

The authors have cloned a novel member of the PRG family that induces filopodia growth in a Cdc42-independent manner. Hence, studies in primary neurons revealed that PRG5 impedes RhoA-mediated axon collapse induced by LPA and Nogo-A. These data reveal a new function of PRG5 with impact on neurite growth in an axonal growth inhibitory environment.
Abstract
Members of the plasticity-related gene (PRG1-4) family are brain-specific integral membrane proteins and implicated in neuronal plasticity, such as filopodia formation and axon growth after brain lesion. Here we report on the cloning of a novel member of the PRG family, PRG5, with high homologies to PRG3. PRG5 is regulated during brain and spinal cord development and is exclusively allocated within the nervous system. When introduced in neurons, PRG5 is distributed in the plasma membrane and induces filopodia as well as axon elongation and growth. Conversely, siRNA mediated knockdown of PRG5 impedes axon growth and disturbs filopodia formation. Here we show that PRG5 induces filopodia growth independently of Cdc42. Moreover, axon collapse and RhoA activation induced by LPA and myelin-associated neurite inhibitor Nogo-A is attenuated in the presence of PRG5, although direct activation of the RhoA-Rho-PIP5K kinase pathway abolishes PRG5 -formed neurites. Thus, we describe here the identification of a novel member of the PRG family that induces filopodia and axon elongation in a Cdc42-independent manner. In addition, PRG5 impedes brain injury-associated growth inhibitory signals upstream of the RhoA-Rho kinase pathway.
INTRODUCTION
Neurite growth and remodeling is a fundamental process for nervous system development and plasticity (Tessier-Lavigne and Goodman, 1996; O'Donnell et al., 2009). During brain development, axons extend over long distances and form contacts with their target structure, facilitating functional connections (Harel and Strittmatter, 2006). These neurite connections become stabilized and restricted during maturation and secure complex brain functions. Conversely, decline of intrinsic axonal remodeling activity and the lesion environment comprising neurite growth inhibiting factors limit regeneration and repair in brain and spinal cord after acquired neurological injury (David and Aguayo, 1981; He and Koprivica, 2004; Silver and Miller, 2004).
Evidence for a neurite growth inhibitory environment in the adult CNS came from axon regeneration studies in vivo through peripheral nerve grafts (David and Aguayo, 1981; Benfey and Aguayo, 1982). Currently several extracellular ligands have been identified which are released in the injured brain (Silver and Miller, 2004). Of those lysophosphatidic acid (LPA) is a bioactive lipid borne from astrocytes and blood stream (Moolenaar, 1994; Kranenburg et al., 1999; Inoue et al., 2004; Savaskan et al., 2007), whereas Nogo has been identified as a myelin-associated inhibitor derived from oligodendrocytes and myelin breakdown products (Chen et al., 2000; GrandPré et al., 2000; Prinjha et al., 2000). Interestingly, although LPA and Nogo act on different receptors (LPA on LPA1–5 receptors; Nogo on NgR and PirB/LILRB2), both ligands converge on RhoA-Rho kinase pathway, mediating neurite retraction and inhibition of axon growth (Jalink et al., 1994; Fournier et al., 2001; Wang et al., 2002; Miao et al., 2006; Atwal et al., 2008). Concomitantly, pharmacological and genetic studies revealed that interfering with Nogo/NgR function or LPA signaling promotes axonal regeneration and functional recovery after CNS injury (Schnell and Schwab, 1990; GrandPré et al., 2002; Bräuer et al., 2003; Inoue et al., 2004).
During development, in an initial step of neuritogenesis neurites are generated out of actin-rich needle-like membrane protrusions, also called filopodia or microspikes. Filopodia are important also for various other fundamental cellular processes such as cell attachment and migration (Mattila and Lappalainen, 2008). Control of these membrane protrusions and their growth is ultimately dependent on cytoskeletal dynamics that to a large extent are regulated by the small-molecular-weight GTPases of the Rho family. In particular, the Rho family GTPase Cdc42 regulates bundled actin filaments extending from the cell periphery and thus forming canonical filopodia of different lengths and widths (Nobes and Hall; 1995a; Etienne-Manneville and Hall, 2002). The best studied signaling pathway initiating filopodia growth currently is centered on the activation of WASP and N-WASP by Cdc42 (Machesky and Insall, 1998; Rohatgi et al., 1999). The concept is that interaction of CDC42 with WASP and N-WASP in the presence of phosphatidylinositol-4,5-bisphosphate (PIP2) leads to activation of the ARP2/3 complex, which acts as a nucleator and growth promoter for actin filaments. However, recent data indicate that other signaling pathways independent of Cdc42 must exist because filopodia are also formed in the absence of N-WASP and WASP (Lommel et al., 2001; Snapper et al., 2001). In addition, N-WASP is often not enriched in filopodia, and it has been reported that even in the absence of Cdc42 or ARP2/3 filopodia growth is not affected (Prehoda et al., 2000; Ridley, 2006). An alternative pathway for filopodia induction requires the formin family of actin regulatory proteins located at the tips of filopodia (Evangelista et al., 2003; Mellor, 2009). Formins act independently of ARP2/3 and recruits the GTPase Rif (Rho in filopodia) instead of Cdc42. Together, molecules regulating the formation of filopodia and growth cone shaping are critically involved in axon growth control and are implicated in overcoming neurite growth inhibition signals (Gallo and Letourneau, 1998; Tanabe et al., 2000; Jain et al., 2004; Matusek et al., 2008).
Recently, we identified a novel class of integral membrane proteins called plasticity-related genes (PRG), which are differentially expressed in the developing brain and reexpressed in regenerating axons (Bräuer et al., 2003; Savaskan et al., 2004). In particular, PRG3 (Plasticity Related Gene, alternatively called LPR1) induces the formation of filopodia and neurite growth (Savaskan et al., 2004). Interestingly, the membrane protein PRG3 acts independently of Cdc42 and does not require VASP family proteins, pointing to an overlooked role of integral membrane proteins in the shaping of membrane curves (Sigal et al., 2007; Savaskan, unpublished observation). We continued our search for more members of the PRG family in the genome and identified a novel gene termed PRG5. Here we report on the integral membrane protein PRG5, which is mostly related to PRG3, and is specifically expressed in brain and spinal cord. PRG5 is distributed in plasma membranes and induces filopodia formation as well as neurite growth. We characterized the required domains for filopodia inducing activity and found that the localization of the carboxyl terminal domain is critical for neurite growth and can overcome retraction responses of common neurite growth inhibitors such as Nogo-A and LPA.
MATERIALS AND METHODS
Northern Blotting
For probing multitissue Northern blots (Takara Biosystems, Heidelberg, Germany) we used the full-length coding sequence of PRG5 for 38P-radioactive labeling (Prime-a-Gene, Promega, Heidelberg, Germany; DuPont-NENDuPont-NEN, Germany) and led the hybridization run overnight at 68°C after which blots were washed in 2× SSC/0.5% SDS and 0.1× SSC/0.1% SDS for 10 min at each step. Exposure was performed on Kodak X-OMAT AR x-ray films (Eastman Kodak, Stuttgart, Germany) for 8–12 h.
The Allan Brain Atlas and NCBI Resources
The Allan Brain Atlas (ABA) Web site (www.brain-map.org) supports gene-specific expression analysis in mouse brain by providing an automated high-throughput in situ hybridization Internet platform. The Brain Explorer 1.3 software distributed from the ABA Web site was used to visualize gene expression three dimensionally and navigate through the brain. Coronal and sagittal data were taken form the Web interface at the appropriate layer. An eight-color scale bar displays relative gene expression signal (from low to high: blue-aqua-turquoise-bright green-yellow-gold-light orange-orange; Lein et al., 2007). Expression levels in different brain regions were downloaded as xml files and were processed with Microsoft Excel 2007 (Richmond, VA). Sagittal planes of spinal cord were taken at approximately level T8 for further analysis of PRG5 expression. For human PRG5 expression analysis we acquired the National Center for Biotechnology Information (NCBI) GeneNote project (NCBI profile graph, http://www.ncbi.nlm.nih.gov/geo/).
Molecular Cloning of PRG5, Sequence Analysis, and Vector Construction
In silico search for homologues of PRG3 revealed homologies to another human-expressed sequence tag. Using reverse transcription-PCR, full-length clones of PRG5 have been amplified. For sequence alignments and homology searches of PRGs we utilized the www.ncbi.nlm.nih.gov database and A Plasmid Editor software (ApE; M. W. Davis, Salt Lake City, UT). Transmembrane domains have been predicted using the Kyte Doolittle algorithm and all orthologous sequences of PRG5 (human, mouse, and rat) are deposited at the NCBI database (human PRG5, GenBank Accession no. FJ472844; Rattus norvegicus PRG5, GenBank Accession no. EU792473; and Mus musculus PRG5, GenBank Accession no. AY512657). For construct cloning we extended single restriction sites at the 5′ and 3′ prime regions of amplified murine PRG5-cDNA fragments by PCR and inserted the resulting amplicons into the pEGFP (Takara Biosystems), p3FLAG (Sigma, Steinheim, Germany) and pmRFP (Kess Jalink, NKI, Amsterdam, the Netherlands) vectors. C-terminal domain of PRG5 was cloned by PCR amplification out of the full-length clone and inserted the amplicon into linearized peGFP vectors.
