 2
The expected percentage recovery is based on the isotope dilution if the methyl group must dissociate before product can be formed, equal to the ratio of [14CH3-CFeSP] to total [CH3-CFeSP]
When the above reaction was repeated with 240 μm unlabeled CH3-CFeSP (80 μm, final concentration), in the CO/CoA solution, the amount of radiolabeled methyl groups collected in the product acetyl-CoA was the same as in the absence of the CH3-CFeSP chase. If the methyl group needed to dissociate in order that CO or CoA bind as the first substrate, one would expect only 7% recovery of radioactivity in the product based on the mass ratio of the labeled to unlabeled methyl groups present in the chase.3 Therefore, minimal isotope dilution occurred in the pulse-chase experiment. When the 14CH3-CFeSP concentration was increased to 10 or 15 μm, the yield of labeled methyl groups increased, and again no isotope dilution was observed in the pulse-chase experiment.
Similar results were obtained with the recombinant M. thermoacetica ACS subunit (ACSMTHT), which lacks an intramolecular tunnel for CO/CO2. When ACSMTHT was incubated with 14CH3-CFeSP (Table 1) and TiCit for ∼100 s and then rapidly mixed with an excess of CO and CoA (1 mm final concentration each), 4.8 μm 14C2-acetyl-CoA (20.8%) was recovered as acetyl-CoA. When a 3-fold excess of CH3-CFeSP (relative to the labeled substrate) was used to chase the 14CH3-CFeSP, an almost identical isotope recovery was obtained, indicating no isotope dilution by the chase with the unlabeled methyl group.
Therefore, with both CODH·ACS and the ACS subunit alone, methyl group transfer from the 14CH3-CFeSP forms a productive CH3-ACS complex that is committed to acetyl-CoA synthesis. These results are consistent with either an ordered mechanism with the methyl group binding first or with a random mechanism and inconsistent with an ordered mechanism with CO or CoA as the first substrate.
14CO Pulse-Chase Reactions—To measure the extent of conversion of 14CO into acetyl-CoA in pulse-chase experiments, we mixed a solution containing CODH·ACS (50 μm, final) and 14CO (42 μm, final) with another solution containing 1 mm TiCit, 1 mm CoA, 50 μm CH3-CFeSP, 1 mm CH3-H4folate, and 11 μm MeTr (all final concentrations) and quenched the reaction after 100 s. Recovery of the 14CO in acetyl-CoA was between 64 and 69% (Table 2). The concentration of active ACS was ∼17 μm, since the fraction of active ACS was equivalent to the fraction of this paramagnetic NiFeC species (0.33 spins per monomeric ACS) (20, 33). The limiting reagent in these reactions was 14CO, since the CoA and methyl groups (in the CH3-CFeSP and CH3-H4folate/MeTr) were both present at 1 mm concentrations. The CoA concentration was about 40-fold higher than the Ki for CoA (25 μm) to ensure that no isotope dilution of the 14C1-acetyl-CoA occurred from carbonyl exchange with free CO. In the pulse-chase experiment with unlabeled CO, to obtain maximal isotope dilution, the quench solution (CODH·ACS and 14CO) was injected into a sealed V-vial containing the same substrates as above (TiCit, CoA, CH3-CFeSP, CH3-H4folate, and MeTr) plus 1 atm CO. As with the methyl group pulse-chase experiments, the amount of 14C incorporated into acetyl-CoA did not diminish when 14CO was chased with unlabeled CO (Table 2). Similarly, when ACS was used instead of CODH·ACS, ∼82% of the 14CO was incorporated into acetyl-CoA in the absence of unlabeled CO and 84% was incorporated when 980 μm unlabeled CO was included in the chase.
TABLE 2.
Pulse-chase experiments for trapping 14CO for CODH/ACS and for ACS with 1 mm TiCit, 1 mm CoA, 50 μm CH3-CFeSP, 1 mm CH3-H4folate, and 11 μm MeTr by hand mixing
The experiments in this table were done by hand mixing with both pulse and chase times set at 100 s. All of the experiments without CO chase were under an N2 atmosphere.
