

Abstract
Survivin is a novel member of the inhibitor of apoptosis (IAP) protein family, and its aberrant expression in cancer cells has been shown to be associated with tumorigenesis, cancer progression, radiation/drug resistance and shorter patient survival. Survivin is also expressed in certain human adult tissues and cells, and has been shown to play a role in physiology. Interestingly, targeting survivin for cancer treatment did not show obvious toxicity to normal tissues and cells. This suggests that the mechanism for the regulation and function of survivin may actually be different in cancer cells as compared to normal cells. This review intends to summarize the most important information about the transcriptional and/or posttranscriptional controls of survivin in cancer cells. Further studies along this line may find essential interfaces for the development of novel approaches for cancer therapeutics.
Keywords: survivin, survivin splice variant, transcription, post-transcription, cancer cells
Introduction
Survivin is a bifunctional protein that has been implicated in the control of apoptosis and the regulation of mitosis (1, 2). Survivin is a 16.5 kDa protein that is cell cycle-regulated with a robust increase in the G2/M phase in cancer cells (1, 3). Many important gene products including signaling molecules, transcription factors and other ligands appear to modulate survivin expression and/or function in cancer cells through transcriptional and/or posttranscriptional mechanisms. The posttranscriptional regulation of survivin expression also takes place at the pre-mRNA splicing level. The differential splicing of survivin pre-mRNA results in several known survivin splice variants which appear to have differential functions in tumorigenesis (4) or other functions (2). Several new survivin splice variants were also recently described (2). Therefore, the elucidation of both transcriptional and posttranscriptional regulations of survivin will be important for cancer prevention and treatment. This review will summarize the most important information about the transcriptional and posttranscriptional controls of survivin in cancer cells. Several recent studies have also revealed a role for survivin in the regulation of gene transcription by acting as either a transcription activator or a transcription co-activator. In addition, we will discuss possible differences in the mechanism that governs the regulation and function of survivin in cancer cells as compared to normal cells.
Methylation and genetic alteration/polymorphism of the survivin gene
One of the major strategies for the modulation of survivin expression is through the regulation of its gene transcription. Several studies have indicated that the survivin gene promoter is essential for the regulation of its gene transcription. Analysis of the survivin gene promoter from humans (5) and mice (6) reveals that the core promoter region lacks a TATA box motif but contains a typical CpG island, which harbors several cell cycle dependent elements (CDE) and one cell cycle homology region (CHR).
The expression of survivin in human cancer is correlated with an unmethylated CpG island in the survivin core promoter. It was initially shown that the survivin gene CpG island is not methylated in both normal and cancerous tissues (5). Studies from normal and cancerous ovarian tissues, however, have indicated that the Hpa II sites located in the core promoter region as well as in exon 1 of the survivin gene were methylated in most normal ovarian tissues but demethylated in most ovarian cancer tissues (7). Recent studies have shown that there was no methylation for the survivin gene in oral tumor tissues, while only 4 of the 9 normal oral tissues without survivin expression showed methylation of the survivin gene (8), suggesting that silencing of survivin in survivin negative tissues or cells may not be necessarily attributed to methylation. Examinations of 17 genes including survivin have consistently displayed a uniformly unmethylated pattern in all the astrocytoma and non-astrocytoma tissues examined (9). Furthermore, methylation was not found in advanced ovarian cancer after chemotherapy and DNA damage (10) nor could 5-aza-dC treatment increase the expression of survivin in Colo-320 and SW1116 colon cancer cells (11). These observations suggest that the survivin gene was not methylated in both cancer tissues and cell lines. However, studies of the methylation status of the CpG island and the exon 1 of the survivin gene in DMBA-induced hamster buccal-pouch squamous-cell carcinomas showed that all the untreated and mineral-oil treated control samples had a survivin-methylated allele, whereas the DMBA-treated cancerous tissues showed no evidence of survivin methylation, determined by a PCR-based methylation assay (12). Together, these observations suggest that survivin is unmethylated in cancer but may be selectively methylated in normal tissues with individual variations. Nevertheless, methylation of the survivin gene promoter appears to be involved in the wild type p53-induced downregulation of survivin transcription in cancer cells (13). P53 interacted with DNA-methyltransferase 1 (DNMT1) and stimulated DNMT1-mediated methylation in vitro. Doxorubicin-induced p53 upregulation resulted in the methylation of the survivin promoter in wild type p53 HCT116 cells but not in DNMT1 null or p53 null cells (13). Endogenous survivin gene repression was relieved by introduction of DNMT1- or p53-specific small inhibitory RNA (siRNA) (13), suggesting a role of methylation in the p53-mediated suppression of survivin transcription.
The survivin gene appears to be a gene with rare mutations in cancer. However, a search for mutations in the survivin promoter from −510 to 40 bp (A in the survivin ATG codon as +1) in various cancerous and normal cell lines by PCR plus sequencing revealed a frequent C to G mutation at the −31 bp position in various cancer cell lines but not in normal cells (14). This mutation is within a CDE motif and involved in the alteration of the DNA-protein interaction in the CDE DNA region that was shown to be associated with increased survivin promoter activity and its endogenous gene expression (14). However, whether the “C to G” mutation at −31 also exists in various cancer tissues is currently unclear and warrants further investigation.
Regulation of survivin expression by various stimuli
Survivin expression appears to be regulated by growth factors, cytokines, hormones, anticancer agents and kinase inhibitors. The antiapoptotic properties of VEGF and IL-11 appear to mediate the induction of survivin in endothelial cells (1). It has been shown that VEGF is strongly associated with the expression of survivin in hepatocellular cancer (15) and breast cancer (16). Furthermore, the induction of survivin expression by VEGF employs a PI3K/Akt pathway in neuroblastoma cells (17). In addition, NVP-LAQ824, a histone deacetylase inhibitor, reduced VEGF and survivin expression as well as angiogenesis in prostate and breast xenograft tumor growth (18). It will be important to determine whether the effect of NVP-LAQ824 on survivin expression takes place through the direct modulation of the histone acetylation on the survivin promoter or in an indirect manner through the reduction of VEGF.
EGFR, a member of the ErbB family of receptor tyrosine kinases, could increase the proliferation, angiogenesis and invasive potential of cancer cells. Studies of the expression of survivin, EGFR and ErbB2/HER2 in invasive breast cancer revealed a strong coexpression (19). The coexpression of HER2 and EGFR were shown to upregulate survivin and increase resistance to etoposide-induced apoptosis in breast cancer cells, whereas the HER2-specific inhibitor, Herceptin has been shown to decrease survivin expression and apoptosis resistance, apparently through a PI3K/Akt, rather than a MEK/Erk signaling pathway (19). Similarly, activated EGFR has been shown to increase survivin expression through the PI3K but not the Erk signaling pathway (20). However, taxol-mediated rapid induction of survivin appeared to employ both PI3K/Akt and MEK/Erk signaling pathways (21), which appear to be associated with taxol-mediated phosphorylation of EGFR (22). These studies provide new perspectives on controlling of survivin expression in cancer cells by inhibiting the important protein kinases that are involved in cell signaling pathways.