Short Interfering RNA Vector Cloning, Protein Isolation, and Expression Analysis
According to the criteria of Naito et al. (2005), three 19-mer short interfering RNAs (siRNAs) were chosen for RNA interference (RNAi) with rodent PRG5 transcripts (GenBank accession no. AB022345). Cloning of the synthetic oligonucleotides into the pSuperGFP vector (pS-GFPneo; OligoEngine, Seattle, WA) was performed by digesting the empty vector with BglII and EcoRI according to the manufacturer's instruction. Cells were transfected at low density (<20,000 cells/cm2) and were harvested 72 h after transfection. Cell lysates were resolved on 4–12% SDS-PAGE gel and electroblotted on nitrocellulose membranes (Schleicher & Schuell, Dassel, Germany). All incubations were done overnight at 4°C in PBST. For equal loading controls, a mouse β-actin mAb (Sigma) was used. SeaBlue protein ladder (Invitrogen, Karlsruhe, Germany) was used as protein weight reference. Immunoblots and Pierce-stained membranes were digitalized using Raytest (Wetzikon, Switzerland) and Aida software or conventional chemiluminescence film development (GE Healthcare, Munich, Germany).
Cell Culture and Transfection
N1E-115 neuronal cells (ATCC, Manassas, VA; CRL-2263) and P19 cells (ATCC, CRL-1825) were routinely maintained at 37°C with 5% CO2 in DMEM (Invitrogen, 31965) supplemented with 10% fetal bovine serum. Chemical transfection with the cloning constructs peGFP-PRG5, peGFP-PRG5CT, p3FLAG, pmRFP, and the GFP control vector peGFP (Takara Biosystems) was performed as followed. Cells were trypsinized, pelleted, resuspended in an adequate volume of medium and counted with a Neubauer cell chamber, and plated out at a final density of 3–4 × 103 cells/cm2 on 8-cm2 Petri dishes for time-lapse imaging. Alternatively, cells were plated on coverslips in 12-well plates for subsequent confocal imaging. Twelve hours after plating, cells were transfected with FuGENE 6 (Roche, Indianapolis, IN) or Lipofectamine (Invitrogen-Invitrogen) according to the supplier's instructions, with 100 μl Optimem (Invitrogen-Invitrogen), 1 μg DNA as indicated in Results, and 3 μl FuGENE 6. The transfection reagent was incubated for 15–45 min. Meanwhile the old medium was discharged and replaced with 700 μl prewarmed fresh medium. The transfection master mix was added dropwise to the dish. Cultured cells were used for experiments after 24–48 h. Hoechst and actin staining was performed as described previously (Savaskan et al., 2004, 2007). Images were taken with a CCD camera on a Zeiss Axioskope2 microscope (Jena, Germany). Digital data were processed with Adobe Photoshop CS4 (Adobe, San Jose, CA). Quantification of number and length of neurites was performed with the ImageJ 1.36 software (http://rsb.info.nih.gov/ij/; National Institutes of Health). The constructs were validated by DNA sequencing prior transfection (BioSynth, Staad, Switzerland).
RhoA GTPase Activation Assays
Active, GTP-bound RhoA fractions were determined by incubating cell lysates with glutathione S-transferase (GST)-Rhotekin Rho-binding domain proteins bound to glutathione-agarose according to the supplier's manual (Millipore, Temecula, CA). After serum starvation for 16 h cells were subsequently stimulated with LPA with various concentrations for 2.5 min and afterward cells were lysed on ice. A small fraction of the total lysate was saved for GTPase normalization. Protein fractions were resolved on a gradient SDS-PAGE, and immunoblots were performed with monoclonal RhoA antibody (clone 55, Millipore).
Primary Neuron Culture, Transfection, and Neurite Analysis
Rat neocortical neurons were isolated from E18 Wistar pups (Charles River Laboratories, Wilmington, MA), cortices freed of meninges were placed in ice-cold HBSS buffer without serum. Cortex tissue was further gently triturated with fire-polished Pasteur pipettes of diminishing tip diameter. Then, minced cortices were trypsinized with 0.25% trypsin for 10 min. After resuspending and centrifugation, neurons were resuspended in Neurobasal medium supplemented with B27 (Invitrogen-Invitrogen) and penicillin/streptomycin. Cortical neurons were plated on poly-l-lysine–coated glass coverslips to a final density of 25,000 per cm2. Transfection of primary neurons was performed with lipid-based reagents (Effectene, Qiagen, Hilden, Germany; Lipofectamine LTX, Invitrogen) according the manufacture's protocol 2–8 h after plating.
Two days after transfection neurons were fixed in 4% paraformaldehyde for 15 min and processed for βIII-tubulin immunostaining (Promega). Measurements for cell morphology were performed on βIII-tubulin positive structures with the ImageJ software (NIH, Bethesda, MD).
Time-Lapse Measurements
N1E-115 mouse neuronal cells were plated on 8-cm2 Petri dishes with a cell density of 4000 cells/cm2. To determine equal transfection efficacy solely green fluorescent protein (GFP)-tagged constructs were used. Control cells were transfected with peGFP-N1 and resulted similar phenotypes as untransfected controls. The temperature of the heating plate was set to 38°C. Camera exposure time was set to 500 ms. Controls of the time-lapse conditions was implemented by monitoring GFP-transfected N1E cells under the same conditions at least for the time period of the assay. N1E-115 cells were cultivated under serum starvation in 1 ml Optimem (Invitrogen) for 1–12 h, 24 h after transfection. Total measurement time was 20 or 40 min, respectively, snapshot images were taken every 15 s. Transfected cells were identified by acquiring fluorescence images at the beginning and end of each experiment. Test substances were added to the culture medium ∼1 min after starting the measurement series.
LPA Collapse Assay
Total time for LPA assays was 20 min, which were performed after at least 4 h of serum-free conditions before the assay. The stock solution of 5 mM was diluted 1/100 in OptiMem (Invitrogen-Invitrogen), and 10 μl of this solution was added to the cells to a final concentration of 0.5 μM LPA.
Nogo-A Retraction Assays
The total time for monitoring cells in this assay was 40 min under serum-free conditions. The stock solution of Nogo-A (12 μl at 0.361 mg/ml) was diluted in 28 μl Optimem. This solution, 10 μl, was added to the cells to a final concentration of 1.05 μg/ml (which is 9.92 × 10−3 μM; NAext = 105811.9 g/mol). For assay controls we used monoclonal mouse IgGs or the monoclonal mouse antibody α-11C7 with NAext assay conditions. The stock solution was solved in OptiMem (Invitrogen-Invitrogen), and this solution was added to the culture dish resulting in a final concentration of approx. 1 μg of recombinant Nogo-A.
Statistical Analysis
Quantitative data were obtained all from experiments as stated in the figure legend. Data from immunoblots and cell culture assays shown in the figures are selected representatives from performed experiments. Analysis was performed using unpaired Student's t test if not otherwise stated (Statview II, Abacus, Berkeley, CA). The level of significance was set at *p < 0.05, **p < 0.01, and ***p < 0.001. Error bars represent ±SD.
RESULTS
Identification of a New Member of the PRG Family
Previously, we described a subtraction hybridization screen for the identification of brain lesion induced genes and thereby identified a novel gene family termed plasticity related genes (PRGs) (Bräuer et al., 2003). Further RT-PCR screenings with degenerated primers revealed so far four members of the PRG family (Savaskan et al., 2004). Homology searches displayed that PRGs belong to the lipid phosphate phosphatase/type 2 phosphatidic acid phosphatase ectoenzyme superfamily. We extended our search for novel genes by in silico analysis of PRG homologues with focus on PRG3 relatives due to its axon growth–promoting activity. This investigation revealed highly homologous expressed sequence tags (ESTs). Assembly of these ESTs resulted in one full-length transcript. On the basis of this assembled sequence, we performed RT-PCR of cDNAs from neurons of various species and cloned the full-length sequence from mouse, rat, and human mRNAs, termed PRG5 (human PRG5, GenBank Accession no. FJ472844; R. norvegicus PRG5, GenBank Accession no. EU792473; and M. musculus PRG5, GenBank Accession no. AY512657). The new member of the PRG family in the human genome is most closely related to PRG3 (GenBank Accession no. NM_017753) with more than 73% homology at the nucleotide level and with 55% identical amino acid residues (AA; 174/315 AA) and 75% positive AA (237/315 AA; no gaps; Figure 1A). The human PRG5 gene is located on chromosome 1, 1p21 (organized in six exons in mammals; rat chromosome 2, 2q41 and mouse chromosome 3, 3G1) and shares high homologies with the membrane-associated PRG family (PRG1-4, mostly with PRG3). The previously cloned splice versions of lipid phosphate phosphatase/type 2d phosphatidic acid phosphatase (Kai et al., 1996, 1997; Kanoh et al., 1997; Sun et al., 2005) were initially annotated as type II phosphatidic acid phosphatases and represent the human orthologue of PRG5. The integral membrane protein PRG5 consists of six transmembrane domains and a 50-AA long C-terminus located putatively toward the cytosol (Figure 1B). Thus, these sequence features make PRG5 a novel member of the PRG family (Figure 1C) within the class of the lipid phosphate phosphatase/phosphatidic acid phosphatase superfamily (Savaskan et al., 2004; Sigal et al., 2005). PRG5 is highly conserved within mammals (>88% homologies within rat, mouse, and human PRG5 transcripts). Another common feature of PRG5 is its high expression in the brain, and there in particular in highly plastic regions such as the hippocampus, and to a lower extent in spinal cord, which is common for many members of the PRG family as well as its expression in testis (Figure 2, A and B). However, the unique C-terminus is 50 AA long and does not share sequence or domain similarities with known transmembrane proteins or with the most closely related member of the PRG family, PRG3, except for the shared motifs CVVXNFKG and PXXESPLE (named PEST sequence) (Figure 1A).
Figure 1.

PRG5 is a novel member of the PRG family. PRG5 is a 322-AA-long protein with six transmembrane domains and a 50-AA-long C-terminal tail domain. (A) Amino acid sequence of human PRG5 (GenBank Accession no. FJ472844) aligned to human PRG3 (GenBank Accession no. NM_017753). Putative transmembrane domains (TM) are boxed in cyan, and homologous sequences are indicated in light green. The C-terminus is marked in orange, and the homologous PEST domain at the C-terminus is boxed in yellow. (B) Proposed orientation of PRG5 in the plasma membrane. The six transmembrane domains are shown in blue, the intracellular N-terminal region, three extracellular, and two intracellular loops are shown in green, and the intracellular C-terminal tail (orange). (C) Phylogenetic analysis of the lipid phosphate phosphatase (LPP) superfamily including the PRG subfamily and PRG5. PRG3 is mostly related to PRG5 within the PRG subfamily.