|
Reaction time |
CODH/ACS |
[14CO] pulse |
Chase |
[14C1-acetyl-CoA] |
Percentage recovery in pulse/chase |
Percentage recovery expecteda |
|---|---|---|---|---|---|---|
| s | μm | μm | μm | % | ||
| 100 | 50 | 42 | 0 | 31 ± 2 | ||
| 980 μm CO | 28 ± 2 | 90 ± 9 | 4 | |||
| 0 | 29 ± 2 | |||||
| 50 μm Mb | 25 ± 2 | 86 ± 9.0 | 0 | |||
| 0 | 27 ± 2 | |||||
| 15 mm CO2 |
1.3 ± 1.5 |
4.8 ± 5.5 |
0 |
|||
|
[ACSHT] |
||||||
| μm | ||||||
| 120 | 24 | 38 | 0 | 31 ± 2 | ||
| 980 μm CO | 32 ± 2 | 100 ± 9 | 4 | |||
| 29 | 0 | 29 ± 2 | ||||
| 40 μm Mb
|
14 ± 1.5 |
48 ± 6 |
0 |
The expected percentage recovery is based on the isotope dilution if CO must dissociate before product can be formed. It is assumed that myoglobin can bind all of the CO
These experiments unambiguously rule out an ordered mechanism in which the methyl group must bind before CO. If the methyl group must bind before CO, the bound 14CO would have undergone displacement to allow the methyl group to bind, diluting the 14CO by 27-fold and yielding only a 4% recovery of 14C.
To capture any 14CO that was not committed to acetyl-CoA synthesis, 50 μm myoglobin was included in a set of pulse-chase experiments. With CODH·ACS as the catalyst, the amount of 14C converted to acetyl-CoA was unaffected by the presence of myoglobin; however, about 50% of the 14C was trapped in experiments with ACS alone. Similarly, when the 14CO chase was carried out with CODH·ACS in the presence of saturating CO2, washout of the 14CO was observed. These results are consistent with previous experiments, which showed that the CO generated from CO2 at the C-cluster of CODH is sequestered within an intersubunit channel that is inaccessible to myoglobin during its incorporation into the carbonyl of acetyl-CoA (49, 50). In ACS alone, this channel would be disabled and incapable of trapping CO.
In order to measure the total acetyl-CoA generated from 14CO in the pulse-chase experiments, a parallel experiment was performed in which no 3H-labeled acetyl-CoA was present. The total amount of acetyl-CoA was quantified based on the UV spectrum of the lyophilized acetyl-CoA peak from the HPLC analysis. The resulting specific radioactivity was 44 dpm/nmol of total product. This is the expected outcome if 14CO + CO were simultaneously added to the reaction (2220 dpm/nmol × 20/1000). At least two explanations can be considered: 1) the isotopes equilibrated much more quickly than the rate of synthesis, or 2) the entire amount of CO was consumed during 100 s, with a correction for the reversibility of the reaction. The first possibility was ruled out by rapid chemical quench experiments that were quenched from 1 to 100 s. For these experiments, the 14CO (pulse) and unlabeled CO (chase) concentrations were 33 and 326 μm, respectively. Thus, full dilution of the 14C would result in about 9% recovery of the radioactivity. However, the recoveries were 41–100% (Table 3), indicating that the ACS-14CO complex has a large commitment to catalysis.
TABLE 3.
Pulse-chase experiments for trapping 14CO with 1 mm TiCit, 50 μm CH3-CFeSP, 1.0 mm CH3-H4folate, 10 μm MeTr, 1 mm CoA, and 0.33 atm of CO by rapid chemical quench
|
Reaction time |
CODH/ACS |
[14CO] pulse |
[CO] chase |
[14C1-acetyl-CoA]a |
Percentage recovery in pulse/chase |
Percentage recovery expectedb |
|---|---|---|---|---|---|---|
| s | μm | μm | μm | μm | % | % |
| 100.05 | 50 | 33 | 0 | 22.8 ± 2.0 | ||
| 326 | 16.0 ± 1.6 | 70 ± 9 | 10 | |||
| 33.05 | 0 | 21.5 ± 2.0 | ||||
| 326 | 10.0 ± 1.0 | 46 ± 6 | 10 | |||
| 10.05 | 0 | 14.8 ± 1.5 | ||||
| 326 | 6.13 ± 1.5 | 41 ± 11 | 10 | |||
| 3.35 | 0 | 6.90 ± 1.5 | ||||
| 326 | 5.44 ± 1.5 | 79 ± 26 | 10 | |||
| 1.05 | 0 | 4.82 ± 1.5 | ||||
| 326 |
5.17 ± 1.5 |
107 ± 46 |
10 |
The background of radioactivity detection was measured using a control without CODH/ACS. It was estimated as 2.15 μm. These experiments were carried out using the rapid chemical quench apparatus
The expected percentage recovery is based on the isotope dilution if CO must dissociate before product can be formed
Dephospho-CoA Pulse-Chase Reactions—To establish whether CoA binds to ACS by an ordered or random mechanism and to establish the order of CoA binding, we used 3′-dephospho-CoA in the pulse, CoA in the chase, and 14CH3-H4folate (0.35 mm) as the source of radioactivity. CoA and dephospho-CoA are analogous substrates and exhibit similar Km values (29). Thus, dephospho-CoA is the “labeled” substrate in the pulse phase of these experiments. The reverse-phase HPLC peak for dephosphoacetyl-CoA elutes 7 min after the acetyl-CoA peak, so these fractions can be easily separated and the 14C-counts can be used to calculate the amount of the pulsed substrate, dephospho-CoA, trapped in the product. Table 4 shows that there is a significant dilution of dephospho-CoA by the CoA chase. The detection limit for the radiolabeled acetyl-dephospho-CoA product is about 2.15 μm; therefore, the recovery of labeled product in the absence of CoA could be accurately measured. However, when CoA was present in the chase, the amount of 14C-acetyl-dephospho-CoA was too low to accurately measure. Thus, the values shown are an upper limit of the percentage recovery when 1 mm CoA is present. It is clear that dephospho-CoA undergoes isotope dilution, which indicates that CoA cannot serve as the first substrate in acetyl-CoA synthesis.