Several studies focused on the regulation of survivin expression by hormones in cancer cells. Consistent with a role of estrogens in enhancing breast cancer cell survival and resisting apoptosis, estradiol (E2) has been shown to upregulate survivin expression in MCF-7 breast cancer cells and to prevent these cells from etoposide-induced apoptosis (23). Mechanistically, E2-activated estrogen receptor (ERα) physically interacted with p53 and inhibited the p53 suppression of survivin transcription at the p53 binding site of the survivin promoter in MCF-7 cells (24). Intriguingly, after re-expressing ERα in ERα-negative MDA-MB-231 breast cancer cells, E2 did not stimulate but suppressed survivin expression and cell proliferation (25). These observations suggest that the function of E2 and its receptor ERα in the regulation of survivin expression and cancer cell proliferation is also dependent on the presence or absence of other protein factors in the particular breast cancer cells. Identification of these differences may lead to novel approaches for cancer treatment. In addition, survivin appears to play a role in androgen receptor (AR)-dependent and AR-independent drug resistance. Androgen has been shown to upregulate the expression of survivin in both AR-positive LNCaP and AR-negative PC-2 and DU145 prostate cancer cells (26). In AR-positive LNCaP cells, upregulation of survivin occurs likely through AR since the anti-androgen therapeutic drug Flutamide could diminish this effect. In AR-negative PC-2 and DU145 cells, upregulation of survivin occurs likely through androgen-mediated activation of Akt and thus is resistant to Flutamide. These studies, as well as others (2), indicate that the upregulation of survivin by various ligands appears to be a universal drug resistant factor, although the mechanism for the control of survivin upregulation by various stimuli may vary. Thus, various approaches to inhibit survivin expression might be developed for cancer treatment.
STAT3 signaling and the constitutive expression of survivin
Based on current information, one possible mechanism for the aberrant expression of survivin in cancer cells is that various events, such as virus infection, p53 mutation, APC mutation, signaling protein activation/mutation or gene amplification, may result in the direct or indirect activation of transcription factors to maintain survivin transcription (1). Growing evidence supports this notion. A strong association between survivin expression and STAT3 activation/phosphorylation was found in cancer tissues (27, 28). Inhibition of constitutive activation of STAT3 signaling in gastric cancer cells by ectopic dominant-negative STAT3 or Janus kinase inhibitor, tyrphostin AG490 markedly reduced survivin expression and induced apoptosis, whereas forced expression of survivin rescued cancer cells from apoptosis induced by STAT3 inhibition (29). Knockdown of STAT3 expression by siRNA downregulated survivin expression and induced apoptosis in several astrocytoma cell lines as well, while there was no effects in primary human astrocytes (30). Importantly, while IGF-1-mediated activation of Akt and NF-κB upregulated many antiapoptotic proteins including survivin in multiple myeloma, IL-6 could not activate NF-κB and only weakly activated Akt. However, IL-6 specifically upregulated survivin expression (31), suggesting that a specific effect of the IL-6/STAT3 pathway on survivin transcription was involved. Consistent with the association of survivin expression with COX-2 expression and cancer recurrence (32), overexpression of COX-2 increased STAT3 phosphorylation and apoptosis resistance in non-small cell lung cancer cells, while siRNA-mediated silencing of STAT3 or IL-6 downregulated survivin and triggered apoptosis in A549 lung cancer cells (33). COX-2 stabilized survivin and made non-small cell lung cancer cells resist apoptosis (34). Elevated levels of phosphorylation of STAT3 were found in many invasive breast tumors, which were significantly associated with the increased expression of survivin as well as other STAT3 target genes in invasive breast cancer tissues (35). Celecoxib, a COX-2 inhibitor, induced malignant mesothelioma cell apoptosis and decreased Akt phosphorylation and survivin expression (36), suggesting a potential inhibition of the Akt-survivin pathway (1). The derivatives of Indirubin, an active component of a traditional Chinese herbal medicine, inhibited constitutive STAT3 signaling and downregulated survivin as well as Mcl-1 expression, followed by induction of apoptosis in human breast and prostate cancer cells (37). Recently, the survivin gene has been identified alternatively as a direct downstream target for STAT3 by microarray, ChIP assays and survivin promoter-reporter assays (38). Similarly, the direct inhibition of STAT3 signaling blocked survivin expression and induced apoptosis in breast cancer cells. Importantly, increased survivin expression has been shown to be strongly associated with elevated STAT3 activity and chemotherapeutic resistance in primary breast tumor specimens (38). These studies may provide important information for the development of novel approaches for cancer treatment.
NF-κB signaling and survivin expression
RC3 renal clear cell carcinomas cells, lacking the von Hippel-Lindau tumor suppressor protein, showed an activation of NF-κB and an upregulation of antiapoptotic proteins including survivin (39). SCH 66336, a Ras farnesyltransferase inhibitor, inhibited NF-κB and downregulated survivin as well as other NF-κB-regulated gene products (40). Angiotensin II activated NF-κB and subsequently increased the production of anti-apoptotic molecules including survivin (41). Transient transfection assays have shown that the survivin promoter was transactivated by HTLV-I Tax via activating NF-κB. On the other hand, the pharmacological inhibition of NF-κB has resulted in the suppression of survivin expression in CTLL-2 cells, a mouse T-cell line expressing HTLV-I Tax (42). Interestingly, while NF-κB activation contributed to both survivin and Bcl-2 expression, the expression of these two markers was mutually exclusive in the vast majority of B-cell lymphomas (43), suggesting their distinct roles. Together, survivin appears to be a downstream target for NF-κB. However, the molecular mechanism for NF-κB-mediated upregulation of survivin remains to be determined.
P53 and survivin gene transcription
It has been shown that survivin expression is closely associated with mutant p53 accumulation in cancer cells, and the introduction of wild type p53 into p53-null human cancer cells has resulted in a significant decrease of survivin expression. P53 protein can bind to the p53 binding site in the survivin core promoter to repress its transcription and expression (1). Recent studies have also showed that the expression of survivin is negatively regulated by wild type p53 in non-small lung cancer cells (44) and positively regulated by mutant p53 in laryngeal squamous cell carcinoma (LSCC) (45) and breast cancer cells (46). Certain tumors without p53 mutation, however, also showed high-level expression of survivin, suggesting that other signaling pathways also contribute to the expression of survivin. For example, as we mentioned above, ERα interacted with p53 and neutralized p53’s role in survivin transcription suppression (24). Interestingly, the expression of survivin and its splice variants, survivin-2B and survivin-ΔEx3, can be differentially regulated by doxorubicin-induced stabilization of p53. Survivin and survivin-ΔEx3 were downregulated by p53, whereas survivin-2B was upregulated by p53 (47), suggesting that the modulation of survivin gene expression by p53 may include both transcriptional and post-transcriptional mechanisms. Survivin-2B has been shown to play a negative role in cancer cell growth and appears to offer a good prognosis for cancer patients (2). Therefore, the differential modulation of survivin and survivin-2B expression by chemotherapeutic agents or other approaches may represent a novel strategy for cancer treatment. Intriguingly, survivin could differentially regulate p53 gene family proteins as well (48), although the detailed mechanism remains to be elucidated.
