

Abstract
Doxorubicin is a commonly used drug to treat various types of cancers. Unfortunately, after a period of administration, tumour cells develop resistance generally under the form of an accelerated drug extrusion. Recently, cell penetrating peptides have been used for their ability to deliver non-permeant compounds into cells. One such cell penetrating peptide, maurocalcine, has been isolated from the venom of a Tunisian scorpion. Herein, we report on the in vitro anti-tumoral efficacy of doxorubicin covalently coupled to an analogue of maurocalcine. On a doxorubicin-sensitive cancer cell line, the maurocalcine-conjugated form appears slightly less efficient than doxorubicin itself. On the contrary, on a doxorubicin-resistant cancer cell line, doxorubicin coupling allows to overcome the drug resistance. This strategy can be generalized to other cell penetrating peptides since Tat and penetratin show similar effects. We conclude that coupling anti-tumoral drugs to cell penetrating peptides represent a valuable strategy to overcome drug resistance.
Keywords: Antibiotics, Antineoplastic; chemistry; metabolism; pharmacology; Breast Neoplasms; metabolism; pathology; Cell Survival; drug effects; Chemistry, Pharmaceutical; Dose-Response Relationship, Drug; Doxorubicin; chemistry; metabolism; pharmacology; Drug Carriers; Drug Compounding; Drug Resistance, Neoplasm; Female; Humans; Scorpion Venoms; chemistry; metabolism; toxicity; Time Factors
Keywords: Drug delivery systems, doxorubicin, cell-penetrating peptide, drug resistance, maurocalcine
Introduction
During the last fifteen years, numerous peptides able to translocate across the plasma membrane within seconds to minutes and termed cell-penetrating peptides1 (CPPs) have been characterized (1). The 60 amino-acid-long homeodomain of the Drosophila transcription factor Antennapedia was the first CPP discovered and shown to serve as a signal for the internalization of other polypeptides (2, 3). Its penetration and translocation properties were further restricted to a peptide of 16 residues, corresponding to the 43–58 third helix of this DNA binding domain and thereafter called penetratin2 (Pen) (4, 5). Now, along with Pen, multiple CPPs (peptides derived from the HIV1-Tat protein, synthetic 7–9 homoarginine peptides, chimera peptides such as transportan, model amphipathic peptide, etc…) are intensively studied to facilitate penetration of various molecules or particles of different sizes inside cells and are considered as important tools in drug delivery (6–8).
Maurocalcine3 (MCa) is a 33-mer toxin derived from the venom of the Tunisian scorpion Scorpio maurus palmatus which activates the ryanodine receptor type 14 (RyR1), an intracellular calcium channel involved in excitation-contraction coupling in skeletal muscle cells (9). Because of its ability to modulate calcium responses of intact skeletal myotubes, it was hypothesized to also behave as a CPP (10). The demonstration of Mca’s vector properties was made by using biotinylated MCa coupled to fluorescent streptavidin. This complex was shown to enter various cell types within minutes and in all cell types tested, a common feature of CPPs (11). Numerous mutants of MCa were then designed in order to unravel the most active residues for its pharmacological and penetration activities (12). MCa folds following an Inhibitor Cystine Knot arrangement with three disulfide bridges (13). Taking into account that studies of the mode of entry of the CPPs have shown that the CPPs 3D-structure is dispensable for their translocation performances (8, 14), a disulfide-less mutant called MCaAbu5 was synthesized by replacing the six cysteine residues of MCa with the L-α-aminobutyric acid residue, Abu. Interestingly, this MCa mutant was devoid of effect on [3H]-ryanodine binding onto RyR1 from sarcoplasmic reticulum vesicles, whereas its penetration activity was mainly retained (15).
Doxorubicin6 (Dox) is one of the most used anticancer drugs, in particular in the treatment of breast cancer patients (16, 17). Unfortunately, resistance to this agent is common and thereby the development of new drugs or alternative drug delivery systems to overcome the unsuccessful outcome of patients treatment is desirable (18, 19). Mechanisms of cell resistance to Dox, as well as those behind enhanced Dox uptake and retention are intensively studied on various cancer cell lines (18, 20, 21). Current models used are cancer cell lines selected in vitro, such as low- and high-invasive breast carcinoma MCF7 and MDA-MB 231 cells, respectively representing Dox-sensitive and Dox-resistant cells (21–24). Using cancer model cell lines, various methods have been developed to improve doxorubicin efficacy and/or delivery. These methods comprise so far: (i) entrapping the drug in submicron carriers like liposomes, (ii) using polymeric micelles, (iii) coupling to nanoparticules or lactosaminated human albumin (25–29).
In this work, to gain insight into the potential of MCa as an efficient CPP for drug delivery and for overriding drug resistance, we have examined the intracellular delivery and subcellular distribution of MCaAbu peptide covalently coupled to Dox into MCF7 and MDA-MB 231 cell lines and studied the cytotoxicity of this complex comparatively to Dox. We compared these properties with similar complexes of Dox linked to two archetypical poly-cationic CPPs, Pen and Tat, a HIV1-Tat derived peptide of ten amino acids. The results obtained indicate that MCaAbu is a good peptide vector for the cell entry of Dox and that the coupling strategy does not prevent Dox toxicity. In addition, coupling of Dox to CPPs permits to overcome the observed Dox resistance of MDA-MB 231 cells.
Materials and Methods
Synthesis and purification of doxorubicin-peptide conjugates
The MCaAbu peptide was synthesized with an additional cysteine residue at its amino terminus by NeoMPS S.A. Similarly modified Tat peptide and Pen were synthesized by UFPeptides s.r.l. Dox (Alexis Biochemicals) was covalently bound to the cysteinylated peptides using the bifunctional cross-linker Succinimidyl 4-[N-maleimidomethyl]cyclohexane-1-carboxylate7 (SMCC, Pierce) according to the method described by Liang et al. (30) with a 3-fold increase in Dox concentration over the original description to allow better yield of coupling. Briefly, Dox.HCl was dissolved in DMSO and then diluted to 1 mg/ml in a 3 ml phosphate buffer solution, pH:8.0. Triethylamine (40 μl) and SMCC (270 μl, 10 mg/ml) were added and left for 2 hrs at room temperature. The pH was adjusted to 5.5, before adding the peptide solution (15 mg/ml, 300 μl) containing MCaAbu, Pen or Tat and the mixture was incubated 2 hrs at room temperature for coupling. Because of its toxicity, all precautions for handling Dox were taken according the Material Safety Data Sheet delivered by the manufacturer. Unreacted reagents were removed by chromatography on a 1 ml HiTrap Heparin HP (GE Healthcare) column operated with an ÄKTApurifier System, at a one ml/min flow rate. According to the manufacturer recommendations, the binding buffer was 10 mM Na2HPO4, pH=7.0. After 10 volumes of washing, the column was eluted with a linear 0–2 M NaCl gradient in binding buffer. Simultaneous monitoring of absorbance at 215 (peptide bond) and 480 nm (intrinsic fluorescence of Dox) wavelengths allowed the detection of fractions containing Dox-CPPs complexes. Fractions were also analyzed by Sodium Dodecyl Sulphate - Polyacrylamide Gel Electrophoresis (SDS-PAGE) of peptides onto 16.5% Ready Gel® Tris-Tricine Gels from Biorad. For all experiments, the Dox-CPPs conjugates were used as such in the cell culture media.