TABLE 4.
Pulse-chase experiments for trapping dephospho-CoA for CODH/ACS with 1 mm TiCit, 50 μm14CH3-CFeSP, 0.35 mm14CH3-H4folate, 10 μm MeTr, and 0.33 atm of CO
|
Reaction time |
CODH/ACS |
[Dephospho-CoA] pulse |
[CoA] chase |
[14C1-dephosphoacetyl-CoA] |
Percentage recovery in pulse/chasea |
Percentage recovery (expected)b |
|---|---|---|---|---|---|---|
| s | μm | μm | μm | μm | % | % |
| 33.05 | 50 | 30 | 0 | 14.2 ± 1.5 | ||
| 1000 | 4.2 ± 1.5a | 30 ± 11 | 3 | |||
| 10.05 | 0 | 15.7 ± 1.5 | ||||
| 1000 | 3.0 ± 1.5a | 19 ± 10 | 3 | |||
| 3.05 | 0 | 10.4 ± 1.5 | ||||
| 1000 |
2.5 ± 1.5a |
24 ± 15 |
3 |
The limit of radioactivity detection was estimated to be 2.15 μm using a control without CODH/ACS. Thus, all samples chased with CoA exhibited background levels of “acetyl-dephospho-CoA” when error limits are considered. Thus, the upper limits of percentage recoveries are shown here. The experiments were performed in the rapid chemical quench apparatus
The expected percentage recovery is based on the isotope dilution if dephospho-CoA must dissociate before product can be formed
DISCUSSION
Demonstration That Acetyl-CoA Synthesis Involves Random Addition of the Methyl and CO Groups Followed by Ordered Binding of CoA—Two controversial and interrelated issues in the ACS reaction mechanism include the order in which the three substrates bind and whether acetyl-CoA synthesis occurs through a paramagnetic or diamagnetic mechanism. Isotope chase experiments constitute a rather straightforward approach for establishing kinetic mechanisms of substrate addition to enzymes and identifying covalent intermediates formed during the catalytic cycle (43, 51). In a two-substrate compulsory ordered mechanism, if the isotopically labeled second substrate is incubated with the enzyme (the pulse phase), it will either not bind or form a dead end complex. Dissociation of this nonproductive complex is required for the enzyme to bind the substrates in the proper order and catalyze product formation (43). If the pulse solution is chased with a solution containing excess unlabeled substrate, the isotope is diluted as the labeled and unlabeled substrates equilibrate. The amount of isotope trapped in the product will depend upon the ratio of the concentrations of the labeled/unlabeled second substrate. On the other hand, the binary complex between the first substrate and enzyme would only undergo isotope dilution if its commitment toward product formation is quite low. For a random mechanism, the amount of isotope that is trapped in the product will depend on the reversibility of the reaction and the extent to which each branch of the mechanism contributes to the formation of the ternary complex. Similar considerations hold for a three-substrate reaction, like acetyl-CoA synthesis.
Three types of pulse-chase experiments (14CH3-CFeSP/12CH3-CFeSP, 14CO/12CO, and dephospho-CoA/CoA) have been used to establish the order in which the three substrates bind to ACS during the synthesis of acetyl-CoA. We performed these experiments with CODH·ACS and with the recombinant ACS subunit because CODH·ACS contains a 70-Å CO channel between the CODH and ACS active sites, which is not present in ACS alone. Thus, we considered that the mechanism and order of CO binding to ACS in the macromolecular complex may differ from that with the ACS subunit alone.