APC/β-catenin/TCF-4 signaling and survivin gene transcription
Transcriptional disruption of survivin expression in cancer cells would represent an important and unique approach for cancer therapeutics, if the differential mechanism for survivin expression in cancer cells, when compared with survivin-positive normal tissues, could be defined. In colorectal cancer, APC mutation represents the initiating genetic alteration. It has been shown that wild type APC progressively represses survivin expression and limits proliferative cell populations in the lower crypt in normal colonic epithelium. The mutant APC, however, lost this function resulting in tumorigenesis. Wild type APC downregulates survivin transcription in colorectal cancer cells via the APC/β-catenin/TCF-4 pathway. The survivin gene appears to be an important target gene for the TCF-4/β-catenin signaling pathway since substantial evidence shows that survivin expression is modulated by this pathway [see review (4)]. Recent studies revealed that CREB binding protein (CBP) is a crucial coactivator for TCF/β-catenin to upregulate survivin transcription (49). Treatment of colon cancer cells with ICG001, a β-catenin/CBP interaction-specific inhibitor, has been shown to downregulate survivin gene transcription and expression (49). Mechanistically, ICG-001 disrupted CBP/β-catenin physical interaction, recruited p300 to the survivin promoter and in turn led to the concomitant recruitment of several transcription repressors, including SUMO-1, HDAC6 and PML, to the protein complex on the survivin promoter (49). These observations suggest distinct roles for CBP and p300 in the control of survivin gene transcription. Together, modulation of survivin expression in colorectal cancer cells by inhibition of this pathway may result in cancer therapeutic values.
Differential regulation of survivin in cancer cells versus normal cells
Previous studies, using the 6.4 kb survivin gene promoter region as a probe to perform the DNA-protein interaction using Southwestern blots, showed that the survivin promoter DNA differentially interacts with nuclear proteins isolated from cancer cells versus normal tissues and cells (1). This observation highlights the possibility that the mechanism for the regulation of survivin expression in cancer tissues and cells might be different from that in normal tissues and cells. Consistent with this notion, some ligands such as progesterone may play opposing effects on the regulation of survivin expression in cancer cells versus normal ones [see recent review (50)]. To further explore whether the regulation of survivin promoter activity is different in normal cells compared to cancerous cells, a series of transcription factors was determined for the involvement of the transcriptional regulation of survivin promoter activity. These studies resulted in the finding that the regulation of survivin promoter activity by Ap-2 transcription factors is different in cancer cells versus normal cells. While Ap-2 strongly inhibited survivin promoter activity in tumor-derived cell lines, including MCF-7 breast cancer cells, HCT-116 colon adenocarcinoma cells and HeLa cells, Ap-2 protein had no significant inhibitory effects on, or even slightly up-regulated, survivin promoter activity in non-transformed breast epithelial MCF-10A and fibroblast NIH3T3 cells (51). These findings point to the likelihood for the differential regulation of the survivin promoter in tumor-derived compared to non-transformed cells, and evoke new perspectives and may represent opportunities for the development of novel approaches for low toxicity cancer-specific treatments.
Modulation of survivin transcription and cancer therapeutics
Previous reports revealed that Sp-1 transcription factor plays an essential role in the constitutive expression of the survivin gene in cancer cells. Mutagenesis of Sp1 sequence at −171 and −151 in the survivin core promoter significantly diminished survivin promoter activity (5). Consistently, tetra-O-methyl nordihydroguaiaretic acid (M4N) was shown to inhibit Sp1-dependent survivin expression and activated the mitochondrial apoptotic pathway in C3 carcinoma cells (52). Similarly, Hedamycin, a GC-rich DNA binding agent, was shown to downregulate survivin expression by abrogating the binding of Sp1 protein to a 21-bp cis-acting DNA element in the human survivin core promoter, and mutagenesis of this region consistently diminished survivin promoter activity (53). These studies suggest that abrogation of Sp1 binding to the survivin core promoter region may represent a novel approach for the control of survivin expression for cancer treatment. These studies also point to a general principle for cancer therapeutics by the control of survivin transcription.
Regulation of survivin at the pre-mRNA splicing level
Alternative splicing of survivin pre-mRNA derives several known survivin-related splice variants (survivin-ΔEx3, survivin-2B, survivin-3B, survivin-2α and survivin-image). Moreover, several new survivin splice variants (survivin-ΔptEx2/3, survivin-ΔptEx1/2, survivin-ΔptEx1/2G/T and survivin-ΔptEx2) were recently described as well (2). Limited studies indicated that survivin splice variants may differentially be regulated and have different roles in tumorigenesis (4) and/or cell cycle/mitosis controls (2).
Recently, it was found that survivin-ΔEx3 could associate directly with survivin by means of heterodimerization. The coexpression of survivin with survivin-ΔEx3 caused alterations of subcellular localization of the protein complexes and synergistically inhibited mitochondrial-dependent apoptosis (54). In contrast, survivin-2B disrupted the mitochondrial membrane potential and induced DNA fragmentation (55). Survivin and survivin-2B immunoprecipitated chromosomal passenger proteins Aurora-B and Borealin, while survivin-ΔEx3 did not interact with borealin (56). The expression of survivin and its isoforms appears to be different in cancer cells as compared to normal tissues and cells. In gastric carcinomas and pediatric medulloblastomas, the expression of survivin, survivin-ΔEx3 and survivin-2B was detected by immunohistochemistry and quantitative PCR. When compared with normal tissues, survivin was the dominant transcript (57, 58). Survivin-ΔEx3 and survivin-2B were detected in lung cancer cells (55), breast tumor cell lines and breast carcinoma (59) and medulloblastoma (58), but not in adjacent normal tissues (60). The question is, should we conclude that survivin-2B and survivin-ΔEx3 are better specific markers for malignancy than survivin itself? While this remains to be investigated, it appears that the expression of survivin-2B in cancer is a favorable prognostic parameter (2, 4). Survivin-3B expression was more frequent in high-grade carcinomas while survivin-2B expression was lower in large tumors than in small ones (59). Interestingly, after neoadjuvant chemotherapy, the expression of survivin and survivin-2B was significantly reduced, while no change is found for survivin-ΔEx3 and survivin-3B (59). In human colorectal carcinomas, while the ratio of survivin-ΔEx3/survivin has shown no difference between tumor and normal samples, the relative expression level of survivin-2B to survivin (survivin-2B/survivin) was significantly higher in the tumor tissue samples than in the normal ones (61), which may indicate that there is a negative feedback loop to counter cancer progression by increasing the expression of survivin-2B. This notion is consistent with the observation that the ratio of survivin-2B/survivin in stage III and IV tumors was lower than these in stage I and II tumor, and a higher ratio of survivin-2B/survivin significantly correlated with a better prognosis (61). Finally, a new survivin splice variant, survivin-ΔEx3-3B1, was recently identified (Fig. 1) from five human cancer cell lines (HeLa, Caco-2, MCF-7, NB4 and ZR-75; corresponding GenBank accession No: DQ310375, DQ310375, DQ310375, DQ310375 and DQ310375). Survivin-ΔEx3-3B has 78 amino acids (aa), the first 74 aa is identical to the known survivin splice variant, survivin-2α followed by additional 4 aa (RELC) at the C-terminal end (Fig. 1C). It was reported that survivin-2α expressed in human malignancies and overexpressed survivin-2α localized on certain subcellular structures (62). It would be interesting to determine whether there are similarities or differences between survivin-ΔEx3-3B and survivin-2α in terms of their functions and subcellular localizations. In sum, while the mechanism for the regulation and function of various survivin splice variants in apoptosis and cell proliferation controls remains to be elucidated, limited observations imply that exploring the regulation and function of survivin splice variants may become an interesting research area in the future and may lead to novel approaches for cancer treatment.