Cells and cell culture
Culture media and supplements were purchased from InVitrogen. All cells were maintained at 37°C, 5% CO2 in a Hera cell 150 humidified incubator (Thermo). MDA-MB 231 cells from ATCC were grown in Leibovitz L15 medium supplemented with 10% (v/v) heat-inactivated fetal bovine serum and 10,000 units/ml streptomycin and penicillin. MCF7 (ATCC) were cultured in DMEM medium supplemented as above with additional bovine insulin (10 μg/ml).
MTT cell viability assay
MCF7, MDA-MB 231 cells were seeded into 96-well plates and treated with various concentrations of free or conjugated Dox for 24 hrs to 72 hrs. The number of living cells in culture was measured with a 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyl-tetrazolium bromide8 (MTT) reduction assay (CellQuanti-MTT™, Gentaur) according to the manufacturer specifications, slightly modified as described in Mabrouck et al. (12), except that the MTT reagent was incubated for 3 hrs at 37°C. Results were plotted as percent of cytotoxicity and dose-response curves were fitted using SigmaPlot10 in order to determine the 50% effective concentration (EC50).
Flow Cytometry
Innate fluorescence of Dox allowed us to use flow cytometry and live cell confocal microscopy to study penetration and localization of free or conjugated-Dox. MDA-MB 231 and MCF7 were cultured overnight in 24 well plates with or without free or CPP-conjugated drug and washed twice with Phosphate Buffered Saline9 (PBS) solution to remove extracellular drugs. Next, cells were treated with 1 mg/ml trypsin (InVitrogen) for 10 min at 37°C to remove remaining cell surface-bound drugs and detach cells from the dish surface. The cell suspension was centrifuged at 500 g and resuspended in PBS. Flow cytometry analyses were performed on live cells by Fluorescence-Activated Cell Sorting10 (FACS) using a FACS-Calibur flow cytometer, BD Biosciences). Live cells were gated by forward/side scattering from a total of 10,000 events. Data obtained were analyzed using the CellQuest software (BD Biosciences).
Confocal microscopy
Cells were grown in 3.5 cm diameter cell culture dishes overnight and incubated with 1 or 5 μM Dox or Dox-CPPs for 2 or 24 hrs as specified in the Results section. Immediately after two PBS washes, 1 μM Syto 40 (Molecular Probes) was added for 20 min for nucleus staining. Cells were washed again with PBS and plasma membrane labeling was performed using five min incubation with 5 μg/ml FITC11-conjugated concanavalin A12 (Con A, Sigma). After a last wash with PBS, cells were immediately analyzed by confocal laser scanning microscopy using a Leica TCS-SP2 operating system with a 20x water immersion objective. FITC (λex = 488 nm, λem = 520 nm), Syto 40 (λex = 405 nm, λem = 450 nm) and Dox (λex = 470 nm, λem = 590 nm) were sequentially excited and each emitted fluorescence was collected in z-confocal planes of 10–15 nm steps. Pseudocolors used were: red for Dox, green for Con A and blue for Syto 40.
Results
Characterization of doxorubicin-CPP complexes
MCa has recently been characterized as a CPP (11). Mutation analyses indicated that an analogue of MCa devoid of disulfide bridges, MCaAbu, which lacks pharmacological activity, represents a promising CPP (15). As a proof of concept that MCaAbu represents a useful vector for the delivery of the anti-tumoral drug Dox and has the ability to overcome drug resistance, MCaAbu was chemically conjugated to Dox. Other CPPs, extensively used in numerous other applications, Tat and Pen, were also used for conjugation to Dox to compare the cell delivery properties and antitumoral activities of the resulting conjugates to those of Dox-MCaAbu. All CPP sequences used in the manuscript are shown in Figure 1A. CPPs were synthesized with an extra amino terminal cysteine residue to allow a strategy of covalent coupling to the reactive amino group of Dox. To this end, the membrane-permeable heterobifunctional SMCC crosslinker was used to conjugate Dox to cysteinylated CPPs according to a procedure previously described by Liang et al. (30). A scheme of the expected Dox-CPPs conjugates is presented in Figure 1B. The coupling conditions were set with a higher Dox concentration over CPP in order to favor completion of the conjugation reaction. All the conjugates were purified onto a heparin column as described in the Materials and Methods section (30) (Figure 2). Unreacted reagents (free Dox and crosslinker) were eliminated at the washing step. Non-conjugated peptides and conjugates were separated by the NaCl linear concentration gradient according to their strength of ionic interaction with heparin (Figure 2A). The peptide and Dox contents of the eluates were monitored by measuring light absorbance at 215 nm wavelength for peptide bonds and at 480 nm for Dox. The conjugation reaction with Pen appeared to be complete since the eluate from the column emerges as a single peak. For Tat, roughly 50% of peptide was found to be bound to Dox, whereas coupling was higher in the case of MCaAbu (75% according to peaks’ surface). SDS-PAGE analysis of the peak fractions under white or UV light further assessed the efficiency of Dox conjugation to the peptides (Figure 2B). Absence of differences in the apparent molecular sizes of the conjugates and free peptides indicates the lack of crosslinking between peptides. UV analysis of the fractions revealed the efficient conjugation of Dox to the various peptides (C fractions). The concentrations of Dox-CPP conjugates in the solutions were estimated by measuring absorption at 480 nm and plotting against a calibration curve with known concentrations of free Dox (Supplementary data).
Figure 1.