Figure 2.

PRG5 is expressed during brain development and induces filopodia and membrane extension growth. (A) Multitissue Northern blot analysis of PRG5 mRNA shows a strong band at 4.4 kb in brain tissue and a slight band in testis. Right, GeneNote analysis (NCBI profile graph) of PRG5 gene expression in various human tissues. Samples are composed from a pool of 10–25 individuals. (B) Representative in situ hybridization images of PRG5 mRNA in the adult brain (left, coronal; right, sagittal). Bottom, representative images of PRG5 mRNA in situ expression in spinal cord at postnatal stage (P4) and in adult (A). Color coded bars illustrates the relative expression level of PRG5 in various adult mouse brain regions. Scales bars in all images, 800 μm. Right, quantitative analysis of PRG5 mRNA expression in various brain regions. CB, cerebellum; CTX, cerebral cortex; ISO, isocortex; OLF, olfactory area; HPF, hippocampal formation; HIP, hippocampal region; STR, striatum; STRd, striatum dorsal region; STRv, striatum ventral region; RHP, retrohippocampal region; PAL, pallidum; sAMY, striatum-like amygdala; HY, hypothalamus; P, pons; LSX, lateral septal complex. (C) PRG5-GFP fusion construct expression (PRG5GFP) in P19 cells compared with GFP-transfected controls. PRG5 induces strong filopodia growth and is localized mainly in the plasma membrane and end tips of filopodia. Solely expressed GFP is predominantly found in the cytosol and nucleus. Actin is given in red; nuclei are shown in blue. Scale bar in top right image, 11.1 μm, bottom image, 20 μm. (D) Quantification of filopodia formation in P19 cells. The values were averaged for at least three independent experiments. Statistical analysis was performed with Student's t test (n = 8); **p ≤ 0.01, ***p ≤ 0.001; mean values are shown; error bars, ±SD.
PRG5 Is Expressed during Neuronal Growth and Developmental Phases
Next, we analyzed PRG5 mRNA pattern by utilizing the ABA data set. When investigated in detail, the hippocampus shows strongest PRG5 hybridization signal, and PRG5 mRNA is highly expressed in the dentate gyrus, the cornu ammonis (CA1-CA3), and the olfactory bulb in adult mouse brain (Figure 2B). Slightly lower signals are detected in the cortex, the stratum radiatum, the cerebellum, and in striatum. Other brain regions such as the thalamus, pons and the hypothalamus showed expression levels below 10% of values of the highest PRG5-expressing regions. Noteworthy, during development PRG5 mRNA is expressed in the spinal cord, i.e., in neuron rich regions such as in medial motor nuclei and posterior funicular gray matter (Figure 2B).
PRG5 Is Localized in the Plasma Membrane, Induces Filopodia Sprouting and Promotes Neurite Growth
We investigated the cellular localization of PRG5 by expressing a C-terminal GFP-PRG5 fusion construct. When PRG5 was expressed in nonneuronal cells (in this case P19 embryonic carcinoma cells), most of the signal appeared in the plasma membrane and formed spiky-like membrane extensions (Figure 2C). Indeed, PRG5-expressing cells showed drastically altered cell morphology in the way that many tiny membrane extensions appeared compared with solely GFP-expressing control cells. As revealed in Figure 2C the soma expanded, and the morphology changed toward a “spiky” morphology, as indicated by filopodia formed at the cell surface in which PRG5 is enriched (34.6 ± 15.9 filopodia compared with 5.5 ± 2.4 filopodia per cell in control P19 cells). Also, the actin cytoskeleton was challenged in PRG5-expressing cells with more F-actin bundles (Figures 2C and 3). We quantified these protrusions in terms of length and number. PRG5 induced significantly more filopodia that were also longer compared with wild type or GFP-expressing controls (Figure 2D). Surprisingly, the PRG5 induced phenotype resembled in particular a similar phenotype also found in PRG3 expressing neurons though not in other PRG family members (Savaskan et al., 2004).
Figure 3.

PRG5 induces filopodia and is located at the plasma membrane and tips of filopodia and neurites. (A) PRG5 expression in neuronal cells induces dramatic neurite and filopodia growth. PRG5GFP (green) is localized at the tips of filopodia and neurites, whereas sole GFP (green) is mainly found in the cytosol. Actin is given in red; nuclei are shown in blue. Scale bar, 15 μm. (B) PRG5 expression in neuronal cells induces massive neurite and filopodia growth independent of the tag. For PRG5 expression a flag-tagged PRG5 (green) construct has been utilized and N1E-115 cells transfected. Immunostained flag-tagged PRG5 is localized at the tips of filopodia, neurites, and internal membrane structures. Actin is given in red. Bottom, higher magnification of framed region in top images. Scale bar, 20 μm. (C) Comparison of localization of RFP-PRG5 fusion protein (red) with PRG5-flag tagged protein (green). Both PRG5 constructs show equal subcellular distribution and phenotype. Scale bar, 20 μm. (D) High-resolution image of PRG5-expressing neuron. Higher magnification of the red boxed area (right) indicates the distribution of PRG5 in single actin-rich filopodia. Scale bar, 50 μm.
The changes in P19 cell morphology resembled stages of early neuronal differentiation comparable to the phenotype found by cultivation in the presence of growth factors and serum withdrawal, which prompted us to continue our investigations in more detail in neuronal cells. When PRG5 was expressed in neurons, cells developed massive filopodia trees with multiple branches and extended neurites (Figure 3). There, PRG5 is located in the plasma membrane and enriched in actin containing end tips of filopodia (Figure 3, A and B). In comparison, GFP is predominantly found in the cytosol (Figure 3A). Because we utilized a PRG5-GFP fusion construct that may raise concerns in terms of that PRG5 induced phenotype might be artificially produced, we further verified the PRG5 phenotype by acquiring two more constructs differing in tag size and orientation (N- vs. C-terminal). Interestingly, neuronal cells expressing a flag-tagged PRG5 construct demonstrated similar morphological alterations and overall similar phenotype as seen with PRG5 red fluorescent protein (RFP) fusion constructs (Figure 3C). Moreover, immunofluorescence analysis of the PRG5-flag allocation revealed a similar subcellular distribution as PRG5-RFP, i.e., both proteins were localized in intracellular membrane structures and in the plasma membrane (Figure 3D). Within the plasma membrane, PRG5 appeared in apical portions of the cell membrane and occurred enriched in end tips of filopodia (Figure 3D). These filopodia were rich in polymerized actin bundles and showed many branches (Figure 3D).
siRNA-mediated Knockdown of PRG5 Disturbs the Formation and Growth of Filopodia and Neurites
Because PRG5 expression induces filopodia formation and promotes neurite growth in different cells, the question remains whether this protein is also required in inducing and maintaining these structures. We went on performing an inverse experimental approach by constructing three independent siRNAs targeting PRG5 transcripts (siRNA322, siRNA373, siRNA1078). Two of the small interfering constructs (siRNA322 and siRNA373) revealed sufficient PRG5 knockdown at the protein level, whereas siRNA1078 was not functional in terms of translational suppression or morphological alteration (Figure 4A). However, when knocking down PRG5 by RNAi, the number of filopodia and neurite length was significantly reduced and was even below the number of wild-type cells (Figure 4C). Further, we challenged the culture condition by serum withdrawal for 16 h, which in wild-type cells promoted neurite growth to 10.8 ± 3.3 μm. Still, even serum-starved neuronal cells did not develop neurites when PRG5 was knocked down (neurite length 3.7 ± 1.6 μm). Together, these data indicate that PRG5 expression is required for filopodia formation and growth.
Figure 4.

siRNA-mediated PRG5 knockdown inhibits filopodia formation and neurite growth. (A) Validation of PRG5 expression in neuronal cells and RNAi-mediated PRG5 knockdown. PRG5-GFP fusion protein runs at ∼60 kDa. RNAi-mediated knockdown of PRG5 with two functional siRNAs (siRNA322 and siRNA373) reduces PRG5 expression significantly, as revealed by immunoblotting. Notably, siRNA1078 construct does not affect PRG5 expression as scramble siRNA and therefore serves as a control siRNA. Actin serves as a control for equal loading. Because GFP and siRNA expression is driven by a bicistronic expression cassette, GFP serves as control for efficient siRNA expression levels independently of RNAi efficacy. (B) siRNA-mediated PRG5 knockdown blocks filopodia formation and neurite growth. Representative images of scramble siRNA (siRNAcon) serving as a control and PRG5 siRNA373 knockdown in neuronal cells. Scale bar, 20 μm. (C) Quantification of neurite length and number of filopodia. Statistical analysis contains values averaged for three independent experiments and was performed with Student's t test; ***p < 0.001; error bars, ±SD of each group.
PRG5 Promotes Neurite Formation and Drives Axon Elongation in Cortical Neurons
To unequivocally demonstrate its neurite growth–promoting effects, we investigated PRG5 and PRG5 knockdown in primary rat cortical neurons. Primary cortical neurons were transfected with GFP or PRG5 and subsequently morphology was analyzed. Neurite length was rapidly enhanced in PRG5-expressing neurons compared with GFP controls (Figure 5). Furthermore, PRG5 induced dramatic neurite formation and branches not seen in GFP-expressing controls (Figure 5). We conducted a series of siRNA experiments in which we silenced PRG5 expression in comparison to scrambled siRNA controls. Scrambled siRNAs affected neither neurite morphology and length (32.6 ± 13.9 μm) nor number of neurites (8.9 ± 3.1) and resulted in similar values comparable to solely GFP-expressing neurons (neurite length 30.5 ± 13.3 μm, number of neurites per cell: 9.0 ± 2.4; Figure 5B). In contrast, PRG5 knockdown attenuated neurite length and number of neurites as evidenced by two independent siRNAs (si322 and si373; Figure 5, A and B). These results indicate that PRG5 renders filopodia induction and axon elongation in primary cortical neurons comparable to what was seen in neuronal cell lines.