Fig. 1.
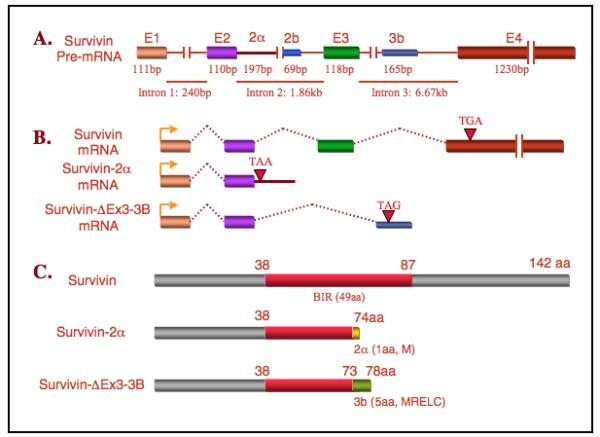
Structural comparison of the new survivin variant, survivin-ΔEx3-3B with survivin and survivin-2α is shown. A. Survivin pre-mRNA structure showing the organization of exons and introns. E1, exon 1; E2, exon 2; 2α, hidden exon 2α; 2b, hidden exon 2b; 3b, hidden exon 3b; and E4, exon 4. B. Comparison of the survivin-ΔEx3-3B mRNA structure with those for survivin and survivin-2α. “→” represents the translation start codon ATG, and “▽” represents the translation stop codon. C. Comparison of the survivin-ΔEx3-3B protein structure with those for survivin and survivin-2α.
Regulation of survivin at the protein level
Phosphorylation of survivin is important for its biological activity and function. The mechanism for survivin to gain or lose its biological function through phosphorylation or dephosphorylation has been extensively studied. Phosphorylation on survivin Thr34 by Cdc2-cyclinB1 was shown to be critical for survivin function in preserving cell viability during cell division (63). Grossman et al. confirmed this result by using a dominant negative mutant (Thr34Ala) survivin, which could trigger apoptosis in vitro and in vivo (64). Purvalanol A or Flavopiridol, cyclin-dependent kinase inhibitors, inhibited Cdc2-mediated survivin Thr34 phosphorylation, which resulted in cancer cell growth inhibition, apoptosis and tumor suppression in vitro and in vivo (65, 66).
Furthermore, at least in some examples, survivin has been shown to interact competitively with the Cdk4/p16 (INK4a) complex. Survivin interacted with Cdk4 and formed survivin/Cdk4 complex, which enhances procaspase-3/p21 formation and results in the suppression of cell death signaling (67). Recently, it has been found that the Thr117 residue phosphorylation status and the K63-linked ubiquitination status on survivin are important for controls of the dynamic association/dissociation of survivin and Aurora B to/from centromeres for normal mitosis in cancer cells [see recent review (2)]. Together, these studies may provide clues for the control of survivin and the development of novel strategies for cancer therapeutics.
Modulation of survivin stability/function by other proteins
Several studies have shown that the interaction of survivin with its cofactor proteins would modify survivin stability or function. The chaperon protein Hsp90 could stabilize survivin protein and prevent protease from its degradation. The inhibition of Hsp90 function or the disruption of the survivin/Hsp90 complex has resulted in survivin degradation (68). The hepatitis B X-interacting protein (HBXIP) was found to be a cofactor for survivin. HBXIP existed in both cancer and nonmalignant liver tissues of human with chronic HBV infection, and formed a complex with survivin. The HBXIP/survivin complex interacted with and prevented precaspase-9 recruitment to Apaf1, and selectively suppressed apoptosis initiated by cytochrome C release (69). Survivin cooperated with X-linked IAP (XIAP) via BIR domain. XIAP stability could be protected in survivin-XIAP complex from ubiquitination/proteasomal degradation, and caspase-9 activity could be more efficiently suppressed by the complex (70). CIAP1, another IAP family member, was shown to interact with survivin in mitotic cells (71). Given that survivin and cIAP1 were colocalized on midbody microtubules at telophase and interacted with each other during mitosis, cytokinesis defects might be resulted from the interference of both the function and proper localization of survivin after cIAP1 overexpression (71). Recent studies have shown that, while introduction of Smac, a mitochondrial activator of caspase, did not cause apoptosis in normal ovarian surface epithelial cells, it did induce apoptosis in ovarian cancer cells (72). Interestingly, Smac interacted with both survivin and XIAP, but it only downregulated survivin via ubiquitination and proteasomal degradation in ovarian cancer cells despite the fact that RNA interference experiments failed to reveal a role of survivin in the inhibition of Smac-mediated apoptosis (72). The later finding is somehow inconsistent with the previous finding that direct interaction between survivin and Smac was essential for the anti-apoptotic activity of survivin during taxol-induced apoptosis (73). Recently, it was also reported that Aurora C, a member of Aurora kinase family, was directly associated with survivin and Aurora B in vivo and bound to survivin but not Aurora B in vitro (74). Interferences in the function of Aurora C resulted in a cytokinesis defective phenotype identical to that which results from the silencing of Aurora B. In contrast to survivin, Aurora C was not only highly expressed in the testis, but also in a broad-spectrum of other normal human tissues (74). Given that the biological function and protein stability of survivin could be modulated by many other partner proteins, further studies of their protein-protein interactions and the function associated with these protein interactions may reveal additional clues for the development of new strategies for cancer treatment.
Survivin as a transcription activator or co-activator
Early studies indicated that survivin and telomerase, an enzyme that plays a critical role in telomere length maintenance and cell immortalization, co-overexpressed in human glioblastoma (75) and head and neck cancers (76). Recent studies revealed that the overexpression of survivin and telomerase was also found in colon cancer and that the forced expression of survivin in LS180 colon cancer cells increased telomerase activity via the upregulation of human telomerase reverse transcriptase (hTERT) (77). Survivin increased the phosphorylation of Sp1 and c-Myc proteins and enhanced the binding of these phosphorylated proteins on the hTERT core promoter while the silencing of survivin by siRNA in SW480 cells decreased Sp1 and c-Myc phosphorylation (77). There are examples in non-mammalian species as well. The siRNA-mediated silencing of BIR-1, the homologue of human survivin in C. elegans, decreased the expression of several gfp transgenes as well as the endogenous genes dpy-7 and hlh-1 (78). BIR-1 could phosphorylate histone H3 in connection with aurora kinase AIR-2 and regulated the RNA Polymerase II-mediated transcription during transcription activation process. Moreover, in a heterologous transfection system, BIR-1 increased thyroid hormone-regulated transcription and has been shown to have an additive effect with SKIP, a factor involved in transcription (78). These observations suggest that BIR-1 functions as transcription activator or co-activator during C. elegan development. Interestingly, SIX, the Xenopus counterpart of human survivin, interacted with RXRα, a nuclear receptor/transcription factor, through the AF2 domain of RXRα in the absence of ligand, which was weakened in the presence of ligand (79). This observation suggests that SIX may act as a co-factor for RXRα and involved in RXRα-mediated gene transcription. Together, these studies point to a role for survivin in gene transcription control. These findings would further expand our understanding of the functional diversity for survivin in cancer as well as other pathophysiology.