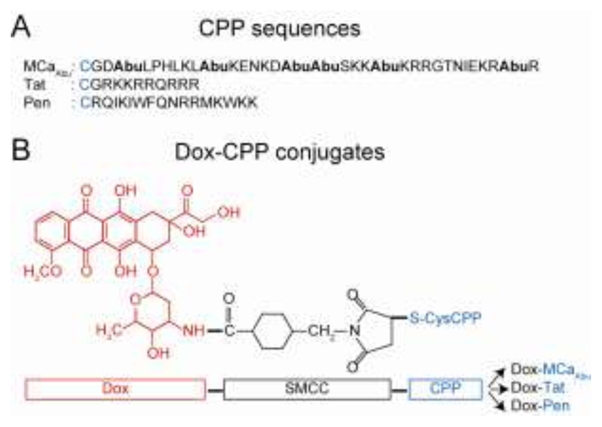
Scheme of the Dox-CPP cell delivery complexes. (A) Amino acid sequences of the CPPs used for conjugation (single letter code). The extra cysteine used for chemical conjugation of Dox is indicated in blue. Mutated cysteine residues of MCa, replaced by Abu residues, are in bold. (B) Chemical structure of Dox (red) and SMCC crosslinker (black) bound to Cys-CPP (blue). Synthesis of peptides and coupling method with Dox are described in Materials and Methods. Resulting conjugates are indicated in the far right.
Figure 2.
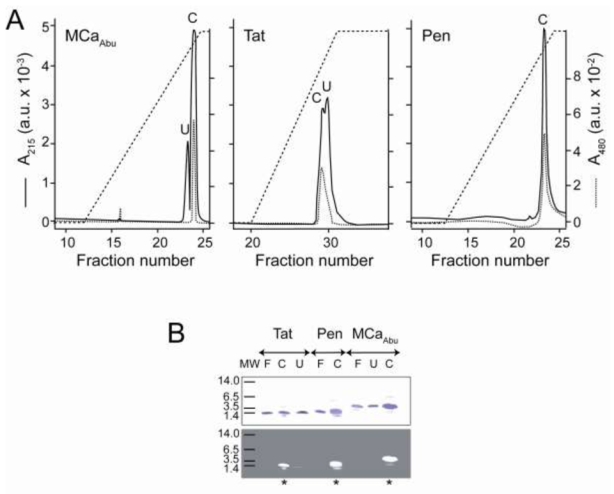
Purification and biochemical characterization of the Dox-CPPs delivery complexes. (A) Heparin chromatography profiles of the three Dox-CPPs. Purification protocol is described in Materials and Methods. Absorbance at 215 nm (A215, ——) of the peptide bonds was monitored during the elution by the 0–2 M NaCl salt gradient (– – –) in parallel to the Dox absorbance at 480 nm (A480, ······). Peak fractions are denoted C and U. (B) SDS-PAGE analysis of peak fractions. Two μl of C or U fractions were analyzed by electrophoresis on 16.5% Tris-tricine gels which were fixed, stained by G-250 Coomassie blue and visualized under white (upper panel) or ultraviolet light (lower panel). Control lanes, labelled F, were loaded with solutions of free peptides. * indicates the C fraction that contains the Dox-CPP conjugate and U lanes show pure uncoupled peptides, with however a slight contamination by Dox coupled peptide in case of Tat.
Cell toxicity of the CPP vectors used
Before examining the cell toxicity of the Dox-CPP conjugates, the cell toxicity of free peptides was investigated on both MCF7 and MDA-MB 231 cell lines (Figure 3). As shown, incubation for 24 hrs of these cell lines with free MCaAbu, Tat, or Pen up to a concentration of 5 μM produces no toxicity; the toxicity values observed were not significant from baseline and lower than 9%. However, above 5 μM, all peptides produced a limited cell toxicity that ranges between 11 and 24%. Interestingly, there was no significant difference observed among the various CPPs indicating that they should all represent equipotent vectors for the delivery of Dox into these cell types.
Figure 3.
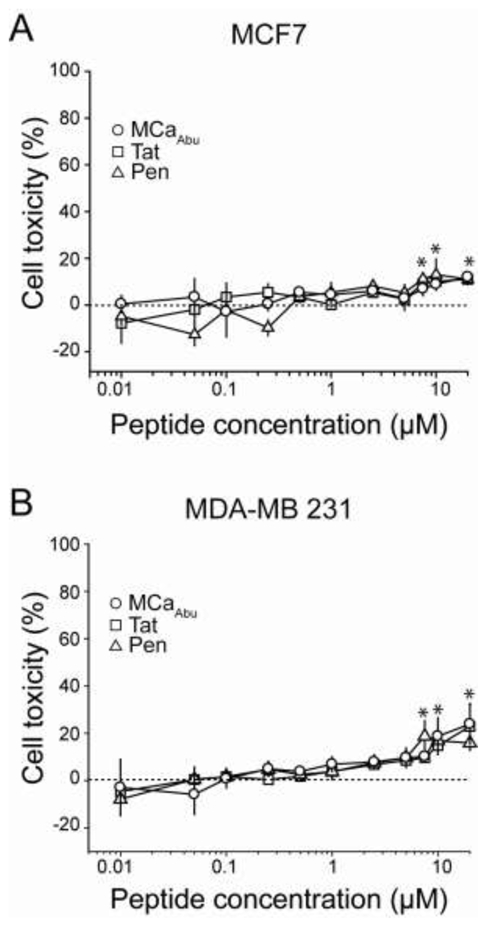
Cell toxicity of MCaAbu, Tat and Pen peptides on MCF7 and MDA-MB 231 cells. Peptides were applied at various concentrations on MCF7 (A) and MDA-MB 231 (B) cells for 24 hrs before performing the MTT assay, as described in Materials and Methods. Asterisks denote significant deviation from baseline (mean value ± 3 SD values).