Figure 5.

PRG5 promotes neurite formation in primary cortical neurons and PRG5 silencing attenuates neurite formation and growth. (A) PRG5GFP expression in E18 wild-type rat cortical neurons promotes neurite formation and neurite growth. GFP alone (green) is mainly found in the cytosol of cell body, whereas PRG5 (green) is mainly localized at the end tips of neurites. The neuronal marker ßIII tubulin is shown in red. Scale bar, 20 μm. (B) Quantification of neurite length and number in cortical neurons. Measurements of number of neurites are given per neuron. Values are averaged for three independent experiments; statistical analysis was performed with Student's t test; ***p < 0.001; error bars, ±SD of each group.
Membrane Targeting of C-terminal PRG5 Domain Transduces Neurite Growth Activity
Sequence alignments for various PRG members revealed that PRG5 shares more than 70% sequence homology with other PRGs. However, PRG5 bears a unique C-terminus with solely two domain features shared by PRG3 (PXXESPLE, and CVVXNFKG), whereas the rest of the C-terminal tail is unique to PRG5 and does not show similarities with defined molecules in public databases (Figure 1A). In addition, the only other member of the PRG family with such axon growth activity is PRG3, whereas other PRGs such as PRG1 or PRG2 do not spontaneously promote axon growth (Savaskan et al., 2004; Savaskan, unpublished data). Thus, we cloned solely the C-terminal domain of PRG5 omitting the membrane spanning domain and the N-terminal region of full-length (wild type) PRG5. To achieve localization in accordance to the wild-type distribution and to prevent artificial protein distribution and folding, we constructed a PRG5-CT expression construct by acquiring the myrostilation consensus sequence of the YES-kinase (for enhanced plasma membrane targeting) fused to the C-terminal domain of PRG5. Expression of PRG5CT resulted in plasma membrane localization of the C-terminus, which resembled distribution similar to that of wild-type PRG5 (Figure 6A). There, C-terminal PRG5 was enriched at distal portions and at the tips of filopodia and neurites (Figure 6A). Examination of total protein lysates of PRG5 wild type, GFP, and PRG5CT-expressing neurons in immunoblots showed that both PRG5 wild type as well as PRG5CT were sufficiently expressed (Figure 6B). Further, we compared the morphological alterations induced by PRG5CT to that found in wild-type PRG5 and controls. PRG5CT-expressing neurons showed equally long axons, whereas those revealed less branches and complexity compared with the wild-type PRG5 (Figure 6, A and C). Thus, the C-terminal domain of PRG5 is sufficient in inducing neurite growth, although subtle differences in terms of branches exist in comparison to full-length PRG5.
Figure 6.

Membrane targeting and localization of PRG5 is essential for axon growth. (A) Neuronal cells were transfected with PRG5 wild-type construct (PRG5GFP, green) and with the PRG5CT consistent of PRG5 C-terminus fused to the myristylation consensus sequence of the YES-kinase for membrane targeting (green). PRG5 and PRG5CT are both localized in neurites and promote similar phenotypes. Arrows indicate branching points. Actin, red. Scale bar, 50 μm. (B) Total cell lysates from neuronal cells transfected with GFP or GFP fusion constructs, PRG5, or PRG5CT were examined by immunoblotting with a GFP-specific antibody. GAPDH serves as a control for equal loading. (C) Quantification of neurite length and number of filopodia in controls, PRG5 and PRG5CT neurons. Statistical analysis contains values averaged for three independent experiments and was performed with Student's t test; *p < 0.01, **p < 0.005; error bars, ±SD of each group.
PRG5 Induces Filopodia Growth Independently of Cdc42 and mDia1
We continued investigating the filopodia inducing activity of PRG5. Cdc42 has been shown to form filopodia or microspikes (Nobes and Hall, 1995b; Kozma et al., 1997) and regulates various actin-remodeling factors (Hall, 1998). We proceeded our analysis and blocked Cdc42 signaling by expressing a dominant negative form of Cdc42 (Cdc42N17). However, PRG5 still induced filopodia as well as neurite elongation, as did PRG5CT in the presence of dominant negative Cdc42 (Figure 7), indicating a function alternatively or downstream of Cdc42. We speculated that PRG5 may act downstream of Cdc42 and focused on downstream effectors of Cdc42. First indications come from earlier work identifying an actin regulatory molecule class termed formins, which are required for filopodia induction (Evangelista et al., 2003; Peng et al., 2003; Mellor, 2009). Interestingly, formins act independently of N-WASP ARP2/3 in inducing filopodia. In mammals at least three Diaphanous-related formins (mDia1,2,3) exist, acting as critical effectors of Rho GTPases (Watanabe et al., 1997; Tominaga et al., 2000) and one of the formins, mDia1 (murine mDia1/Drf1/DFNA1) can bind to the primary Cdc42 effector and filopodia regulator IRSp53 and facilitate filopodia formation and axon elongation (Fujiwara et al., 2000; Arakawa et al., 2003; Peng et al., 2003). Further because dependent on context, mDia1 can stimulate filopodia independent of mDia2 (Carramusa et al., 2007; Sarmiento et al., 2008), we tested its role in PRG5 dependent cytoskeletal remodeling. For this we expressed dominant negative mDia1 (mDia1ΔN2) in neuronal cells and investigated cell shape to PRG5-mDia1ΔN2–coexpressing neurons. Although mDia1ΔN2 induced cell rounding and blocked filopodia formation, PRG5-mDia1ΔN2–expressing neurons still showed filopodia growth (Supplementary Figure S1), revealing that PRG5 acts independently of mDia1 and Cdc42.
Figure 7.

PRG5 promotes filopodia formation and neurite growth independently of Cdc42. (A) Neuronal cells were transfected with dominant negative Cdc42 (pIRES-GFP-Cdc42N17), and neurite length and filopodia were monitored. Representative images of Cdc42N17-expressing neurons and cotransfected neurons are shown. PRG5-expressing neurons cotransfected with Cdc42N17 show normal neurite length and filopodia number (mean neurite length 18 ± 5.9 μm; 54.6 ± 5.9 filopodia/cell) comparable to solely PRG5RFP-expressing cells (mean neurite length 21.6 ± 7.5 μm; 64.7 ± 8 filopodia/cell). Bottom images, neurons coexpressing Cdc42N17 (green) and C-terminal tail of PRG5 (PRG5CT, red). Scale bar, 20 μm. (B) Quantification of neurite length and number of filopodia in transfected neurons compared with Cdc42N17-expressing neurons. Measurements of number of filopodia are given per neuron. Statistical analysis was performed with Student's t test; ***p < 0.001; error bars, ±SD.
PRG5 Attenuates Axon Collapsing Activity of Different Neurite Growth Inhibitors
Next we studied the functional implications of PRG5 induced neurites and asked whether PRG5 induced neurites are to the same extent affected or even more sensitive than wild-type neurons toward neurite growth inhibitory signals. Therefore, we exposed PRG5-expressing neurons to a axon repellent environment by facilitating two neurite growth inhibitors, which are commonly present in vivo after brain and spinal cord injury. In addition, both neurite inhibitors mediate their effects similarly by the activation of the RhoA-Rho kinase pathway. First, time-lapse measurements were performed and neurite length was monitored after addition of LPA, a well-established axon-collapsing ligand (Jalink et al., 1994; Kranenburg et al., 1999). In controls, LPA induced a robust neurite retraction response within minutes (Figure 8). There, neurons retracted neurites rapidly and showed dramatic cell rounding (Figure 8A). In contrast, PRG5-expressing neurons displayed resistance toward LPA-induced collapse, and neurite length remained stable over the time monitored (Figure 8, A and B). Also, expressing the carboxyl terminal domain of PRG5 led to resistance toward LPA-mediated collapse (Figure 8, A and B). These data indicate that the presence of PRG5 attenuates LPA-induced axon collapse (Figure 8).
Figure 8.

PRG5 inhibits lysophosphatidic acid (LPA)-induced rapid neurite retraction. (A) Time-lapse measurements of neurons transfected with GFP, PRG5, and PRG5CT. PRG5-positive cells and PRG5CT do not show changes in neurite length after LPA treatment, whereas controls display neurite collapse. Arrows indicate retracting neurites in representative examples. Neurons were monitored for 20 min after treatment. Scale bar, 50 μm. (B) Quantification of neurite length in LPA-treated neurons. Statistical analysis was performed with Student's t test (n = 3); values are mean ± SD; ***p < 0.001.
We continued investigating PRG5 interaction with another well-established neurite growth inhibitor present in myelin. When applying the extracellular domain of Nogo-A (NAext) to the culture bath, wild-type neurons retracted their neurites, and maximal effects were found within 40 min (Figure 9). However, addition of Nogo-A did not provoke significant neurite retraction in PRG5-expressing neurons even when considering relative neurite length (Figure 9B). Concomitantly, when expressing the carboxyl terminal domain of PRG5 (PRG5CT), Nogo-A—induced axon retraction response was omitted (Figure 9, A and B). To control for a general retraction response and for monitoring neurite motility, we used control IgG proteins within the same and even higher concentration and incubated neurons in the same manner. Adding solely IgGs to neurons did not affect neurite length nor alter general morphology of these cells (Figure 9C). Thus, expression of PRG5 counteracts those two common neurite growth inhibitors.
Figure 9.