Last remarks
The transcriptional and posttranscriptional regulation of survivin is summarized in Table 1. Survivin appears to be aberrantly expressed in cancer cells and plays important roles in antiapoptosis and promotion of cell division (1, 2). Survivin is also expressed in certain normal human adult tissues or cells, however, and plays a role in physiology (50). While this fact raises concerns as to the toxicity to normal human tissues or cells when targeting the survivin gene for cancer treatment, studies have showed that the interference of survivin in vitro and in vivo for cancer treatment induces little toxicity to normal tissues or cells (2, 50). This low risk of toxicity may be partially attributable to the fact that most adult human tissues do not express survivin and that the expression of survivin in certain normal human tissues or cells shows a very low level and is strictly controlled. Growing evidence suggests different mechanisms for the regulation and function of survivin in cancer versus normal cells (2, 50). For example, the survivin gene is the downstream target for many survival signal molecules including STAT3 and Akt in cancer, but these survival signals are usually very weak in normal human adult tissues or cells. On the other hand, in contrast to many cancer cells with mutant p53 that upregulates survivin expression, normal cells possess the wild-type p53 gene that downregulates survivin expression. Moreover, downregulation of survivin by wild-type p53 in cancer cells may also be different from that in normal cells. In addition, many survivin associated proteins including Hsp90 and XIAP may preferably interact with survivin in cancer cells but not in normal cells that express survivin. It is highly possible that the DNA-protein interactions involving the control of survivin transcription and the protein-protein interactions that involve the control of survivin function as well as the signaling upstream of these actions in cancer cells are qualitatively and/or quantitatively different from those in normal cells. Investigation of the mechanisms for transcriptional and post-transcriptional controls of survivin in cancer cells would reveal clues for the development of novel approaches for cancer-specific treatment.
Table 1.
Summary of the transcriptional and posttranscriptional regulation of survivin
| Mechanism | Pathway | Key information and current status |
|---|---|---|
| Epigenetic Genetic |
Methylation, base mutation |
The survivin gene is not methylated in cancer and in most normal tissues; it is rarely mutated. C to G mutation at −31 in various cancer cell lines but unknown for cancer tissues |
| Transcription Posttranscription |
Various pathways |
Survivin expression could be regulated by growth factors, cytokines, hormones, anticancer agents and kinase inhibitors. Detailed mechanisms for most findings remain to be determined |
| Transcription Posttranscription |
STAT3 Cox-2 |
IL-6-induced survivin production is likely transcriptional through STAT3 activation; but Cox-2 modulation of survivin is more complex |
| Transcription | NF-κB | NF-κB appears to regulate survivin but remains to be investigated |
| Transcription | P53 | P53 transcriptionally downregulates survivin. Survivin also posttranscriptionally modulates p53 family proteins |
| Transcription | APC/ β-catenin/ TCF-4 |
APC downregulates survivin by inhibiting β-catenin/TCF-4. It is not clear whether survivin expression in normal epithelial cells vs. cancer cells employs different approaches involving β-catenin/TCF-4 |
| Transcription | Protein-DNA | The survivin promoter DNA interacts differentially with cancer nuclear proteins vs. normal cell nuclear proteins; Some ligands appear to differentially regulate survivin in cancer vs. normal cells |
| Transcription | Sp1-DNA | Disruption of Sp1-survivin promoter interaction downregulates survivin, which may have therapeutic values |
| Posttranscription | Pre-mRNA splicing |
Survivin pre-mRNA splicing produces five known survivin variants and several new variants have been described recently |
| Posttranscription | Protein modification |
Dynamic phosphorylation and ubiquitination of survivin is important for survivin stability and function properly |
| Posttranscription | Protein- protein |
Several partner proteins (Hsp90, HBXIP, XIAP, cIAP1, Smac) interact with survivin and modulate survivin stability and/or function |
| Transcription | Protein-DNA | Survivin may act as a transcription activator or a transcription co-activator to control gene transcription |
Acknowledgement
We would like to thank Dr. Paul Spengler (RPCI) for critical reading of the manuscript. We apologize that some relevant reports could not be cited due to the space limitation. This work was sponsored in part by the Shanghai Pu Jiang fund PJ[2005]00746, and a NIH R01 Grant (CA109481).
Footnotes
The current nomenclature for survivin splice variants reflects the survivin exon used. Survivin-ΔEx3-3B is consistent with this rule.
References
- 1.Li F. Survivin study: what is the next wave? J Cell Physiol. 2003;197(1):8–29. doi: 10.1002/jcp.10327. [DOI] [PubMed] [Google Scholar]
- 2.Li F, Ling X. Survivin Study: An update of “What is the next wave?”. J Cell Physiol. 2006 doi: 10.1002/jcp.20634. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Altieri DC, Marchisio PC, Marchisio C. Survivin apoptosis: an interloper between cell death and cell proliferation in cancer. Lab Invest. 1999;79(11):1327–33. [PubMed] [Google Scholar]
- 4.Li F. Role of survivin and its splice variants in tumorigenesis. Br J Cancer. 2005;92(2):212–6. doi: 10.1038/sj.bjc.6602340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li F, Altieri DC. Transcriptional analysis of human survivin gene expression. Biochem J. 1999;344(Pt 2):305–11. doi: 10.1042/0264-6021:3440305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li F, Altieri DC. The cancer antiapoptosis mouse survivin gene: characterization of locus and transcriptional requirements of basal and cell cycle-dependent expression. Cancer Res. 1999;59(13):3143–51. [PubMed] [Google Scholar]