Difference in Dox toxicity between MCF7 and MDA-MB 231 cell lines
In control experiments, the suitability of the two cell lines for the study of Dox resistance was validated by studying the cell toxicity of free Dox (Figure 4). MCF7 and MDA-MB 231 cells were incubated for 24 hrs with increasing concentrations of free Dox whose toxicity was evaluated with an MTT cell viability assay. As expected, MCF7 and MDA-MB 231 cells show strong differences in the drug sensitivity (Figure 4A). For MCF7 cells, cytotoxicity appeared at 20 nM Dox and increased to 83.5 ± 8.5% at 5 μM. In contrast, MDA-MB 231 cell death occurred at concentrations above 200 nM and efficient killing of 79.1 ± 1.5% was observed at 20 μM Dox. The EC50 for Dox toxicity is estimated at 0.1 ± 0.02 μM for MCF7, whereas it reaches 2.7 ± 1.4 μM for MDA-MB 231. MDA-MB 231 cells thus show a 27-fold relative resistance to Dox. To examine whether this difference in sensitivity to Dox is linked to a defect in Dox accumulation or distribution, the entry of Dox in both cell lines was investigated both by confocal microscopy and by FACS. Using an intermediate concentration of 1 μM of Dox, incubated for 24 hrs as in the toxicity experiments, Dox was found to concentrate in the nuclei of MCF7 cells after analysis by confocal microscopy. In contrast, Dox is barely visible at this concentration in MDA-MB 231 cells (Figure 4B). However, when using a higher concentration of Dox, 5 μM, at which both cell lines undergo cell death, Dox was not only visible in MCF7 cells, but also in the nuclei of MDA-MB 231 cells (Figure 4C). In both cell lines, and at toxic concentration, Dox appears mainly in the nuclei of cells, but some fluorescence is also detected in cytoplasmic patches in some cells. One should keep in mind that this distribution is observed on mainly dying cells and requires confirmation at times of Dox incubation where no toxicity is observed (see Figure 5). In any case, these data indicate that higher concentrations of Dox are required to detect its presence in MDA-MB 231 cells, a property that may be at the basis of part of Dox resistance of this cell line. To confirm that Dox accumulation in MDA-MB 231 cells is limited compared to the one in MCF7 cells, a quantitative study was initiated by FACS. As shown in Figure 4D, the mean fluorescence of Dox measured at 1 μM after 24 hrs of incubation is negligible in MDA-MB 231 cells, whereas it represents 35.8% of the maximal achievable fluorescence value in MCF7 cells. In addition, the maximal achievable Dox fluorescence value is 2.2-fold higher in MCF7 cells than in MDA-MB 231 cells (as measured at 10 μM). These data indicate that MCF7 cells more readily accumulate intracellular Dox than MDA-MB 231 cells, and that this accumulation occurs at slightly lower concentrations (EC50 value of 1.5 ± 0.2 μM for MCF7 versus 2.9 ± 0.3 μM for MDA-MB 231 cells). The EC50 values of Dox accumulation into cells, measured by FACS, should not be compared to the EC50 values determined for toxicity. There is possibly a threshold of toxicity induction by Dox that is lower than the threshold of fluorescence detection by FACS. However, the combined information indicates that these two cell lines are adequate to study the effects of Dox-CPP on drug resistance of both cell lines.
Figure 4.
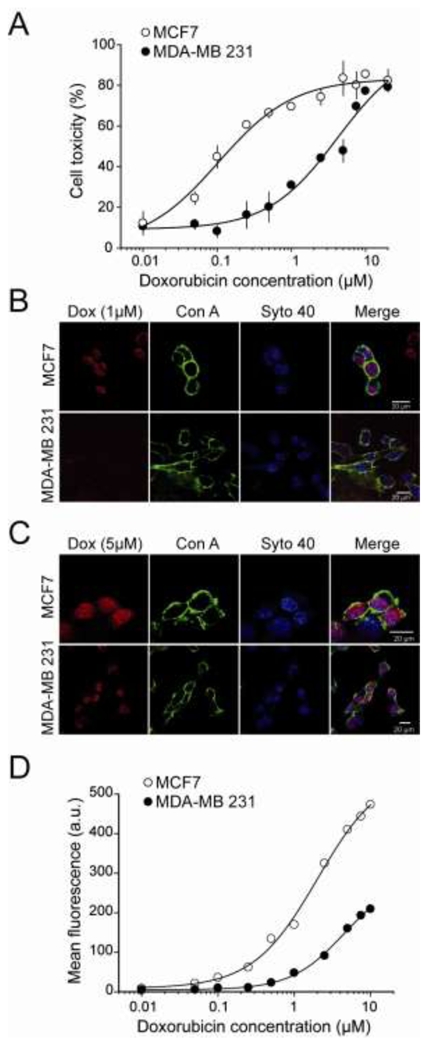
Study of free Dox cytotoxicity and accumulation in MCF7 and MDA-MB 231 cell lines. (A) Dose-response curve evaluating Dox cytotoxicity by the MTT assay on MCF7 and MDA-MB 231 cells. Dox incubation time with cells was 24 hrs. Data were fitted with sigmoid functions and yield EC50 values of 0.10 ± 0.02 μM (MCF7) or 2.7 ± 1.4 μM (MDA-MB 231) and maximal toxicity of 83.4 ± 5.0% (MCF7) and 90.7 ± 17.1% (MDA-MB 231). (B) Representative confocal images of MCF7 (upper panels) and MDA-MB 231 cells (lower panels) showing Dox (red), Con A (green) and Syto 40 (blue) labelling, as well as merge pictures. Incubation time of 24 hrs with 1 μM Dox. (C) Same as in (B) except for a Dox concentration of 5 μM. (D) Dose-response curve evaluating Dox accumulation in cells by FACS analysis of MCF7 and MDA-MB 231 cells. Dox incubation time with cells was 24 hrs. Data were fitted with sigmoid functions and yield EC50 values of 1.56 ± 0.21 μM (MCF7) or 2.92 ± 0.28 μM (MDA-MB 231) and maximal fluorescence values of 442 ± 20 a.u. (MCF7) and 200 ± 9 a.u. (MDA-MB 231).
Figure 5.