PRG5 attenuates Nogo-A–induced axon collapse. (A) Time-lapse measurements of neurons after Nogo-A treatment. Representative examples of time-lapse images are shown. Note, that PRG5GFP positive cells and PRG5CT remain their neurite length after Nogo-A treatment, whereas neurites from controls (GFP-transfected cells) collapsed rapidly. Arrows indicate retracting neurites in controls. Time scale of treatment was 40 min. Scale bar, 50 μm. (B) Quantification of neurite length after Nogo-A treatment of neurons. Statistical analysis was performed with Student's t test (n = 3), values are mean ± SD; *p < 0.05. (C) Quantification of neurite length after control IgG-antibody treatment in neuronal cells. Statistical analysis was performed with Student's t test (n = 3); *p < 0.01, error bar, ±SD.
Neurite Growth Activity of PRG5 Occurs Upstream of RhoA-Rho-PIP5K Kinase Pathway
In two neurite-retraction and cell-rounding paradigms, i.e., LPA and Nogo-A, PRG5 was sufficient to attenuate both axon collapse effects at similar quantitative levels. We followed up these findings and asked whether PRG5 interferes with downstream signals induced by LPA and Nogo-A. Indeed, both Nogo-A and LPA induce signal cascades converging on the RhoA-Rho kinase pathway. Thus, we tested whether PRG5 lies within this pathway interfering with one of the downstream targets of those neurite growth inhibitors. Transfection of constitutively active RhoA (RhoAV14) completely abolished neurite growth and reduced dramatically filopodia numbers (Figure 10). In most cases, RhoA induced rapid cell rounding. Coexpression of PRG5 or PRG5CT and RhoAV14 completely suppressed the PRG5 phenotype and neurons showed dramatically reduced neurite length (Figure 10), indicating that neurite growth activity of PRG5 may act upstream of RhoA. However, in cotransfected cells filopodia numbers were still increased (on average 34 filopodia per cell) compared with solely RhoA-expressing cells (8 filopodia per cell; Figure 10B) to ∼50% of the level found in solely PRG5-expressing cells (65 filopodia per cell). We investigated whether this filopodia formation activity is independent of the Rho-Rho kinase pathway. Cotransfection of PRG5 and ROCK revealed dramatic neurite retraction and cell rounding similar to solely ROCK-transfected cells (Figure 11A). Further investigations on downstream signals of the Rho-Rho kinase pathway led us test the effects of PIP5Kβ (type 1 phosphatidylinositol 4-phosphate 5-kinase β) on PRG5-induced neurites. When PRG5 was coexpressed with PIP5Kβ, the filopodia-inducing phenotype of PRG5 was significantly abolished (Figure 11B). These data indicate that PRG5 induced neurite-elongation and filopodia-forming activity act upstream of the RhoA-ROCK-PIP5Kβ kinase pathway.
Figure 10.

PRG5-dependent effects on neurites are impeded by RhoA signaling. (A) Representative images of neuronal cells expressing constitutive active RhoA (RhoAV14, green). RhoAV14-expressing neurons show massive reduction in neurite length and retraction of filopodia. RhoV14 (green)- and PRG5 (red)-coexpressing neurons show dramatically reduced neurite length (6.2 ± 2.7 μm) and filopodia number (34.2 ± 7.7 filopodia per cell) compared with solely PRG5-expressing neurons (neurite length 21.6 ± 7.5 μm, 64.7 ± 10.9 filopodia per cell). Bottom images, neurons coexpressing RhoAV14 (green) and C-terminal tail of PRG5 (PRG5CT, red). Scale bar, 20 μm. (B) Quantification of number of filopodia and neurite length in transfected neurons compared with RhoAV14-expressing cells. Measurements of number of filopodia are given per neuron. Statistical analysis was performed with Student's t test; ***p < 0.001; error bars, ±SD.
Figure 11.

PRG5 acts upstream from RhoA-ROCK-PIP5K pathway and interferes with LPA-induced RhoA activation. (A) Neuronal cells were transfected with ROCK and PRG5, and neurite growth and filopodia formation was monitored. Solely ROCK-expressing neurons show dramatic cell rounding. Note, that neurons expressing both PRG5 (red) and ROCK (green) also display round cell shape. (B) Neurons transfected with PRG5 and PIP5Kβ (type 1 phosphatidylinositol 4-phosphate 5-kinase β) show a phenotype comparable to solely PIP5K1β (green)-expressing neurons. Nuclei in A and B are shown in blue. Scale bar, 20 μm. (C) Neurons were transfected with either GFP or PRG5 as indicated and stimulated with various concentrations of LPA and levels of activated GTP-RhoA were determined by Rhotekin-binding assay (top panel). Total RhoA and actin served as controls for equal loading (bottom panels). Details on the Rhotekin-binding assay are described in Materials and Methods.
Because PRG5 induced neurites are resistant toward LPA- and Nogo-A—induced axon collapse, we analyzed whether the downstream activation of these neurite growth inhibitors is affected in PRG5-expressing neurons. In PRG5-expressing neurons, the amount of active RhoA was already lower compared with control neurons under control conditions (Figure 11C). Hence, LPA treatment at 0.5 or 5 μM led to massive elevation of the active RhoA pool (Figure 11C). In contrast, PRG5-positive neurons showed only slightly increased GTP-RhoA after 0.5 μM LPA treatment, a LPA concentration at which PRG5-expressing neurons remained their axons and overall morphology (Figure 11C). However, higher concentrations of LPA (5 μM) induced increased RhoA activation in PRG5 neurons comparable to controls.
DISCUSSION
Our results reveal a novel member of PRG family named PRG5, which is an integral membrane protein with six transmembrane domains and a carboxyl terminal tail located putatively in the cytoplasm. Homology searches uncover that PRG5 is the fifth representative of the PRG family with overall homology to the bona fide lipid phosphate phosphatases (LPPs). Members of the PRG family are characterized by variable amino carboxyl domains and catalytic domain residues in which amino acids crucial for the enzymatic activity in other family members such as PRG1 are replaced by nonconservative substitutions (Bräuer et al., 2003; Savaskan et al., 2004). Comparable to the closest relative PRG3 (Savaskan et al., 2004), overexpression of PRG5 induced dramatic morphological changes in neuronal cells as well as in various other cell types. PRG5 expression resulted in a pronounced induction of actin-enriched protrusions and growth in both nonneuronal cells as well as in neurons in an Cdc42-independent way. Concomitantly, knockdown of PRG5 impeded the formation of filopodia and neurite growth, which points to an unexpected role of a plasma membrane protein in the formation of Cdc42 independent filopodia growth.
PRG5 during Brain and Spinal Cord Development
We started our investigation on PRG5 with its transcriptional appearance in brain and other organs. First, Northern blot data revealed that PRG5 is mainly expressed in brain tissue comparable to PRG1, which is in stark contrast to PRG3 distribution giving a wider tissue allocation (Savaskan et al., 2004). Further, PRG5 transcripts show a strong developmental regulation, with high expression levels during postnatal stages. There, PRG5 mRNA is mainly found in the hippocampus and in cortex. Hence, PRG5 transcripts are tightly regulated as exemplified by expression in spinal cord during postnatal stages and almost no expression signals in the adult spinal cord. In contrast, other members of the PRG family such as PRG1 and PRG3 disclose a different expression dynamic with readily identifiable transcription even in discrete regions of the adult brain. In this way, PRG5 expression during spinal cord development resembles those of many other axon guidance molecules and their receptors, such as semaphorins and NgR (i.e., Sema3A, Sema3C, and their receptor neuropilin and neogenin; Steup et al., 1999, 2000; GrandPré et al., 2002), netrin-1/DCC (Steup et al., 2000), ephrins (ephrin A3, A7; Stein et al., 1999), and robo-slits (Whitford et al., 2002). However, when expressed, none of the aforementioned axon guidance molecules induce massive filopodia in cis-interaction, i.e., on neurites at the same plasma membrane domain.
What are the functional implications of the transient expression pattern? Indeed, PRG5 is expressed in early postnatal stages and there PRG5 transcripts are present within the spinal cord. Interestingly, this time window of expression is characterized by enhanced neuronal outgrowth and wiring and an overall high activity of structural plasticity of neuronal connections. It is tempting to speculate that PRG5 may play a role in these processes, especially when considering the morphological effects induced by PRG5 in neurons. However, this interpretation awaits further functional in vivo data. Also, whether PRG5 is regulated during synaptic activity as PRG3 is or directly participates in synaptic transmission as PRG1 does (Trimbuch et al., 2009), needs to be further investigated. One interpretation backed up by expression analysis is that PRG5 distribution in the adult is restricted solely to neuronal layers where structural plasticity is required.