In the first set of pulse-chase experiments, 14CH3-CFeSP was reacted with CODH·ACS or ACS in the presence of 1 mm CO, TiCit, and CoA under conditions where the amount of active enzyme was slightly higher (17 μm) than the amount of 14CH3-CFeSP. When the reaction was repeated in the presence of 80 μm unlabeled CH3-CFeSP (the chase), we observed almost complete recovery of the 14C bound to the enzyme. These results demonstrate that, when 14CH3-CFeSP is provided as the first substrate, the methyl group is bound productively in a CH3-ACS complex that is committed to product formation. If 14CH3-CFeSP did not bind to the ACS subunit during the pulse phase or if it bound in a nonproductive manner, then the chase would have diluted the 14C content of the acetyl-CoA by ∼5-fold (Table 1). These results are inconsistent with an ordered mechanism in which CO or CoA must bind before CH3-CFeSP.
When a similar set of experiments was performed with 14CO in the pulse phase and with either a 20- or 10-fold (Tables 2 and 3, respectively) excess of unlabeled CO in the chase, most of the label is recovered in the product. Thus, as with the pulse-chase experiment with 14CH3-CFeSP, insignificant isotope dilution occurs. Since MeTr-catalyzed methylation of CFeSP by CH3-H4folate proceeds at a rate of 120 μm s-1 (half-life of 0.29 s at 50 μm CH3-CFeSP) (47) and methylation of ACS occurs at a rate of 2 s-1 (half-life of 0.35 s-1), methylation of ACS-CO was complete in the time course of these pulse-chase experiments. Moreover, for all of the mixtures containing the chase, the visible spectrum of the CFeSP showed full methylation prior to starting the experiments (absorption maximum at 450 nm, not shown). Thus, the pulse-chase experiments with 14CO/12CO demonstrate that CO binds productively to form an ACS-CO complex and rule out an ordered mechanism with 14CH3-CFeSP (or CoA) as the first substrate to bind to ACS.
Similar isotope-trapping experiments were performed with dephospho-CoA in the pulse phase and a 33-fold excess of unlabeled CoA in the chase (Table 4). The dephospho-CoA showed significant “isotope” dilution by the CoA, demonstrating that CoA does not bind productively as the first substrate.
Thus, the combined results using 14CH3-CFeSP or 14CO in the pulse clearly demonstrate that, whether CODH·ACS or the recombinant ACS subunit is the catalyst, either the ACS-CH3 or the ACS-CO form of the enzyme is committed to product formation. Therefore, CO and the CH3-CFeSP bind randomly during the first step of acetyl-CoA synthesis to generate the carbonylated or the methylated enzyme. The combined results of these three complementary series of pulse-chase experiments rule out a compulsory ordered mechanism for the ACS reaction and strongly indicate that either CH3-CFeSP or CO can bind randomly as the first substrate, whereas CoA must be the last substrate to bind, as shown in Fig. 3. Random binding of the methyl group and CO had been suggested earlier (28), because both the carbonylated and the methylated forms of ACS can react with the other two substrates to form acetyl-CoA (8, 33, 52).
FIGURE 3.
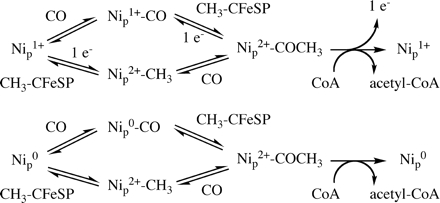
Random Mechanism of acetyl-CoA synthesis by ACS and CODH·ACS according to the paramagnetic (top) and diamagnetic (bottom) proposals.
Our results indicate that CoA binds to an acetylated form of ACS (i.e. after CO and methyl group binding), which is consistent with stopped flow and kinetic simulation experiments (53) and with studies demonstrating that ACS (or CODH·ACS) can be methylated (8, 31) or carbonylated (36, 54) in the absence of CoA. Furthermore, studies of the CoA/acetyl-CoA exchange reaction (Reaction 5) demonstrate that synthesis/cleavage of the C–S bond of acetyl-CoA occurs much faster than methylation or carbonylation of ACS or synthesis/cleavage of the C–C bond (27, 29).