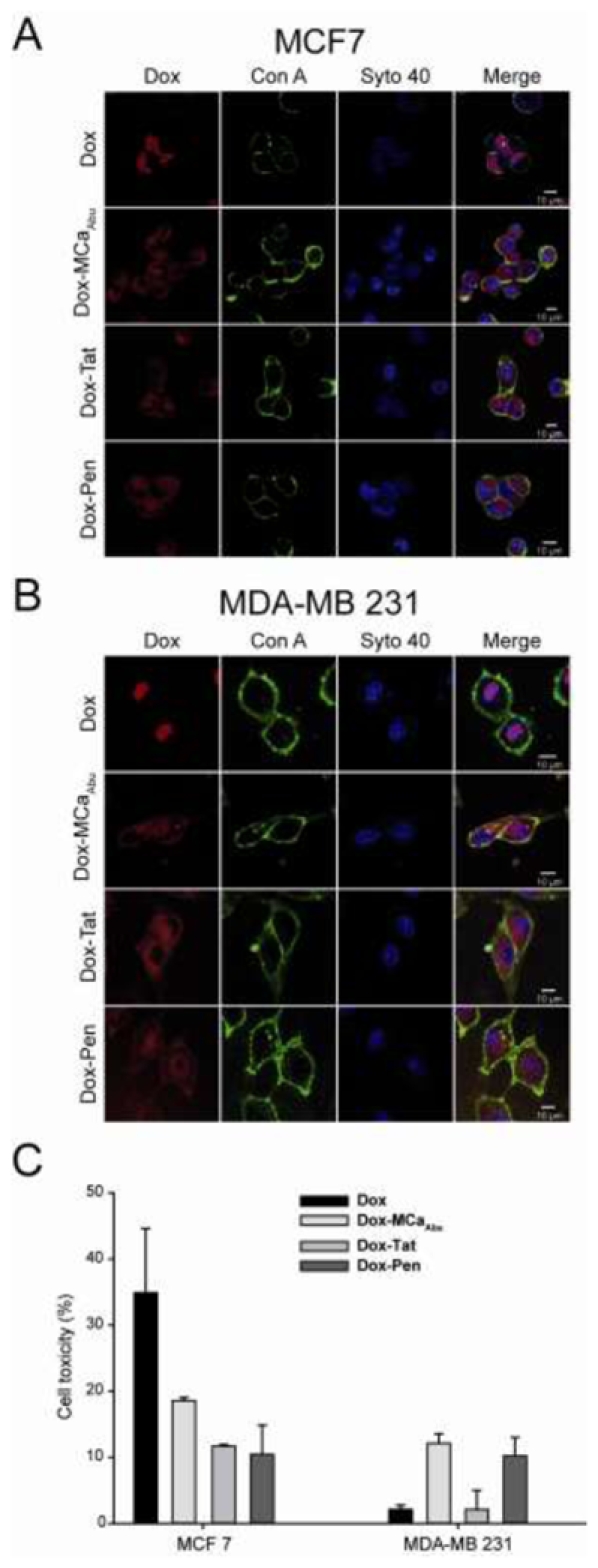
Subcellular localization and cytotoxicity of free or conjugated Dox in MCF7 and MDA-MB 231 cells after short time treatments. (A) Confocal images of living MCF7 cells comparing the distribution of Dox fluorescence for free Dox (upper panels) or CPP-conjugated Dox (three lower panels) after a 2 hrs incubation. Cells were incubated with a drug concentration of 5 μM. (B) Same as in (A) but for MDA-MB 231 cells. (C) Cell toxicity of Dox and Dox-CPP conjugates evaluated with the MTT test in the same experimental conditions as in (A) and (B).
Comparative cell distribution and cytotoxicity between Dox and Dox-CPPs after short term cell exposure
The effect of Dox conjugation on cell distribution and cytotoxicity was evaluated on short term exposure (2 hrs). A concentration of 5 μM was chosen to ensure Dox fluorescence detection by confocal microscopy (Figure 5). As shown, MCF7 or MDA-MB 231 cells exhibit similar patterns of cell distribution of Dox or Dox-CPP conjugates. In both cell lines, there was a marked difference in cell distribution of Dox-CPP comparatively to free drug. As observed in other conditions (Figure 4), Dox concentrates mainly in the nuclei in both cell lines (Figure 5A,B). In sharp contrast, Dox-CPP conjugates were found diffuse into the cytoplasm of both cell lines (Figure 5A, B). Obviously, conjugation of Dox to CPPs prevented the accumulation of Dox into the nuclei of the cells. However, since Dox fluorescence is intrinsically low, we cannot exclude that a fraction of Dox-CPP reaches the nucleus but remains undetected. Next, the cytotoxic effects of 5 μM Dox or Dox-CPPs on these two cells lines were investigated using the MTT test after this 2 hrs of drug incubation (Figure 5C). Interestingly, 2 hrs of incubation with 5 μM Dox MCF7 cells is sufficient to induce 34.9 ± 9.7% of MCF7 cell killing (Figure 5C). This cytotoxicity value is decreased to 18.6 ± 0.5, 11.7 ± 0.2 and 10.4 ± 4.4% when using Dox conjugated to MCaAbu, Tat or Pen, respectively, instead of Dox. This reduced efficacy may be related to the change in Dox localization upon conjugation to CPPs as the primary mode of Dox action is related to nuclear functions (31). In contrast however, a 2 hrs incubation of MDA-MB 231 cells with 5 μM Dox produces almost no cell toxicity (2.1 ± 0.6%) further confirming the cell resistance to Dox. This toxicity value was significantly enhanced to 12.1 ± 1.4% for Dox-MCaAbu conjugate. A similar enhancement was noticed for Dox-Pen conjugate (10.2 ± 2.8%). No significant change was observed when using the Dox-Tat conjugate. These data seem to indicate that using CPP conjugates of Dox can reverse the cell resistance to Dox. This may possibly be due to a reduced cell extrusion of Dox when conjugated to CPP (32). Of note, the levels of the observed Dox-CPPs toxicities are quite low and at a concentration where some cell toxicity of CPPs alone was observed for longer exposure times (Figure 3). In order to be more conclusive, another set of experiments were designed with longer exposure times.