PRG5 Forms Filopodia and Axon Elongation Independently of Cdc42
When expressed, PRG5 drastically challenged cell morphology of various cell types. Formation of fine and long filopodia and membrane protrusions positive for PRG5 was observed with complex branching, a phenotype also referred to hedgehog-like in contrast to the Cdc42 phenotype referred to as starfish-like (Passey et al., 2004). Conversely, RNAi knockdown of PRG5 impeded filopodia formation and neurite outgrowth resulting in neurite length below the level of controls. The morphology of PRG5 induced filopodia resembled those of PRG3 filopodia, which are in stark contrast to the short and thick filopodia characteristic for Cdc42 (Savaskan et al., 2004; Passey et al., 2004; Sigal et al., 2007). This is of particular interest because control of these membrane protrusion is driven by cytoskeletal dynamics, which to a large extent are regulated by the small-molecular-weight GTPases of the Rho family (Nobes and Hall; 1995a; Etienne-Manneville and Hall, 2002). So far, the Rho family GTPase Cdc42 takes center stage in regulating actin filament extensions and forming filopodia of different lengths and widths (Nobes and Hall, 1995b; Kozma et al., 1997). In this respect PRG5 forms canonical filopodia characterized by organized F-actin fibers in membrane extensions as Cdc42 does (Faix et al., 2009). Within Cdc42 signaling events, activation of WASP and N-WASP by Cdc42 in the presence of PIP2 leads to ARP2/3 activation, which acts as a nucleator and growth promoter for actin filaments, leading to the concept of “convergent-elongation” model of filopodia formation (Machesky and Insall, 1998; Rohatgi et al., 1999; Svitkina et al., 2003). We proceeded our investigations on PRG5-induced filopodia growth and tested whether PRG5 is also capable in filopodia growth promotion when Cdc42 signaling is disrupted. Indeed, when blocking Cdc42 by expressing a dominant negative form of Cdc42, PRG5-expressing cells still displayed dramatic filopodia growth comparable to the extent that PRG3 does (Sigal et al., 2007). This may point to an alternative pathway or downstream pathway of Cdc42 for filopodia formation (Figure 12A). First indications for alternative pathways came from earlier work identifying a class of actin regulatory molecules termed formins required for filopodia induction (Watanabe et al., 1997; Tominaga et al., 2000; Mellor, 2009). Interestingly, a subfamily of formins referred to Diaphanous-related formins (Dia/Drfs) acts independently of the Cdc42 effectors N-WASP and ARP2/3 and recruits another filopodia regulator (IRSp53) for promoting filopodia growth (Fujiwara et al., 2000; Arakawa et al., 2003; Peng et al., 2003). In particular, mDia1 (murine Dia1/Drf1/DFNA1) can bind to the primary Cdc42 effector and filopodia regulator IRSp53 and facilitate filopodia formation and axon elongation (Fujiwara et al., 2000; Arakawa et al., 2003; Peng et al., 2003). Hence, mDia1 has also been found to stimulate filopodia growth independent of mDia2 (Carramusa et al., 2007; Sarmiento et al., 2008), which motivated us to test its role in PRG5-dependent cytoskeletal remodeling. Interestingly, when mDia1 pathway and subsequently mDia2 was blocked by dominant negative mDia1 (Peng et al., 2003), PRG5 was still capable in inducing filopodia growth. These results indicate that PRG5 acts independently of mDia1 and Cdc42 in forming filopodia (summarized in Figure 12). Whether PRG5 activates an alternative pathway is open, however, its closest member PRG3 has been reported to act independent of the formin mDia2 (Sigal et al., 2007).
Figure 12.
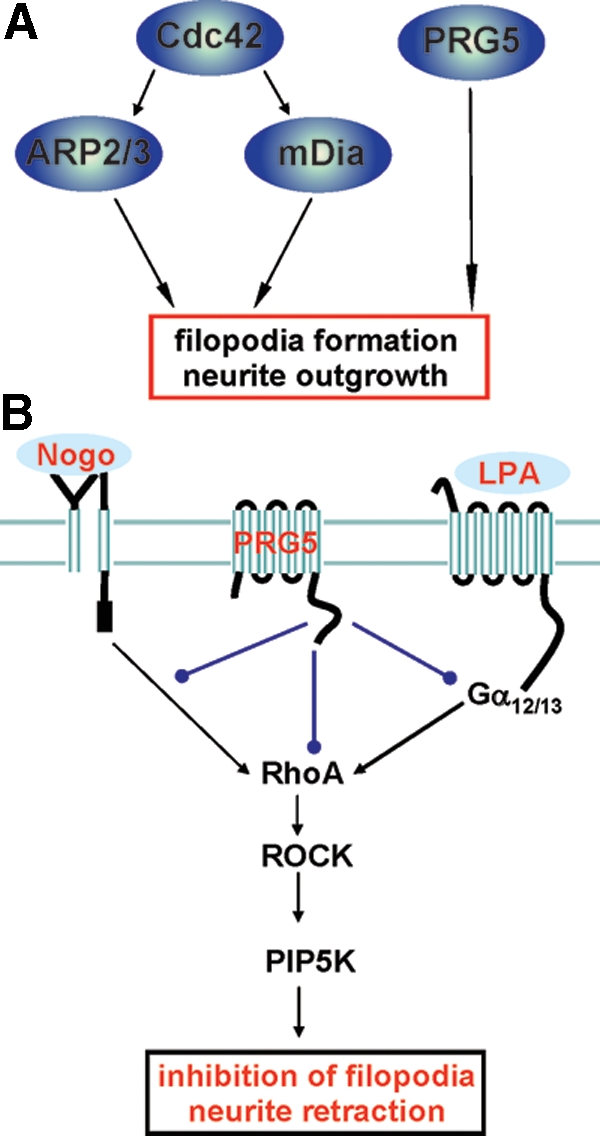
PRG5 signaling in axon growth and neurite retraction. PRG5 induces filopodia and axon growth independent of Cdc42 and impedes RhoA-dependent neurite retraction. (A) Proposed model of PRG5 action on filopodia and axon growth. (B) Summary and proposed model of PRG5 signaling in the context of neurite growth inhibitors. The common neurite growth inhibitors Nogo-A and LPA (underscored light blue) act on their receptors, and their signaling pathway converge at the RhoA-Rho kinase pathway. PRG5 is localized in the plasma membrane and attenuates LPA and Nogo-A—induced neurite retraction and alleviates basal RhoA activation, whereas direct RhoA, ROCK or PIP5Kβ activation overcomes PRG5 induced neurites. Thus, PRG5 may act upstream of RhoA interfering with Nogo-A and LPA receptor signaling or alternatively PRG5 directly balances or impacts RhoA activation.
Furthermore what our experiments reveal is that localization of PRG5 is essential for the execution of filopodia formation. Remarkably, when comparing different truncated PRG5 constructs with wild-type PRG5, the activity for filopodia formation as well as neurite growth is almost preserved when expressing the carboxy terminal tail of PRG5. Moreover, the 50-AA-long PRG5 C-terminus give slightly longer neurites compared with the wild-type PRG5. Notably, plasma membrane targeting is essential for this neurite growth activity, which questions the function of the amino terminal six integral membrane domains. So far, we do not have any indications for ectoenzymatic activity as is the case for PRG3 (Savaskan et al., 2004), and thus the integral membrane domains may primarily serve for safe plasma membrane integration. Another explanation is that due to the topology PRG5 may serve as a sensor or receptor, and therefore integral membrane domains are required for the exposure of putative extracellular binding domains to the outer surface. We asked whether there are similar features or consensus sequences in PRG5 and PRG3 when looking at the carboxyl terminal domain. So far, PRG5 shares only two obvious motifs also present in PRG3, namely CVVXNFKG and PXXESPLE, also known as PEST domain sequence, whereas the rest of the 50 AA are disparate and do not give clear similarities to known protein domains. Ongoing work now centers on putative interaction partners with the C-terminus of PRG5.
PRG5 Acts Upstream of RhoA and Overcomes Neurite Growth Inhibition
Importantly, formation of filopodia and growth cone shaping are critically involved in axon growth control and are implicated in neuritogenesis (Gallo and Letourneau, 1998; Tanabe et al., 2000; Abe et al., 2003; Jain et al., 2004; Korobova and Svitkina, 2008; Matusek et al., 2008). This is in particular the case for PRG5, which induces neurite growth already in culture conditions not optimized for differentiation and axon growth. Conversely, knockdown of PRG5 by RNAi impedes the growth of neurites, pointing to an essential role of PRG5 in neurite elongation. Indeed, PRG5 is mainly localized at the plasma membrane and there is enriched at the tips of filopodia and neurites. We asked whether the promoted neurite growth activity has functional implications for, i.e., regenerative axon growth. During development formation of neuronal circuits is highly controlled by inhibitory signals. PRG5 expression is temporally restricted, with highest levels in early postnatal stages, emerging in parallel to the repellent extracellular space at these stages (Savaskan and Nitsch, 2001; Harel and Strittmatter, 2006). We tested two prominent neurite growth inhibitors in this setting, LPA and Nogo-A. Surprisingly, PRG5-expressing neurites abolished axon collapsing activities of both ligands. To explicitly test whether PRG5 may induces “pathological” neurites with impeded motility rendering axons insensitive toward extracellular signals, we analyzed this feature in time-lapse microscopy observing no differences to wild-type neurons. Because the C-terminus of PRG5 was equally efficient in blocking neurite retraction, we exclude any direct antagonization at the ligand-receptor side. Common to both ligands, LPA and Nogo-A signaling converge on activation of the RhoA-ROCK-PIP5K kinase pathway, leading eventually to actin depolymerization and ultimately axon collapse (Figure 12B). It is tempting to speculate that PRG5 interferes at this level or alternatively, that PRG5 signaling can override such cascade under the conditions we used. However, when testing directly the consequences of RhoA activation and its downstream effectors ROCK and PIP5K1β kinase in the context of PRG5, neurite elongation as well as formation of filopodia were dramatically inhibited at the level of RhoA downwards. Thus, these data are consistent with the notion that under neurite retracting conditions PRG5 acts upstream of the RhoA-ROCK-PIP5K pathway interfering or counterbalancing the fine tuned signaling complex leading to RhoA coupling (Figure 12B). However, whether this scenario also happens in vivo in the developing or injured brain needs to be investigated in the future. So far, we propose that PRG5 promotes filopodia initiation and neurite growth during the critical periods of brain development and may assist in events of axon fine tuning finalizing neuronal circuits by interfering with signals coming from a repelling microenvironment.