- 7.Hattori M, Sakamoto H, Satoh K, Yamamoto T. DNA demethylase is expressed in ovarian cancers and the expression correlates with demethylation of CpG sites in the promoter region of c-erbB-2 and survivin genes. Cancer Lett. 2001;169(2):155–64. doi: 10.1016/s0304-3835(01)00499-2. [DOI] [PubMed] [Google Scholar]
- 8.Tanaka C, Uzawa K, Shibahara T, Yokoe H, Noma H, Tanzawa H. Expression of an inhibitor of apoptosis, survivin, in oral carcinogenesis. J Dent Res. 2003;82(8):607–11. doi: 10.1177/154405910308200807. [DOI] [PubMed] [Google Scholar]
- 9.Yu J, Zhang H, Gu J, et al. Methylation profiles of thirty four promoter-CpG islands and concordant methylation behaviours of sixteen genes that may contribute to carcinogenesis of astrocytoma. BMC Cancer. 2004;4(1):65. doi: 10.1186/1471-2407-4-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Teodoridis JM, Hall J, Marsh S, et al. CpG island methylation of DNA damage response genes in advanced ovarian cancer. Cancer Res. 2005;65(19):8961–7. doi: 10.1158/0008-5472.CAN-05-1187. [DOI] [PubMed] [Google Scholar]
- 11.Fang JY, Chen YX, Lu J, et al. Epigenetic modification regulates both expression of tumor-associated genes and cell cycle progressing in human colon cancer cell lines: Colo-320 and SW1116. Cell Res. 2004;14(3):217–26. doi: 10.1038/sj.cr.7290222. [DOI] [PubMed] [Google Scholar]
- 12.Chen YK, Hsue SS, Lin LM. Survivin expression is regulated by an epigenetic mechanism for DMBA-induced hamster buccal-pouch squamous-cell carcinomas. Arch Oral Biol. 2005;50(6):593–8. doi: 10.1016/j.archoralbio.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 13.Esteve PO, Chin HG, Pradhan S. Human maintenance DNA (cytosine-5)-methyltransferase and p53 modulate expression of p53-repressed promoters. Proc Natl Acad Sci U S A. 2005;102(4):1000–5. doi: 10.1073/pnas.0407729102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu Y, Fang F, Ludewig G, Jones G, Jones D. A mutation found in the promoter region of the human survivin gene is correlated to overexpression of survivin in cancer cells. DNA Cell Biol. 2004;23(9):527–37. doi: 10.1089/dna.2004.23.527. [DOI] [PubMed] [Google Scholar]
- 15.Zhu H, Chen XP, Zhang WG, Luo SF, Zhang BX. Expression and significance of new inhibitor of apoptosis protein survivin in hepatocellular carcinoma. World J Gastroenterol. 2005;11(25):3855–9. doi: 10.3748/wjg.v11.i25.3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ryan BM, Konecny GE, Kahlert S, et al. Survivin expression in breast cancer predicts clinical outcome and is associated with HER2, VEGF, urokinase plasminogen activator and PAI-1. Ann Oncol. 2006 Jan 10; doi: 10.1093/annonc/mdj121. [DOI] [PubMed] [Google Scholar]
- 17.Beierle EA, Nagaram A, Dai W, Iyengar M, Chen MK. VEGF-mediated survivin expression in neuroblastoma cells. J Surg Res. 2005;127(1):21–8. doi: 10.1016/j.jss.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 18.Qian DZ, Wang X, Kachhap SK, et al. The histone deacetylase inhibitor NVP-LAQ824 inhibits angiogenesis and has a greater antitumor effect in combination with the vascular endothelial growth factor receptor tyrosine kinase inhibitor PTK787/ZK222584. Cancer Res. 2004;64(18):6626–34. doi: 10.1158/0008-5472.CAN-04-0540. [DOI] [PubMed] [Google Scholar]
- 19.Asanuma H, Torigoe T, Kamiguchi K, et al. Survivin expression is regulated by coexpression of human epidermal growth factor receptor 2 and epidermal growth factor receptor via phosphatidylinositol 3-kinase/AKT signaling pathway in breast cancer cells. Cancer Res. 2005;65(23):11018–25. doi: 10.1158/0008-5472.CAN-05-0491. [DOI] [PubMed] [Google Scholar]
- 20.Wang Q, Greene MI. EGFR enhances Survivin expression through the phosphoinositide 3 (PI-3) kinase signaling pathway. Exp Mol Pathol. 2005;79(2):100–7. doi: 10.1016/j.yexmp.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 21.Ling X, Bernacki RJ, Brattain MG, Li F. Induction of survivin expression by taxol (paclitaxel) is an early event which is independent on taxol-mediated G2/M arrest. J Biol Chem. 2004;279(15):15196–203. doi: 10.1074/jbc.M310947200. [DOI] [PubMed] [Google Scholar]
- 22.Qiu L, Di W, Jiang Q, et al. Targeted inhibition of transient activation of the EGFR-mediated cell survival pathway enhances paclitaxel-induced ovarian cancer cell death. Int J Oncol. 2005;27(5):1441–8. [PubMed] [Google Scholar]
- 23.Frasor J, Danes JM, Komm B, Chang KC, Lyttle CR, Katzenellenbogen BS. Profiling of estrogen up- and down-regulated gene expression in human breast cancer cells: insights into gene networks and pathways underlying estrogenic control of proliferation and cell phenotype. Endocrinology. 2003;144(10):4562–74. doi: 10.1210/en.2003-0567. [DOI] [PubMed] [Google Scholar]
- 24.Konduri SD, Sayeed A, Liu W, Bansal S, Li F, Das GM. Estrogen receptor abrogates transcriptional repression by p53 tumor suppressor protein. In: AACR, editor. 96th American Association for Cancer Research (AACR) Annual Meeting; Anaheim, CA. 2005 April 16-20; 2005. pp. 861–2. Anaheim, CA. [Google Scholar]
- 25.Moggs JG, Murphy TC, Lim FL, et al. Anti-proliferative effect of estrogen in breast cancer cells that re-express ERalpha is mediated by aberrant regulation of cell cycle genes. J Mol Endocrinol. 2005;34(2):535–51. doi: 10.1677/jme.1.01677. [DOI] [PubMed] [Google Scholar]
- 26.Zhang M, Latham DE, Delaney MA, Chakravarti A. Survivin mediates resistance to antiandrogen therapy in prostate cancer. Oncogene. 2005;24(15):2474–82. doi: 10.1038/sj.onc.1208490. [DOI] [PubMed] [Google Scholar]
- 27.Schlette EJ, Medeiros LJ, Goy A, Lai R, Rassidakis GZ. Survivin expression predicts poorer prognosis in anaplastic large-cell lymphoma. J Clin Oncol. 2004;22(9):1682–8. doi: 10.1200/JCO.2004.10.172. [DOI] [PubMed] [Google Scholar]
- 28.Diaz N, Minton S, Cox C, et al. Activation of Stat3 in Primary Tumors from High-Risk Breast Cancer Patients Is Associated with Elevated Levels of Activated Src and Survivin Expression. Clin Cancer Res. 2006;12(1):20–8. doi: 10.1158/1078-0432.CCR-04-1749. [DOI] [PubMed] [Google Scholar]
- 29.Kanda N, Seno H, Konda Y, et al. STAT3 is constitutively activated and supports cell survival in association with survivin expression in gastric cancer cells. Oncogene. 2004;23(28):4921–9. doi: 10.1038/sj.onc.1207606. [DOI] [PubMed] [Google Scholar]