Long-term exposure to Dox-CPP of MDA-MB 231 cells overcomes Dox-resistance
Exposure of cells to 5 μM Dox-CPPs for 24 hrs yields strong Dox fluorescence in the cytoplasm of both cell types (Figure 6A, B). This pattern of distribution is similar to that observed for 2 hrs incubation (Figure 5). However, the greater apparent intensities in fluorescence seem to indicate that longer exposure times to the conjugated drug allow a greater accumulation in cells. A quantification by FACS analysis of Dox fluorescence of the Dox-CPP conjugates indicate that both cell lines accumulate significant amounts of Dox-CPP with a similar concentration threshold (around 500 nM) with a saturation close to 10 μM (Figure 6C, D). In MCF7 cells, this accumulation occurs at higher concentration for Dox-CPPs than for free Dox indicating CPP conjugation does not represent an advantage in this cell line (Figure 6C). In contrast, accumulation of Dox-CPPs is slightly improved over free Dox accumulation in MDA-MB 231 cells, suggesting that Dox conjugation could be interesting for this cell line (Figure 6D). Next, dose-responses curves of cytotoxicity of Dox-CPP conjugates were investigated. As suggested by the FACS data, Dox-CPP conjugates turned out to be less efficient to promote cell death of MCF7 cells (Figure 6E). Dox-MCaAbu was the most efficient Dox-CPP conjugate leading to a maximal cell toxicity of 67.6 ± 3.4% of cell death and with an EC50 of 0.37 ± 0.09 μM. Other Dox-CPPs behaved similarly to Dox-MCaAbu but with lower toxicities (on average 54% of maximal toxicity for Dox-Tat). In comparison, the EC50 value for free Dox was 0.1 ± 0.02 μM and the maximal cell toxicity was 83.5 ± 8.5% (Figure 4A). These data indicate that for MCF7 cells, conjugation of Dox to CPPs significantly reduces the efficacy of Dox both in terms of effective concentration and maximal effect. In contrast, in MDA-MB 231 cells, Dox-CPP conjugates were more toxic than free Dox (Figure 6F). Dox-MCaAbu induced cell death with an EC50 value of 0.32 ± 0.10 μM and with a maximal effect of 84.9 ± 3.6%. In comparison, Dox was far less efficient with an EC50 of 2.7 ± 1.4 μM and a maximal effect of 79.1 ± 1.5% (Figure 4B). Thus, in conclusion, conjugation of Dox to CPPs produces similar dose-responses for toxicity in MCF7 and MDA-MB 231 cells but achieves better efficiency in MDA-MB 231 cells than in MCF7. The observed improvement in cytotoxicity observed for Dox-CPP conjugates over free Dox in MDA-MB 231 cells clearly indicates that this strategy for Dox delivery and efficacy is an advantage in this cell line. A striking observation made upon analysis of the cytotoxicity of Dox-CPPs is that the conjugates appear to possess a cytotoxic effect even at very low concentrations (10 nM). This was particularly evident for MDA-MB 231 cells (Figure 6F). This observation suggested that very long exposures at minimal Dox-CPP concentration may represent a viable strategy for inducing cell death.
Figure 6.
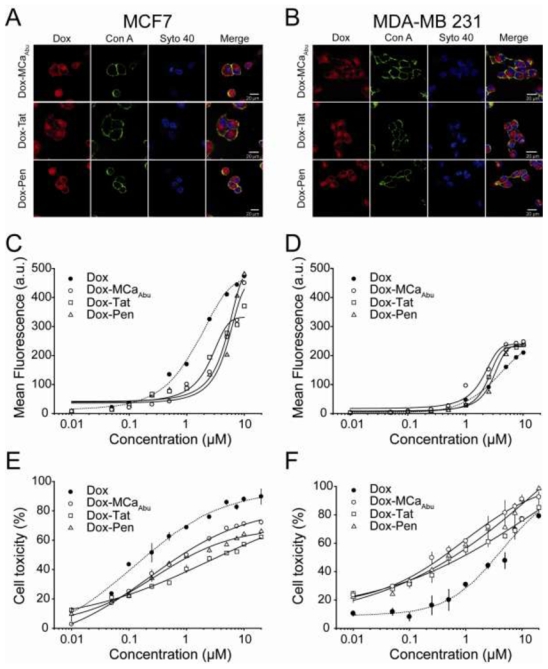
Localization, penetration and cytotoxicity of CPP-conjugated Dox in MCF7 and MDA-MB 231 cells after 24 hrs treatment. (A) Confocal images illustrating Dox-CPP distribution in MCF7 cells for each CPP after 24 hrs incubation at 5 μM. (B) Same as in (A) but in MDA-MB 231 cells. (C) FACS analyses of dose-dependent cell penetration of Dox-CPPs after 24 hrs incubation with MCF7 cells. The dose-dependence of Dox is given in dashed line for comparison (data from Figure 4D). Data were fitted by sigmoid functions yielding EC50 values of 4.6 ± 0.8 μM (Dox-MCaAbu), 2.3 ± 0.4 μM (Dox-Tat) and 5.8 ± 0.9 μM (Dox-Pen), and maximal fluorescence values of 451 ± 50 a.u. (Dox-MCaAbu), 332 ± 20 a.u. (Dox-Tat) and 568 ± 77 a.u. (Dox-Pen). (D) Same as in (C) for MDA-MB 231 cells with EC50 values of 1.9 ± 0.2 μM (Dox-MCaAbu), 2.4 ± 0.1 μM (Dox-Tat) and 3.3 ± 0.1 μM (Dox-Pen), and maximal fluorescence values of 242 ± 12 a.u. (Dox-MCaAbu), 231 ± 2 a.u. (Dox-Tat) and 240 ± 3 a.u. (Dox-Pen). (E) Cell cytotoxicity of Dox-CPPs determined by MTT assays after 24 hrs incubation of MCF7 cells with Dox conjugate. Data were fitted by sigmoid functions yielding EC50 values of 0.37 ± 0.09 μM (Dox-MCaAbu), 0.37 ± 0.11 μM (Dox-Tat) and 0.25 ± 0.05 μM (Dox-Pen), and maximal toxicity values of 67.6 ± 3.4% (Dox-MCaAbu), 54.5 ± 2.2% (Dox-Tat) and 58.9 ± 2.3% (Dox-Pen). Dashed line represents cell toxicity of free Dox (data from Figure 4A). (F) Same as in (E) for MDA-MB 231 cells with EC50 values of 0.32 ± 0.10 μM (Dox-MCaAbu), 0.25 ± 0.11 μM (Dox-Tat) and 0.41 ± 0.15 μM (Dox-Pen), and maximal toxicity values of 84.9 ± 3.6% (Dox-MCaAbu), 71.8 ± 3.3% (Dox-Tat) and 83.2 ± 4.4% (Dox-Pen).
Cytotoxic effects of long-term exposures of MDA-MB 231 cells to low concentrations of free Dox and Dox-CPPs
Long-term exposures of MDA-MB 231 cells to 10 nM Dox or one of the three Dox-CPP conjugate was investigated with the MTT test (Figure 7). As shown, 72 hrs incubation with 10 nM free Dox produces 29.0 ± 4.2% of cell toxicity for MDA-MB 231 cells, compared to 10.5 ± 1.9% after 24 hrs (see Figure 6F). Again, exposure of MDA-MB 231 cells to Dox-CPP conjugates turned out to induce more efficient toxic effects than free Dox and kills an average of 50% MDA-MB 231 cells after 72 hrs (Figure 7). These data show also that long-term exposure to low concentrations of a Dox-MCaAbu conjugate is as efficient as the long-term exposure to Dox-Tat and Pen conjugates to induce MDA-MB 231 cell death.
Figure 7.