In summary, PRG5 is an integral membrane protein that localizes to the plasma membrane and is highly enriched at end tips of filopodia and neurites. PRG5 expression promotes the extension of neurites and its growth promoting activity is located at the carboxy terminus, which needs to be localized near the plasma membrane. Our results reveal that PRG5 prevents RhoA-dependent axon collapse in the presence of common neurite growth inhibitors. In conclusion our data suggest that PRG5 contributes to a Cdc42-independent pathway for filopodia formation and axon growth and contributes to interference mechanisms overcoming LPA- and Nogo-A—induced RhoA activation and subsequently neurite retraction.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to members of the kinase-protease expertise platform at Novartis (Basel, Switzerland) for the support with material and helpful discussions. We thank Trudi Hengeveld (NKI, Amsterdam, Netherlands) for support with the time-lapse set up. Jan Csupor is acknowledged for technical support. We thank the labs of Wouter Moolenaar, Kees Jalink, and Nullin Divecha (former H3 floor, NKI, Amsterdam, The Netherlands) for providing Rho, PIP5K, and ROCK constructs; Rob van der Kammen and John Collard (NKI, Amsterdam, The Netherlands) for providing the pIRES cassette and Cdc42N17 constructs; Arthur Alberts lab for various mDia1 constructs; and members of the Schwab lab (ETH and University Zurich, Switzerland) for support with the initial recombinant protein purifications. Lisa Schnell (ART & Science, Zurich, Switzerland) is acknowledged for sharing details on monoclonal Nogo antibodies. We further thank Alan Hall (MSKCC, New York, NY) and Annette Self (formerly MRC London, United Kingdom) for providing the Cdc42V12 constructs. This study was supported by the International Human Frontiers Science Program Organisation (HFSP, N.E.S.), GENNSA Tech Germany (N.E.S.), SFB 665 (DFG grant to R.N.), and Novartis intramural research grant (T.B.).
Footnotes
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E09-06-0506) on December 23, 2009.
REFERENCES
- Abe T., Kato M., Miki H., Takenawa T., Endo T. Small GTPase Tc10 and its homologue RhoT induce N-WASP-mediated long process formation and neurite outgrowth. J. Cell Sci. 2003;116:155–168. doi: 10.1242/jcs.00208. [DOI] [PubMed] [Google Scholar]
- Arakawa Y., Bito H., Furuyashiki T., Tsuji T., Takemoto-Kimura S., Kimura K., Nozaki K., Hashimoto N., Narumiya S. Control of axon elongation via an SDF-1alpha/Rho/mDia pathway in cultured cerebellar granule neurons. J. Cell Biol. 2003;161:381–391. doi: 10.1083/jcb.200210149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwal J. K., Pinkston-Gosse J., Syken J., Stawicki S., Wu Y., Shatz C., Tessier-Lavigne M. PirB is a functional receptor for myelin inhibitors of axonal regeneration. Science. 2008;322:967–970. doi: 10.1126/science.1161151. [DOI] [PubMed] [Google Scholar]
- Benfey M., Aguayo A. J. Extensive elongation of axons from rat brain into peripheral nerve graft. Nature. 1982;296:150–152. doi: 10.1038/296150a0. [DOI] [PubMed] [Google Scholar]
- Bräuer A. U., Savaskan N. E., Kühn H., Prehn S., Ninnemann O., Nitsch R. A new phospholipid phosphatase, PRG-1, is involved in axon growth and regenerative sprouting. Nat. Neurosci. 2003;6:572–578. doi: 10.1038/nn1052. [DOI] [PubMed] [Google Scholar]
- Carramusa L., Ballestrem C., Zilberman Y., Bershadsky A. D. Mammalian diaphanous-related formin Dia1 controls the organization of E-cadherin-mediated cell-cell junctions. J. Cell Sci. 2007;120:3870–3882. doi: 10.1242/jcs.014365. [DOI] [PubMed] [Google Scholar]
- Chen M. S., Huber A. B., van der Haar M. E., Frank M., Schnell L., Spillmann A. A., Christ F., Schwab M. E. Nogo-A is a myelin-associated neurite outgrowth inhibitor and an antigen for monoclonal antibody IN-1. Nature. 2000;403:434–439. doi: 10.1038/35000219. [DOI] [PubMed] [Google Scholar]
- David S., Aguayo A. J. Axonal elongation into peripheral nervous system “bridges” after central nervous system injury in adult rats. Science. 1981;214:931–933. doi: 10.1126/science.6171034. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville S., Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- Evangelista M., Zigmond S., Boone C. Formins: signaling effectors for assembly and polarization of actin filaments. J. Cell Sci. 2003;116:2603–2611. doi: 10.1242/jcs.00611. [DOI] [PubMed] [Google Scholar]
- Faix J., Breitsprecher D., Stradal T. E., Rottner K. Filopodia: complex models for simple rods. Int. J. Biochem. Cell Biol. 2009;41:1656–1664. doi: 10.1016/j.biocel.2009.02.012. [DOI] [PubMed] [Google Scholar]
- Fournier A. E., GrandPre T., Strittmatter S. M. Identification of a receptor mediating Nogo-66 inhibition of axonal regeneration. Nature. 2001;409:341–346. doi: 10.1038/35053072. [DOI] [PubMed] [Google Scholar]
- Fujiwara T., Mammoto A., Kim Y., Takai Y. Rho small G-protein-dependent binding of mDia to an Src homology 3 domain-containing IRSp53/BAIAP2. Biochem. Biophys. Res. Commun. 2000;271:626–629. doi: 10.1006/bbrc.2000.2671. [DOI] [PubMed] [Google Scholar]
- Gallo G., Letourneau P. C. Localized sources of neurotrophins initiate axon collateral sprouting. J. Neurosci. 1998;18(14):5403–5414. doi: 10.1523/JNEUROSCI.18-14-05403.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GrandPré T., Nakamura F., Vartanian T., Strittmatter S. M. Identification of the Nogo inhibitor of axon regeneration as a Reticulon protein. Nature. 2000;403:439–444. doi: 10.1038/35000226. [DOI] [PubMed] [Google Scholar]
- GrandPré T., Li S., Strittmatter S. M. Nogo-66 receptor antagonist peptide promotes axonal regeneration. Nature. 2002;417:547–551. doi: 10.1038/417547a. [DOI] [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- Harel N. Y., Strittmatter S. M. Can regenerating axons recapitulate developmental guidance during recovery from spinal cord injury? Nat. Rev. Neurosci. 2006;7(8):603–616. doi: 10.1038/nrn1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Z., Koprivica V. The Nogo signaling pathway for regeneration block. Annu. Rev. Neurosci. 2004;27:341–368. doi: 10.1146/annurev.neuro.27.070203.144340. [DOI] [PubMed] [Google Scholar]
- Inoue M., Rashid M. H., Fujita R., Contos J. J., Chun J., Ueda H. Initiation of neuropathic pain requires lysophosphatidic acid receptor signaling. Nat. Med. 2004;10(7):712–718. doi: 10.1038/nm1060. [DOI] [PubMed] [Google Scholar]
- Jain A., Brady-Kalnay S. M., Bellamkonda R. V. Modulation of Rho GTPase activity alleviates chondroitin sulfate proteoglycan-dependent inhibition of neurite extension. J. Neurosci. Res. 2004;77(2):299–307. doi: 10.1002/jnr.20161. [DOI] [PubMed] [Google Scholar]
- Jalink K., van Corven E. J., Hengeveld T., Morii N., Narumiya S., Moolenaar W. H. Inhibition of lysophosphatidate- and thrombin-induced neurite retraction and neuronal cell rounding by ADP ribosylation of the small GTP-binding protein Rho. J. Cell Biol. 1994;126(3):801–810. doi: 10.1083/jcb.126.3.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kai M., Wada I., Imai S., Sakane F., Kanoh H. Identification and cDNA cloning of 35-kDa phosphatidic acid phosphatase (type 2) bound to plasma membranes. Polymerase chain reaction amplification of mouse H2O2-inducible hic53 clone yielded the cDNA encoding phosphatidic acid phosphatase. J. Biol. Chem. 1996;271:18931–18938. doi: 10.1074/jbc.271.31.18931. [DOI] [PubMed] [Google Scholar]
- Kai M., Wada I., Imai S., Sakane F., Kanoh H. Cloning and characterization of two human isozymes of Mg2+-independent phosphatidic acid phosphatase. J. Biol. Chem. 1997;272:24572–24578. doi: 10.1074/jbc.272.39.24572. [DOI] [PubMed] [Google Scholar]
- Kanoh H., Kai M., Wada I. Phosphatidic acid phosphatase from mammalian tissues: discovery of channel-like proteins with unexpected functions. Biochim. Biophys. Acta. 1997;1348:56–62. doi: 10.1016/s0005-2760(97)00094-5. [DOI] [PubMed] [Google Scholar]
- Korobova F., Svitkina T. Arp2/3 complex is important for filopodia formation, growth cone motility, and neuritogenesis in neuronal cells. Mol. Biol. Cell. 2008;19(4):1561–1574. doi: 10.1091/mbc.E07-09-0964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozma R., Sarner S., Ahmed S., Lim L. Rho family GTPases and neuronal growth cone remodelling: relationship between increased complexity induced by Cdc42Hs, Rac1, and acetylcholine and collapse induced by RhoA and lysophosphatidic acid. Mol. Cell. Biol. 1997;17:1201–1211. doi: 10.1128/mcb.17.3.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranenburg O., Poland M., van Horck F. P., Drechsel D., Hall A., Moolenaar W. H. Activation of RhoA by lysophosphatidic acid and Galpha12/13 subunits in neuronal cells: induction of neurite retraction. Mol. Biol. Cell. 1999;10(6):1851–1857. doi: 10.1091/mbc.10.6.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein E. S., et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Lommel S., Benesch S., Rottner K., Franz T., Wehland J., Kühn R. Actin pedestal formation by enteropathogenic Escherichia coli and intracellular motility of Shigella flexneri are abolished in N-WASP-defective cells. EMBO Rep. 2001;2:850–857. doi: 10.1093/embo-reports/kve197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machesky L. M., Insall R. H. Scar1 and the related Wiskott-Aldrich syndrome protein, WASP, regulate the actin cytoskeleton through the Arp2/3 complex. Curr. Biol. 1998;8(25):1347–1356. doi: 10.1016/s0960-9822(98)00015-3. [DOI] [PubMed] [Google Scholar]