Implications of the Pulse-Chase Experiments for the Intermediacy of the NiFeC Species and for the Proposed Paramagnetic and Diamagnetic Mechanisms of Acetyl-CoA Synthesis—The catalytic intermediacy of the paramagnetic NiFeC species in the ACS reaction has been a source of considerable debate. Based mainly on detection of a diamagnetic product of the reaction of CH3-ACS with CO, it has been argued that the NiFeC species is not a true catalytic intermediate in acetyl-CoA synthesis, that it may be an inhibitory state, and that the Ni(0) state is the catalytically relevant state (16, 18). This Ni(0) proposal also requires that methylation occurs prior to carbonylation.4
However, that acetyl-CoA synthesis occurs by a random mechanism of methyl and CO addition provides further substantiation for the intermediacy of a paramagnetic NiFeC species in the mechanism. This is because our results require that carbonylation of free ACS is an active branch on the catalytic pathway of acetyl-CoA synthesis. These experiments complement recent parallel stopped flow infrared and freeze-quench EPR experiments, which demonstrated that the only metal-carbonyl species observed by FTIR when ACS is reacted with CO forms at the same rate as that of formation of the NiFeC species (17). In these experiments, any carbonylated species that is formed after the 10-ms dead time would have been observed. The IR stretching frequency of the M-CO species (1996 cm-1) was identical to that assigned earlier to the NiFeC species (55). A caveat of the stopped flow infrared/freeze-quench-EPR experiments is that they only provide definitive evidence for the intermediacy of the NiFeC species if CO can bind to ACS before the methyl group. For example, one could argue that if acetyl-CoA synthesis follows an ordered mechanism with methyl binding first, the metal-carbonyl species could be mechanistically irrelevant.5 Thus, the pulse-chase experiments described here, by demonstrating an active path with CO as the first substrate, provide unambiguous evidence supporting the NiFeC species as a central intermediate in acetyl-CoA synthesis.
The conclusion that the NiFeC species is a key intermediate in a paramagnetic mechanism is consistent with stopped flow and freeze-quench EPR experiments, which have demonstrated the kinetic competence of a paramagnetic NiFeC species (17, 33, 36, 54), i.e. that it forms and decays (when reacted with the CH3-CFeSP) at least as fast as the steady-state rate of acetyl-CoA synthesis. In addition, this paramagnetic ACS-CO complex is formed by reaction of CODH·ACS with all of the substrates that can serve as donors of the carbonyl group of acetyl-CoA (CO, CO2, or pyruvate) (54, 56) or from acetyl-CoA via reversal of acetyl-CoA synthesis (57). Furthermore, there are drawbacks of the diamagnetic mechanism (e.g. Ni0p has not been observed by EXAFS and L-edge spectroscopies (58), nor was Ni0p-CO (or Ni2+p-CO) detected by SF-IR of reduced ACS and CO) (17). The diamagnetic mechanism is supported by one set of theoretical computations (59) but not by another (60).
Paramagnetic Mechanism of Acetyl-CoA Synthesis—There is a theoretical challenge that accompanies acceptance of the paramagnetic mechanism. It is clear that when the paramagnetic NiFeC state of ACS is reacted with the methyl donor, the EPR signal disappears (8, 33, 61). The loss of the EPR signal appears to conflict with the paramagnetic mechanism because the methyl group of the CH3-CFeSP appears to be transferred in an SN2 mechanism as a methyl cation to ACS (10, 11, 30, 31). The reaction of a paramagnet (NiFeC species) with a diamagnetic methyl cation should yield another paramagnet.
Fig. 3 offers an explanation for how to incorporate the paramagnetic NiFeC species and a diamagnetic acetyl-enzyme as intermediates in catalysis. On the upper branch of Fig. 3, after reductive activation of the A-cluster (8, 28, 29, 62), which may involve electron transfer from CODH in the CODH·ACS complex (63) or an external reductant with the recombinant ACS subunit (21, 64), ACS undergoes carbonylation to generate the paramagnetic NiFeC species. Based on density function theory computations, the NiFeC species is best described as a [4Fe-4S]2+ cluster bridged to an Ni1+ site (Nip) (S = ½) that is thiolate-bridged to another Ni2+ ion in a thiolato- and carboxamido-type N2S2 coordination environment in which CO binds as a terminal carbonyl to the Nip(I) center (60) (see Fig. 3, top).
We propose that, after transmethylation (Reaction 7) an electron is transferred from some one-electron shuttle to the A-cluster to rapidly reduce the methyl-Ni(III) product to a diamagnetic methyl-Ni(II) state, as described in Reaction 8. This proposal would be consistent with the model studies of methyl transfer described by Riordan (65, 66). Another solution would be to transfer a methyl radical, which would generate a CH3-Ni(II)-CO complex and Co(II) (Reaction 9). However, we dismiss a methyl radical transfer because rapid kinetic studies demonstrate that Co(I) is the product of this reaction on the CFeSP, indicating that a methyl cation is transferred to ACS (Reaction 7), as in cobalamin-dependent methyltransferases (67). As with the activation reaction, it is important to identify the source of the electron, since a stopped flow study of the methyl transfer appears to rule out participation of the [Fe4S4]2+ cluster component of the A-cluster (32).