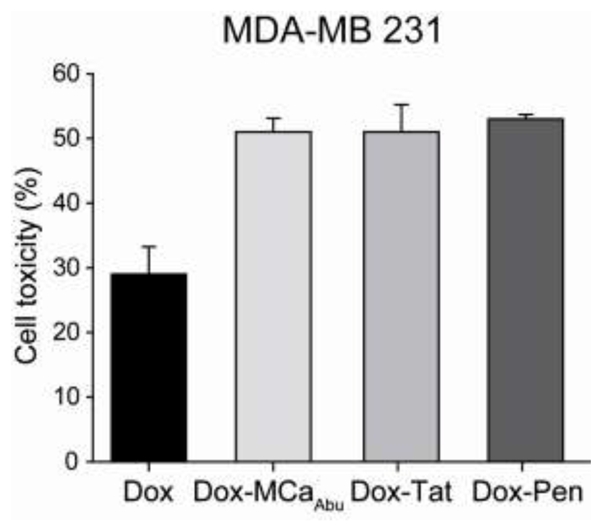
Long-term cytotoxicity of 10 nM free Dox and Dox-CPP conjugates in MDA-MB 231 cells. MTT assay for MDA-MB 231 cells after 72 hrs of incubation with 10 nM Dox or Dox-CPPs.
Discussion
In this study, we demonstrate that an analogue of MCa acts as a potent vector for the cell delivery of Dox into Dox-resistant and Dox-sensitive cell lines. Covalent coupling of Dox to several CPPs is reported, and their use in overcoming Dox-resistance in MDA-MB 231 cells evaluated. The data indicate that three CPPs (MCa analogue, Tat, and Pen) are equipotent both in terms of concentration-dependence and maximal efficacy to induce cell death in both cell lines. Various aspects of the data gathered within this report are discussed hereunder.
To perform this study we have used two cell lines that differ in their sensitivity to Dox. Reasons for a difference in Dox sensitivity of cancer cell lines are multiple. The most studied mechanism concerns the heterogeneous level of multidrug resistance that is mediated by ABC-transporters such as p-glycoprotein (P-gp), multidrug resistance related protein 1 (MRP1) or breast cancer resistance protein (BCRP). These proteins favour the efflux of chemotherapeutic agents (33, 34). Less-well studied mechanisms include (35): (i) subcellular localization of the drug (36), (ii) detoxification reactions that involve glutathione and glutathione-dependent enzymes such as glutathione S-transferase, glutathione peroxydase or reductase (37), (iii) alteration in topoisomerase II activity, (iv) increasing DNA repair to drug-induced damage, and (v) disruption in apoptotic signalling pathways. In our experiments, MDA-MB 231 cells had a lower tendency to accumulate free Dox than MCF7 cells suggesting that greater efflux might be at the basis of the differences in Dox sensitivity of both cell lines. Our data indicate that coupling Dox to CPPs significantly alters the sensitivity of both cell lines to Dox. MCF7 cells become less sensitive, whereas MDA-MB 231 cells acquire a greater sensitivity to Dox. Interestingly, both cell types become almost equally sensitive to Dox when the drug is coupled to any one of the three CPPs used herein. These data may indicate that coupling of Dox to CPPs may alter the balance between the multiple resistance pathways. One evident change was the subcellular localization of the drug which shifted from a nuclear distribution to a predominant cytoplasmic one. It should be mentioned however that such shift in cellular distribution is presumably not expected to increase cell sensitivity to Dox since the main target of cytotoxic action of Dox is thought to be the nuclear topoisomerase II. Since Dox fluorescence is visible only at high concentration by confocal microscopy, we suggest that CPPs could also deliver Dox to the nucleus, but at a lower not detectable concentration. Indeed, we observed that CPPs also overcome MDA-MB 231 Dox resistance at 10 nM, a concentration at which free Dox is not detectable in cells.
CPPs are cell penetration peptides that tend to accumulate into cells because of their basic properties. Besides they possess DNA binding abilities (38) that may come as a synergistic factor for DNA targeting. Our data indicate that cell distribution of Dox is not a reliable indicator of its toxicity effect. Other possible reasons for the greater efficacy of Dox coupled to CPPs in MDA-MB 231 cells may include alterations in efflux pathways or in detoxification reactions. We favour a less efficient efflux of the drug that remains trapped into cells due to its coupling to CPP. CPPs thus appear as useful vectors not only for their cell penetration efficacies but also to counteract efflux mechanisms. Definitive proof that alteration of Dox efflux pathways is responsible for increased Dox sensitivity will require additional experiments.
The results obtained herein underline the value of CPPs as delivery vectors for bioactive molecules. Generally, CPPs are used for their ability to carry across the cell membrane an important number of impermeable compounds including drugs, peptides, proteins such as antibodies, oligonucleotides, peptide nucleic acids, DNA or some inorganic compounds (for exemple nanoparticles) (6, 8). For these compounds, the use of CPPs has evident advantages since they overcome the limited availability of these products inside cells. This technological advantage has permitted the development of numerous applications of basic, therapeutic, diagnostic, imaging or technical importance (6, 8). The use of CPPs to promote the cell entry of compounds that are already membrane permeable (such as Dox) is obviously less evident. As a matter of fact, their use in this case is seldom reported. However, our data illustrate the benefit of using CPPs for altering the stability, efficacy and cell compartment targeting of the drug. Improvements could be brought to this strategy by grafting additional signal sequences to CPPs for their targeting to defined cell compartments. Enhanced nuclear targeting and enhanced efficacy of a Dox-CPP complex may be obtained by adding a Nuclear Localization Sequence (NLS) to the peptide sequence. This chimera strategy may be extended to cell targeting sequence for in vivo application where Dox-CPP delivery could be directed to tumors rather than being diffusely taken up by the entire organism. Such an application has successfully been designed for Dox alone coupled to a cyclic pentapeptide (39).
In this study, we have further validated the use of MCa as cargo delivery vector. A biotinylated derivative of MCa has been used for the cell entry of streptavidin in a various number of experimental conditions (11, 40). Since then, an analogue of MCa, devoid of disulfide bridges, MCaAbu, has been synthesized and shown to be pharmacologically inert on RyR1 but efficient for the cell delivery of cargoes6. Because this analogue was shown to be less efficient than wild-type MCa for cell delivery of streptavidin, one could suggest that better analogues than MCaAbu may still be designed and tested (12, 15). Nevertheless, the data obtained indicate that MCaAbu is no less efficient than Tat or Pen for the intracellular delivery of Dox. Similar concentration-dependence for Dox delivery was obtained by FACS and equivalent cell toxicity on both MCF7 and MDA-MB 231 cells were observed. One important observation made herein during this comparative analysis of CPPs is that Dox-CPP is delivered diffusely into the cytoplasm regardless of the CPP sequence used. This is an important indication that all three CPP complexes use similar routes of cell entry in both cell lines. This observed cytoplasmic distribution is in sharp contrast to the punctuate distribution of streptavidin when MCa is used as vector. Combined, these observations indicate that the nature and/or size of the cargo plays an important role in the mechanism of cell entry; Dox-CPP entering presumably via translocation across the lipid bilayer. Alternatively, we cannot exclude that Dox-CPP enter cells by macropinocytosis, but with an addition step consisting of endosomal escape and accumulation into the cytoplasm.