- Mattila P. K., Lappalainen P. Filopodia: molecular architecture and cellular functions. Nat. Rev. Mol. Cell Biol. 2008;9(6):446–454. doi: 10.1038/nrm2406. [DOI] [PubMed] [Google Scholar]
- Matusek T., Gombos R., Szécsényi A., Sánchez-Soriano N., Czibula A., Pataki C., Gedai A., Prokop A., Raskó I., Mihály J. Formin proteins of the DAAM subfamily play a role during axon growth. J. Neurosci. 2008;28(49):13310–13319. doi: 10.1523/JNEUROSCI.2727-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellor H. The role of formins in filopodia formation. Biochim. Biophys. Acta. 2009 doi: 10.1016/j.bbamcr.2008.12.018. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Miao R. Q., Gao Y., Harrison K. D., Prendergast J., Acevedo L. M., Yu J., Hu F., Strittmatter S. M., Sessa W. C. Identification of a receptor necessary for Nogo-B stimulated chemotaxis and morphogenesis of endothelial cells. Proc. Natl. Acad. Sci. USA. 2006;103(29):10997–11002. doi: 10.1073/pnas.0602427103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moolenaar W. H. LPA: a novel lipid mediator with diverse biological actions. Trends Cell Biol. 1994;4(6):213–219. doi: 10.1016/0962-8924(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Naito Y., Yamada T., Matsumiya T., Ui-Tei K., Saigo K., Morishita S. dsCheck: highly sensitive off-target search software for double-stranded RNA-mediated RNA interference. Nucleic Acids Res. 2005;33:W589–W591. doi: 10.1093/nar/gki419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobes C. D., Hall A. Rho, rac and cdc42 GTPases: regulators of actin structures, cell adhesion and motility. Biochem. Soc. Trans. 1995a;23:456–459. doi: 10.1042/bst0230456. [DOI] [PubMed] [Google Scholar]
- Nobes C. D., Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995b;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- O'Donnell M., Chance R. K, Bashaw G. J. Axon growth and guidance: receptor regulation and signal transduction. Annu. Rev. Neurosci. 2009;32:383–412. doi: 10.1146/annurev.neuro.051508.135614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passey S., Pellegrin S., Mellor H. What is in a filopodium? Starfish versus hedgehogs. Biochem. Soc. Trans. 2004;32(Pt 6):1115–1117. doi: 10.1042/BST0321115. [DOI] [PubMed] [Google Scholar]
- Peng J., Wallar B. J., Flanders A., Swiatek P. J., Alberts A. S. Disruption of the Diaphanous-related formin Drf1 gene encoding mDia1 reveals a role for Drf3 as an effector for Cdc42. Curr. Biol. 2003;13:534–545. doi: 10.1016/s0960-9822(03)00170-2. [DOI] [PubMed] [Google Scholar]
- Prehoda K. E., Scott J. A., Mullins R. D., Lim W. A. Integration of multiple signals through cooperative regulation of the N-WASP-Arp2/3 complex. Science. 2000;290:801–806. doi: 10.1126/science.290.5492.801. [DOI] [PubMed] [Google Scholar]
- Prinjha R., Moore S. E., Vinson M., Blake S., Morrow R., Christie G., Michalovich D., Simmons D. L., Walsh F. S. Inhibitor of neurite outgrowth in humans. Nature. 2000;403:383–384. doi: 10.1038/35000287. [DOI] [PubMed] [Google Scholar]
- Ridley A. J. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol. 2006;16(10):522–529. doi: 10.1016/j.tcb.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Rohatgi R., Ma L., Miki H., Lopez M., Kirchhausen T., Takenawa T., Kirschner M. W. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell. 1999;97(2):221–231. doi: 10.1016/s0092-8674(00)80732-1. [DOI] [PubMed] [Google Scholar]
- Sarmiento C., et al. WASP family members and formin proteins coordinate regulation of cell protrusions in carcinoma cells. J. Cell Biol. 2008;180:1245–1260. doi: 10.1083/jcb.200708123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savaskan N. E., Nitsch R. Molecules involved in reactive sprouting in the hippocampus. Rev. Neurosci. 2001;12(3):195–215. doi: 10.1515/revneuro.2001.12.3.195. [DOI] [PubMed] [Google Scholar]
- Savaskan N. E., Brauer A. U., Nitsch R. Molecular cloning and expression regulation of PRG-3, a new member of the plasticity-related gene family. Eur. J. Neurosci. 2004;19(1):212–220. doi: 10.1046/j.1460-9568.2003.03078.x. [DOI] [PubMed] [Google Scholar]
- Savaskan N. E., et al. Autotaxin (NPP-2) in the brain: cell type-specific expression and regulation during development and after neurotrauma. Cell Mol. Life Sci. 2007;64(2):230–243. doi: 10.1007/s00018-006-6412-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkina T. M., Bulanova E. A., Chaga O. Y., Vignjevic D. M., Kojima S., Vasiliev J. M., Borisy G. G. Mechanism of filopodia initiation by reorganization of a dendritic network. J. Cell Biol. 2003;160(3):409–421. doi: 10.1083/jcb.200210174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell L., Schwab M. E. Axonal regeneration in the rat spinal cord produced by an antibody against myelin-associated neurite growth inhibitors. Nature. 1990;343:269–272. doi: 10.1038/343269a0. [DOI] [PubMed] [Google Scholar]
- Sigal Y. J., McDermott M. I., Morris A. J. Integral membrane lipid phosphatases/phosphotransferases: common structure and diverse functions. Biochem. J. 2005;387:281–293. doi: 10.1042/BJ20041771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigal Y. J., Quintero O. A., Cheney R. E., Morris A. J. Cdc42 and ARP2/3-independent regulation of filopodia by an integral membrane lipid-phosphatase-related protein. J. Cell Sci. 2007;120:340–352. doi: 10.1242/jcs.03335. [DOI] [PubMed] [Google Scholar]
- Silver J., Miller J. H. Regeneration beyond the glial scar. Nat. Rev. Neurosci. 2004:146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- Snapper S. B., et al. N-WASP deficiency reveals distinct pathways for cell surface projections and microbial actin-based motility. Nat. Cell Biol. 2001;3:897–904. doi: 10.1038/ncb1001-897. [DOI] [PubMed] [Google Scholar]
- Stein E., Savaskan N. E., Ninnemann O., Nitsch R., Zhou R., Skutella T. A role for the Eph ligand ephrin-A3 in entorhino-hippocampal axon targeting. J. Neurosci. 1999;19(20):8885–93. doi: 10.1523/JNEUROSCI.19-20-08885.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steup A., Ninnemann O., Savaskan N. E., Nitsch R., Püschel A. W., Skutella T. Semaphorin D acts as a repulsive factor for entorhinal and hippocampal neurons. Euro. J. Neurosci. 1999;11:729–734. doi: 10.1046/j.1460-9568.1999.00517.x. [DOI] [PubMed] [Google Scholar]
- Steup A., Lohrum M., Hamscho N., Savaskan N. E., Ninnemann O., Nitsch R., Fujisawa H., Püschel A. W., Skutella T. Sema3C and netrin-1 differentially affect axon growth in the hippocampal formation. Mol. Cell Neurosci. 2000;15(2):141–155. doi: 10.1006/mcne.1999.0818. [DOI] [PubMed] [Google Scholar]
- Sun L., et al. Cloning and characterization of a novel human phosphatidic acid phosphatase type 2, PAP2d, with two different transcripts PAP2d_v1 and PAP2d_v2. Mol. Cell Biochem. 2005;272(1–2):91–96. doi: 10.1007/s11010-005-7640-6. [DOI] [PubMed] [Google Scholar]
- Tanabe K., Tachibana T., Yamashita T., Che Y. H., Yoneda Y., Ochi T., Tohyama M., Yoshikawa H., Kiyama H. The small GTP-binding protein TC10 promotes nerve elongation in neuronal cells, and its expression is induced during nerve regeneration in rats. J. Neurosci. 2000;20(11):4138–4144. doi: 10.1523/JNEUROSCI.20-11-04138.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessier-Lavigne M., Goodman C. S. The molecular biology of axon guidance. Science. 1996;274:1123–1133. doi: 10.1126/science.274.5290.1123. [DOI] [PubMed] [Google Scholar]
- Tominaga T., Sahai E., Chardin P., McCormick F., Courtneidge S. A., Alberts A. S. Diaphanous-related formins bridge Rho GTPase and Src tyrosine kinase signaling. Mol. Cell. 2000;5:13–25. doi: 10.1016/s1097-2765(00)80399-8. [DOI] [PubMed] [Google Scholar]
- Trimbuch T., et al. Synaptic PRG-1 modulates excitatory transmission via lipid phosphate-mediated signaling. Cell. 2009;138(6):1222–1235. doi: 10.1016/j.cell.2009.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Chun S. J., Treloar H., Vartanian T., Greer C. A., Strittmatter S. M. Localization of Nogo-A and Nogo-66 receptor proteins at sites of axon-myelin and synaptic contact. J. Neurosci. 2002;22(13):5505–5515. doi: 10.1523/JNEUROSCI.22-13-05505.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe N., Madaule P., Reid T., Ishizaki T., Watanabe G., Kakizuka A., Saito Y., Nakao K., Jockusch B. M., Narumiya S. p140mDia, a mammalian homolog of Drosophila diaphanous, is a target protein for Rho small GTPase and is a ligand for profilin. EMBO J. 1997;16:3044–3056. doi: 10.1093/emboj/16.11.3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitford K. L., Dijkhuizen P., Polleux F., Ghosh A. Molecular control of cortical dendrite development. Annu. Rev. Neurosci. 2002;25:127–149. doi: 10.1146/annurev.neuro.25.112701.142932. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.