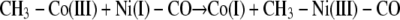 |
 |
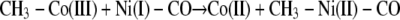 |
Considering the lower branch of the random pathway (where methyl binds before CO), paramagnetic intermediates have not been identified when the methylated enzyme is reacted with CO and CoA. As shown in Fig. 3, it is likely that paramagnetic intermediates are not detected when the methyl group binds first, because C–C bond formation and electron transfer are too fast for a proposed paramagnetic methylated intermediate to accumulate. When methylation occurs first, then the second electron is presumably taken up in that same step.
C–C bond formation would generate the diamagnetic acetyl-Ni(II) intermediate, which then can undergo deacetylation by the third substrate, CoA, to generate acetyl-CoA. This thiolytic deacetylation in the last step of the reaction is an addition-elimination reaction, which would lead to two-electron reduction of the enzyme. One of these electrons could regenerate that active form of ACS, and the other would return to the one-electron shuttle for the next methylation cycle. It is important to design experiments that would identify the one-electron shuttle in the synthesis. This is intriguing, since stopped-flow studies suggest that the most likely candidate, the [Fe4S4] component of the A cluster, is not involved in redox reactions during the synthesis (32).
Supplementary Material
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.

The on-line version of this article (available at http://www.jbc.org) contains supplemental “Experimental Procedures.”
The abbreviations used are: CODH, carbon monoxide dehydrogenase; ACS, acetyl-CoA synthase; CFeSP, corrinoid iron-sulfur protein; MeTr, methyltetrahydrofolate:corrinoid iron-sulfur protein methyltransferase; CH3-H4folate, methyltetrahydrofolate; ACSHT, recombinant acetyl-CoA synthase containing a C-terminal His tag; MES, 2-(N-morpholino)ethanesulfonic acid; TiCit, titanium citrate solution.
Since only 60% of the methyl groups were recovered in the control experiment (pulse phase only), it is likely that only 4% of the methyl groups would have been recovered.
The diamagnetic mechanism does not have an absolute requirement for Ni0. An alternative is an Ni2+ mechanism with the two activation electrons being provided by a reduced disulfide-Nip-Nid electron donor.
However, one should consider that the NiFeC species has firm experimental evidence, and the putative Ni(0) state on ACS has never been observed.
References
- 1.Ragsdale, S. W. (2004) Crit. Rev. Biochem. Mol. Biol 39, 165-195 [DOI] [PubMed] [Google Scholar]
- 2.Ragsdale, S. W. (2006) Chem. Rev. 106, 3317-3337 [DOI] [PubMed] [Google Scholar]
- 3.Drennan, C. L., Heo, J., Sintchak, M. D., Schreiter, E., and Ludden, P. W. (2001) Proc. Natl. Acad. Sci. U. S. A. 98, 11973-11978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dobbek, H., Svetlitchnyi, V., Gremer, L., Huber, R., and Meyer, O. (2001) Science 203, 1281-1285 [DOI] [PubMed] [Google Scholar]
- 5.Doukov, T. I., Hemmi, H., Drennan, C. L., and Ragsdale, S. W. (2007) J. Biol. Chem. 282, 6609-6618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Menon, S., and Ragsdale, S. W. (1998) Biochemistry 37, 5689-5698 [DOI] [PubMed] [Google Scholar]
- 7.Ragsdale, S. W., Lindahl, P. A., and Münck, E. (1987) J. Biol. Chem. 262, 14289-14297 [PubMed] [Google Scholar]
- 8.Barondeau, D. P., and Lindahl, P. A. (1997) J. Am. Chem. Soc. 119, 3959-3970 [Google Scholar]
- 9.Lebertz, H., Simon, H., Courtney, L. F., Benkovic, S. J., Zydowsky, L. D., Lee, K., and Floss, H. G. (1987) J. Am. Chem. Soc. 109, 3173-3174 [Google Scholar]