- 30.Konnikova L, Kotecki M, Kruger MM, Cochran BH. Knockdown of STAT3 expression by RNAi induces apoptosis in astrocytoma cells. BMC Cancer. 2003;3:23. doi: 10.1186/1471-2407-3-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mitsiades CS, Mitsiades N, Poulaki V, et al. Activation of NF-kappaB and upregulation of intracellular anti-apoptotic proteins via the IGF-1/Akt signaling in human multiple myeloma cells: therapeutic implications. Oncogene. 2002;21(37):5673–83. doi: 10.1038/sj.onc.1205664. [DOI] [PubMed] [Google Scholar]
- 32.Barnes N, Haywood P, Flint P, Knox WF, Bundred NJ. Survivin expression in in situ and invasive breast cancer relates to COX-2 expression and DCIS recurrence. Br J Cancer. 2006;94(2):253–8. doi: 10.1038/sj.bjc.6602932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dalwadi H, Krysan K, Heuze-Vourc’h N, et al. Cyclooxygenase-2-dependent activation of signal transducer and activator of transcription 3 by interleukin-6 in non-small cell lung cancer. Clin Cancer Res. 2005;11(21):7674–82. doi: 10.1158/1078-0432.CCR-05-1205. [DOI] [PubMed] [Google Scholar]
- 34.Krysan K, Dalwadi H, Sharma S, Pold M, Dubinett S. Cyclooxygenase 2-dependent expression of survivin is critical for apoptosis resistance in non-small cell lung cancer. Cancer Res. 2004;64(18):6359–62. doi: 10.1158/0008-5472.CAN-04-1681. [DOI] [PubMed] [Google Scholar]
- 35.Hsieh FC, Cheng G, Lin J. Evaluation of potential Stat3-regulated genes in human breast cancer. Biochem Biophys Res Commun. 2005;335(2):292–9. doi: 10.1016/j.bbrc.2005.07.075. [DOI] [PubMed] [Google Scholar]
- 36.Catalano A, Graciotti L, Rinaldi L, et al. Preclinical evaluation of the nonsteroidal anti-inflammatory agent celecoxib on malignant mesothelioma chemoprevention. Int J Cancer. 2004;109(3):322–8. doi: 10.1002/ijc.11710. [DOI] [PubMed] [Google Scholar]
- 37.Nam S, Buettner R, Turkson J, et al. Indirubin derivatives inhibit Stat3 signaling and induce apoptosis in human cancer cells. Proc Natl Acad Sci U S A. 2005;102(17):5998–6003. doi: 10.1073/pnas.0409467102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gritsko T, Williams A, Turkson J, et al. Persistent Activation of Stat3 Signaling Induces Survivin Gene Expression and Confers Resistance to Apoptosis in Human Breast Cancer Cells. Clin Cancer Res. 2006;12(1):11–9. doi: 10.1158/1078-0432.CCR-04-1752. [DOI] [PubMed] [Google Scholar]
- 39.Qi H, Ohh M. The von Hippel-Lindau tumor suppressor protein sensitizes renal cell carcinoma cells to tumor necrosis factor-induced cytotoxicity by suppressing the nuclear factor-kappaB-dependent antiapoptotic pathway. Cancer Res. 2003;63(21):7076–80. [PubMed] [Google Scholar]
- 40.Takada Y, Khuri FR, Aggarwal BB. Protein farnesyltransferase inhibitor (SCH 66336) abolishes NF-kappaB activation induced by various carcinogens and inflammatory stimuli leading to suppression of NF-kappaB-regulated gene expression and up-regulation of apoptosis. J Biol Chem. 2004;279(25):26287–99. doi: 10.1074/jbc.M400963200. [DOI] [PubMed] [Google Scholar]
- 41.Amaya K, Ohta T, Kitagawa H, et al. Angiotensin II activates MAP kinase and NF-kappaB through angiotensin II type I receptor in human pancreatic cancer cells. Int J Oncol. 2004;25(4):849–56. [PubMed] [Google Scholar]
- 42.Kawakami H, Tomita M, Matsuda T, et al. Transcriptional activation of survivin through the NF-kappaB pathway by human T-cell leukemia virus type I tax. Int J Cancer. 2005;115(6):967–74. doi: 10.1002/ijc.20954. [DOI] [PubMed] [Google Scholar]
- 43.Tracey L, Perez-Rosado A, Artiga MJ, et al. Expression of the NF-kappaB targets BCL2 and BIRC5/Survivin characterizes small B-cell and aggressive B-cell lymphomas, respectively. J Pathol. 2005;206(2):123–34. doi: 10.1002/path.1768. [DOI] [PubMed] [Google Scholar]
- 44.Nakano J, Huang CL, Liu D, Ueno M, Sumitomo S, Yokomise H. Survivin gene expression is negatively regulated by the p53 tumor suppressor gene in non-small cell lung cancer. Int J Oncol. 2005;27(5):1215–2. [PubMed] [Google Scholar]
- 45.Pizem J, Cor A, Gale N. Survivin expression is a negative prognostic marker in laryngeal squamous cell carcinoma and is associated with p53 accumulation. Histopathology. 2004;45(2):180–6. doi: 10.1111/j.1365-2559.2004.01925.x. [DOI] [PubMed] [Google Scholar]
- 46.Tsuji N, Furuse K, Asanuma K, et al. Mutations of the p53 gene and loss of heterozygosity at chromosome 17p13.1 are associated with increased survivin expression in breast cancer. Breast Cancer Res Treat. 2004;87(1):23–31. doi: 10.1023/B:BREA.0000041575.73262.aa. [DOI] [PubMed] [Google Scholar]
- 47.Zhu N, Gu L, Findley HW, Li F, Zhou M. An alternatively spliced survivin variant is positively regulated by p53 and sensitizes leukemia cells to chemotherapy. Oncogene. 2004;23(45):7545–51. doi: 10.1038/sj.onc.1208038. [DOI] [PubMed] [Google Scholar]
- 48.Wang Z, Fukuda S, Pelus LM. Survivin regulates the p53 tumor suppressor gene family. Oncogene. 2004;23(49):8146–53. doi: 10.1038/sj.onc.1207992. [DOI] [PubMed] [Google Scholar]
- 49.Ma H, Nguyen C, Lee KS, Kahn M. Differential roles for the coactivators CBP and p300 on TCF/beta-catenin-mediated survivin gene expression. Oncogene. 2005;24(22):3619–31. doi: 10.1038/sj.onc.1208433. [DOI] [PubMed] [Google Scholar]
- 50.Li F, Brattain MG. Role of the survivin gene in pathophysiology. American Journal of Pathology. 2006 doi: 10.2353/ajpath.2006.060121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Spaulding B, Pan D, Ghadersohi A, et al. Characterization of the 12C4 survivin monoclonal antibody and insight into the expression of survivin in human adult tissues. 2006. [DOI] [PMC free article] [PubMed]
- 52.Chang CC, Heller JD, Kuo J, Huang RC. Tetra-O-methyl nordihydroguaiaretic acid induces growth arrest and cellular apoptosis by inhibiting Cdc2 and survivin expression. Proc Natl Acad Sci U S A. 2004;101(36):13239–44. doi: 10.1073/pnas.0405407101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu J, Ling X, Pan D, et al. Molecular mechanism of inhibition of survivin transcription by the GC-rich sequence-selective DNA binding antitumor agent, hedamycin: evidence of survivin down-regulation associated with drug sensitivity. J Biol Chem. 2005;280(10):9745–51. doi: 10.1074/jbc.M409350200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Caldas H, Jiang Y, Holloway MP, et al. Survivin splice variants regulate the balance between proliferation and cell death. Oncogene. 2005;24(12):1994–2007. doi: 10.1038/sj.onc.1208350. [DOI] [PubMed] [Google Scholar]