- 10.Roberts, D. L., James-Hagstrom, J. E., Smith, D. K., Gorst, C. M., Runquist, J. A., Baur, J. R., Haase, F. C., and Ragsdale, S. W. (1989) Proc. Natl. Acad. Sci. U. S. A. 86, 32-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Darnault, C., Volbeda, A., Kim, E. J., Legrand, P., Vernede, X., Lindahl, P. A., and Fontecilla-Camps, J. C. (2003) Nat. Struct. Biol. 10, 271-279 [DOI] [PubMed] [Google Scholar]
- 12.Svetlitchnyi, V., Dobbek, H., Meyer-Klaucke, W., Meins, T., Thiele, B., Romer, P., Huber, R., and Meyer, O. (2004) Proc. Natl. Acad. Sci. U. S. A. 101, 446-451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lindahl, P. A. (2002) Biochemistry 41, 2097-2105 [DOI] [PubMed] [Google Scholar]
- 14.Xia, J. Q., Dong, J., Wang, S. K., Scott, R. A., and Lindahl, P. A. (1995) J. Am. Chem. Soc. 117, 7065-7070 [Google Scholar]
- 15.Xia, J., Hu, Z., Popescu, C. V., Lindahl, P. A., and Munck, E. (1997) J. Am. Chem. Soc. 119, 8301-8312 [Google Scholar]
- 16.Bramlett, M. R., Stubna, A., Tan, X., Surovtsev, I. V., Munck, E., and Lindahl, P. A. (2006) Biochemistry 45, 8674-8685 [DOI] [PubMed] [Google Scholar]
- 17.George, S. J., Seravalli, J., and Ragsdale, S. (2005) J. Am. Chem. Soc. 127, 13500-13501 [DOI] [PubMed] [Google Scholar]
- 18.Lindahl, P. A. (2004) J. Biol. Inorg. Chem. 9, 516-524 [DOI] [PubMed] [Google Scholar]
- 19.Shin, W., Stafford, P. R., and Lindahl, P. A. (1992) Biochemistry 31, 6003-6011 [DOI] [PubMed] [Google Scholar]
- 20.Seravalli, J., Xiao, Y., Gu, W., Cramer, S. P., Antholine, W. E., Krymov, V., Gerfen, G. J., and Ragsdale, S. W. (2004) Biochemistry 43, 3944-3955 [DOI] [PubMed] [Google Scholar]
- 21.Xia, J. Q., and Lindahl, P. A. (1996) J. Am. Chem. Soc. 118, 483-484 [Google Scholar]
- 22.Ragsdale, S. W., Clark, J. E., Ljungdahl, L. G., Lundie, L. L., and Drake, H. L. (1983) J. Biol. Chem. 258, 2364-2369 [PubMed] [Google Scholar]
- 23.Pezacka, E., and Wood, H. G. (1984) Proc. Natl. Acad. Sci. U. S. A. 81, 6261-6265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ragsdale, S. W., and Wood, H. G. (1985) J. Biol. Chem. 260, 3970-3977 [PubMed] [Google Scholar]
- 25.Ragsdale, S. W., Wood, H. G., and Antholine, W. E. (1985) Proc. Natl. Acad. Sci. U. S. A. 82, 6811-6814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raybuck, S. A., Bastian, N. R., Orne-Johnson, W. H., and Walsh, C. T. (1988) Biochemistry 27, 7698-7702 [DOI] [PubMed] [Google Scholar]
- 27.Ramer, S. E., Raybuck, S. A., Orme-Johnson, W. H., and Walsh, C. T. (1989) Biochemistry 28, 4675-4680 [DOI] [PubMed] [Google Scholar]
- 28.Lu, W. P., and Ragsdale, S. W. (1991) J. Biol. Chem. 266, 3554-3564 [PubMed] [Google Scholar]
- 29.Bhaskar, B., De Moll, E., and Grahame, D. A. (1998) Biochemistry 37, 14491-14499 [DOI] [PubMed] [Google Scholar]
- 30.Lu, W.-P., Harder, S. R., and Ragsdale, S. W. (1990) J. Biol. Chem. 265, 3124-3133 [PubMed] [Google Scholar]
- 31.Tan, X. S., Sewell, C., and Lindahl, P. (2002) J. Am. Chem. Soc. 124, 6277-6284 [DOI] [PubMed] [Google Scholar]
- 32.Tan, X. S., Sewell, C., Yang, Q., and Lindahl, P. A. (2003) J. Am. Chem. Soc. 125, 318-319 [DOI] [PubMed] [Google Scholar]
- 33.Seravalli, J., Kumar, M., and Ragsdale, S. W. (2002) Biochemistry 41, 1807-1819 [DOI] [PubMed] [Google Scholar]
- 34.Maynard, E. L., Sewell, C., and Lindahl, P. A. (2001) J. Am. Chem. Soc. 123, 4697-4703 [DOI] [PubMed] [Google Scholar]
- 35.Maynard, E. L., and Lindahl, P. A. (2001) Biochemistry 40, 13262-13267 [DOI] [PubMed] [Google Scholar]
- 36.Kumar, M., Lu, W.-P., Liu, L., and Ragsdale, S. W. (1993) J. Am. Chem. Soc. 115, 11646-11647 [Google Scholar]
- 37.Volbeda, A., and Fontecilla-Camps, J. C. (2004) J. Biol. Inorg. Chem. 9, 525-532 [DOI] [PubMed] [Google Scholar]
- 38.Siegel, I. H. (1975) Enzyme Kinetics: Behavior and Analysis of Rapid Euilibrium and Steady-state Enzyme Systems, 1st Ed., John Wiley & Sons, Inc., New York
- 39.Andi, B., West, A. H., and Cook, P. F. (2004) Biochemistry 43, 11790-11795 [DOI] [PubMed] [Google Scholar]
- 40.Seravalli, J., Brown, K. L., and Ragsdale, S. W. (2001) J. Am. Chem. Soc. 123, 1786-1787 [DOI] [PubMed] [Google Scholar]
- 41.Errey, J. C., and Blanchard, J. S. (2006) Biochemistry 45, 3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu, M., de Carvalho, L. P., Cook, P. F., and Blanchard, J. S. (2006) Biochemistry 45, 14788-14794 [DOI] [PubMed] [Google Scholar]
- 43.Rose, I. A. (1980) Methods Enzymol. 64, 47-59 [DOI] [PubMed] [Google Scholar]
- 44.Andreesen, J. R., Schaupp, A., Neurater, C., Brown, A., and Ljungdahl, L. G. (1973) J. Bacteriol. 114, 743-751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lundie, L. L., and Drake, H. L. (1984) J. Bacteriol. 159, 700-703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Doukov, T. I., Seravalli, J., Stezowski, J. J., and Ragsdale, S. W. (2000) Structure 8, 817-830 [DOI] [PubMed] [Google Scholar]
- 47.Seravalli, J., Zhao, S., and Ragsdale, S. W. (1999) Biochemistry 38, 5728-5735 [DOI] [PubMed] [Google Scholar]
- 48.Zehnder, A. J. B., and Wuhrmann, K. (1976) Science 194, 1165-1166 [DOI] [PubMed] [Google Scholar]
- 49.Maynard, E. L., and Lindahl, P. A. (1999) J. Am. Chem. Soc. 121, 9221-9222 [Google Scholar]
- 50.Seravalli, J., and Ragsdale, S. W. (2000) Biochemistry 39, 1274-1277 [DOI] [PubMed] [Google Scholar]
- 51.Wilkinson, K. D., and Rose, I. A. (1979) J. Biol. Chem. 254, 12567-12572 [PubMed] [Google Scholar]
- 52.Pezacka, E., and Wood, H. G. (1988) J. Biol. Chem. 263, 16000-16006 [PubMed] [Google Scholar]
- 53.Tan, X., Surovtsev, I. V., and Lindahl, P. A. (2006) J. Am. Chem. Soc. 128, 12331-12338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gorst, C. M., and Ragsdale, S. W. (1991) J. Biol. Chem. 266, 20687-20693 [PubMed] [Google Scholar]
- 55.Kumar, M., and Ragsdale, S. W. (1992) J. Am. Chem. Soc. 114, 8713-8715 [Google Scholar]
- 56.Menon, S., and Ragsdale, S. W. (1996) Biochemistry 35, 12119-12125 [DOI] [PubMed] [Google Scholar]
- 57.Russell, W. K., and Lindahl, P. A. (1998) Biochemistry 37, 10016-10026 [DOI] [PubMed] [Google Scholar]
- 58.Gu, W., Gencic, S., Grahame, D. A., and Cramer, S. P. (2003) J. Am. Chem. Soc. 125, 15343-15351 [DOI] [PubMed] [Google Scholar]
- 59.Webster, C., Darensbourg, M., Lindahl, P., and Hall, M. B. (2004) J. Am. Chem. Soc. 126, 3410-3411 [DOI] [PubMed] [Google Scholar]
- 60.Schenker, R. P., and Brunold, T. C. (2003) J. Am. Chem. Soc. 125, 13962-13963 [DOI] [PubMed] [Google Scholar]
- 61.Grahame, D. A., Khangulov, S., and Demoll, E. (1996) Biochemistry 35, 593-600 [DOI] [PubMed] [Google Scholar]
- 62.Lu, Z., and Crabtree, R. H. (1995) J. Am. Chem. Soc. 117, 3994-3998 [Google Scholar]
- 63.Shin, W., and Lindahl, P. A. (1992) Biochemistry 31, 12870-12875 [DOI] [PubMed] [Google Scholar]
- 64.Loke, H. K., Tan, X., and Lindahl, P. A. (2002) J. Am. Chem. Soc. 124, 8667-8672 [DOI] [PubMed] [Google Scholar]
- 65.Ram, M. S., and Riordan, C. G. (1995) J. Am. Chem. Soc. 117, 2365-2366 [Google Scholar]
- 66.Ram, M. S., Riordan, C. G., Yap, G. P. A., LiableSands, L., Rheingold, A. L., Marchaj, A., and Norton, J. R. (1997) J. Am. Chem. Soc. 119, 1648-1655 [Google Scholar]
- 67.Banerjee, R., and Ragsdale, S. W. (2003) Annu. Rev. Biochem. 72, 209-247 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.