- 55.Ling X, Yang J, Tan D, et al. Differential expression of survivin-2B and survivin-DeltaEx3 is inversely associated with disease relapse and patient survival in non-small-cell lung cancer (NSCLC) Lung Cancer. 2005;49(3):353–61. doi: 10.1016/j.lungcan.2005.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Noton EA, Colnaghi R, Tate S, et al. Molecular analysis of survivin isoforms: evidence that alternatively spliced variants do not play a role in mitosis. J Biol Chem. 2006;281(2):1286–95. doi: 10.1074/jbc.M508773200. [DOI] [PubMed] [Google Scholar]
- 57.Krieg A, Mahotka C, Krieg T, et al. Expression of different survivin variants in gastric carcinomas: first clues to a role of survivin-2B in tumour progression. Br J Cancer. 2002;86(5):737–43. doi: 10.1038/sj.bjc.6600153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fangusaro JR, Jiang Y, Holloway MP, et al. Survivin, Survivin-2B, and Survivin-deItaEx3 expression in medulloblastoma: biologic markers of tumour morphology and clinical outcome. Br J Cancer. 2005;92(2):359–65. doi: 10.1038/sj.bjc.6602317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vegran F, Boidot R, Oudin C, Riedinger JM, Lizard-Nacol S. Distinct expression of Survivin splice variants in breast carcinomas. Int J Oncol. 2005;27(4):1151–7. [PubMed] [Google Scholar]
- 60.Ryan B, O’Donovan N, Browne B, et al. Expression of survivin and its splice variants survivin-2B and survivin-DeltaEx3 in breast cancer. Br J Cancer. 2005;92(1):120–4. doi: 10.1038/sj.bjc.6602314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Suga K, Yamamoto T, Yamada Y, Miyatake S, Nakagawa T, Tanigawa N. Correlation between transcriptional expression of survivin isoforms and clinicopathological findings in human colorectal carcinomas. Oncol Rep. 2005;13(5):891–7. [PubMed] [Google Scholar]
- 62.Caldas H, Honsey LE, Altura RA. Survivin 2alpha: a novel Survivin splice variant expressed in human malignancies. Mol Cancer. 2005;4(1):11. doi: 10.1186/1476-4598-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.O’Connor DS, Grossman D, Plescia J, et al. Regulation of apoptosis at cell division by p34cdc2 phosphorylation of survivin. Proc Natl Acad Sci U S A. 2000;97(24):13103–7. doi: 10.1073/pnas.240390697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Grossman D, Kim PJ, Schechner JS, Altieri DC. Inhibition of melanoma tumor growth in vivo by survivin targeting. Proc Natl Acad Sci U S A. 2001;98(2):635–40. doi: 10.1073/pnas.230450097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.O’Connor DS, Wall NR, Porter AC, Altieri DC. A p34(cdc2) survival checkpoint in cancer. Cancer Cell. 2002;2(1):43–54. doi: 10.1016/s1535-6108(02)00084-3. [DOI] [PubMed] [Google Scholar]
- 66.Wall NR, O’Connor DS, Plescia J, Pommier Y, Altieri DC. Suppression of survivin phosphorylation on Thr34 by flavopiridol enhances tumor cell apoptosis. Cancer Res. 2003;63(1):230–5. [PubMed] [Google Scholar]
- 67.Suzuki A, Ito T, Kawano H, et al. Survivin initiates procaspase 3/p21 complex formation as a result of interaction with Cdk4 to resist Fas-mediated cell death. Oncogene. 2000;19(10):1346–53. doi: 10.1038/sj.onc.1203429. [DOI] [PubMed] [Google Scholar]
- 68.Fortugno P, Beltrami E, Plescia J, et al. Regulation of survivin function by Hsp90. Proc Natl Acad Sci U S A. 2003;100(24):13791–6. doi: 10.1073/pnas.2434345100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Marusawa H, Matsuzawa S, Welsh K, et al. HBXIP functions as a cofactor of survivin in apoptosis suppression. Embo J. 2003;22(11):2729–40. doi: 10.1093/emboj/cdg263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dohi T, Okada K, Xia F, et al. An IAP-IAP Complex Inhibits Apoptosis. J Biol Chem. 2004;279(33):34087–90. doi: 10.1074/jbc.C400236200. [DOI] [PubMed] [Google Scholar]
- 71.Samuel T, Okada K, Hyer M, Welsh K, Zapata JM, Reed JC. cIAP1 Localizes to the nuclear compartment and modulates the cell cycle. Cancer Res. 2005;65(1):210–8. [PubMed] [Google Scholar]
- 72.McNeish IA, Lopes R, Bell SJ, et al. Survivin interacts with Smac/DIABLO in ovarian carcinoma cells but is redundant in Smac-mediated apoptosis. Exp Cell Res. 2005;302(1):69–82. doi: 10.1016/j.yexcr.2004.08.029. [DOI] [PubMed] [Google Scholar]
- 73.Song Z, Yao X, Wu M. Direct interaction between survivin and Smac is essential for the anti-apoptotic activity of survivin during Taxol-induced apoptosis. J Biol Chem. 2003;278(25):23130–40. doi: 10.1074/jbc.M300957200. [DOI] [PubMed] [Google Scholar]
- 74.Yan X, Cao L, Li Q, et al. Aurora C is directly associated with Survivin and required for cytokinesis. Genes Cells. 2005;10(6):617–26. doi: 10.1111/j.1365-2443.2005.00863.x. [DOI] [PubMed] [Google Scholar]
- 75.Kleinschmidt-DeMasters BK, Heinz D, McCarthy PJ, et al. Survivin in glioblastomas. Protein and messenger RNA expression and comparison with telomerase levels. Arch Pathol Lab Med. 2003;127(7):826–33. doi: 10.5858/2003-127-826-SIG. [DOI] [PubMed] [Google Scholar]
- 76.Sharma H, Sen S, Mathur M, Bahadur S, Singh N. Combined evaluation of expression of telomerase, survivin, and anti-apoptotic Bcl-2 family members in relation to loss of differentiation and apoptosis in human head and neck cancers. Head Neck. 2004;26(8):733–40. doi: 10.1002/hed.20059. [DOI] [PubMed] [Google Scholar]
- 77.Endoh T, Tsuji N, Asanuma K, Yagihashi A, Watanabe N. Survivin enhances telomerase activity via up-regulation of specificity protein 1- and c-Myc-mediated human telomerase reverse transcriptase gene transcription. Exp Cell Res. 2005;305(2):300–11. doi: 10.1016/j.yexcr.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 78.Kostrouchova M, Kostrouch Z, Saudek V, Piatigorsky J, Rall JE. BIR-1, a Caenorhabditis elegans homologue of Survivin, regulates transcription and development. Proc Natl Acad Sci U S A. 2003;100(9):5240–5. doi: 10.1073/pnas.0730770100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Song KH, Kim TM, Kim HJ, et al. Molecular cloning and characterization of a novel inhibitor of apoptosis protein from Xenopus laevis. Biochem Biophys Res Commun. 2003;301(1):236–42. doi: 10.1016/s0006-291x(02)03013-9. [DOI] [PubMed] [Google Scholar]