In conclusion, we have demonstrated that MCaAbu is as effective as other CPPs for the efficient delivery of Dox into cells. This vector-based delivery can overcome the reduced Dox sensitivity observed in MDA-MB 231 cells over MCF7 cells. Future research avenues will be developed by designing tumor-targeting Dox-CPPs and/or specific cell compartmentalized Dox-CPP analogues with improved cell toxicities. The efficacy of Dox-CPP will be evaluated in in vivo tumor models with a special emphasis to low concentrations of Dox-CPPs which were shown to be efficient in vitro during long-term exposures. Other applications are envisioned such as evaluating in vitro and in vivo the efficacy of drugs grafted to MCaAbu and that possess different cellular targets (41–44).
Acknowledgments
This work was funded by Inserm and a grant from the Life Sciences Division innovation program of the Commissariat à l’Energie Atomique. SA acknowledges the support of the Délégation Générale de la Recherche Scientifique et Technique (Tunisia) and the University of Monastir and University Joseph Fourier for their joint PhD training program.
Footnotes
CPP: Cell Penetrating Peptide
Pen: Penetratin
MCa: Maurocalcine
RyR1: Ryanodinee Receptor type 1
MCaAbu: Maurocalcine analogue with cysteine residues replaced with L-α-aminobutyric acid
Dox: Doxorubicine
SMCC: Succinimidyl 4-[N-Maleimidomethyl]Cyclohexane-1-Carboxylate
MTT: 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyl-tetrazolium bromide
PBS: Phosphate Buffered Saline
FACS: Fluorescence Activated Cell Sorting
FITC: Fluoresceine Iso ThioCyanate
Con A: Concanavalin A
References
- 1.Howl J, Nicholl ID, Jones S. The many futures for cell-penetrating peptides: how soon is now? Biochem Soc Trans. 2007;35:767–769. doi: 10.1042/BST0350767. [DOI] [PubMed] [Google Scholar]
- 2.Perez F, Joliot A, Bloch-Gallego E, Zahraoui A, Triller A, Prochiantz A. Antennapedia homeobox as a signal for the cellular internalization and nuclear addressing of a small exogenous peptide. J Cell Sci. 1992;102(Pt 4):717–722. doi: 10.1242/jcs.102.4.717. [DOI] [PubMed] [Google Scholar]
- 3.Joliot A, Pernelle C, Deagostini-Bazin H, Prochiantz A. Antennapedia homeobox peptide regulates neural morphogenesis. Proc Natl Acad Sci U S A. 1991;88:1864–1868. doi: 10.1073/pnas.88.5.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Derossi D, Joliot AH, Chassaing G, Prochiantz A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J Biol Chem. 1994;269:10444–10450. [PubMed] [Google Scholar]
- 5.Derossi D, Chassaing G, Prochiantz A. Trojan peptides: the penetratin system for intracellular delivery. Trends Cell Biol. 1998;8:84–87. [PubMed] [Google Scholar]
- 6.Mae M, Langel U. Cell-penetrating peptides as vectors for peptide, protein and oligonucleotide delivery. Curr Opin Pharmacol. 2006;6:509–514. doi: 10.1016/j.coph.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 7.Zorko M, Langel U. Cell-penetrating peptides: mechanism and kinetics of cargo delivery. Advanced drug delivery reviews. 2005;57:529–545. doi: 10.1016/j.addr.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 8.Temsamani J, Vidal P. The use of cell-penetrating peptides for drug delivery. Drug discovery today. 2004;9:1012–1019. doi: 10.1016/S1359-6446(04)03279-9. [DOI] [PubMed] [Google Scholar]
- 9.Fajloun Z, Kharrat R, Chen L, Lecomte C, Di Luccio E, Bichet D, El Ayeb M, Rochat H, Allen PD, Pessah IN, De Waard M, Sabatier JM. Chemical synthesis and characterization of maurocalcine, a scorpion toxin that activates Ca(2+) release channel/ryanodine receptors. FEBS Lett. 2000;469:179–185. doi: 10.1016/s0014-5793(00)01239-4. [DOI] [PubMed] [Google Scholar]
- 10.Esteve E, Smida-Rezgui S, Sarkozi S, Szegedi C, Regaya I, Chen L, Altafaj X, Rochat H, Allen P, Pessah IN, Marty I, Sabatier JM, Jona I, De Waard M, Ronjat M. Critical amino acid residues determine the binding affinity and the Ca2+ release efficacy of maurocalcine in skeletal muscle cells. J Biol Chem. 2003;278:37822–37831. doi: 10.1074/jbc.M305798200. [DOI] [PubMed] [Google Scholar]
- 11.Esteve E, Mabrouk K, Dupuis A, Smida-Rezgui S, Altafaj X, Grunwald D, Platel JC, Andreotti N, Marty I, Sabatier JM, Ronjat M, De Waard M. Transduction of the scorpion toxin maurocalcine into cells. Evidence that the toxin crosses the plasma membrane. J Biol Chem. 2005;280:12833–12839. doi: 10.1074/jbc.M412521200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mabrouk K, Ram N, Boisseau S, Strappazzon F, Rehaim A, Sadoul R, Darbon H, Ronjat M, De Waard M. Critical amino acid residues of maurocalcine involved in pharmacology, lipid interaction and cell penetration. Biochim Biophys Acta. 2007;1768:2528–2540. doi: 10.1016/j.bbamem.2007.06.030. [DOI] [PubMed] [Google Scholar]
- 13.Mosbah A, Kharrat R, Fajloun Z, Renisio JG, Blanc E, Sabatier JM, El Ayeb M, Darbon H. A new fold in the scorpion toxin family, associated with an activity on a ryanodine-sensitive calcium channel. Proteins. 2000;40:436–442. doi: 10.1002/1097-0134(20000815)40:3<436::aid-prot90>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 14.Drinand G, Temsamani J. Translocation of protegrin I through phospholipid membranes: role of peptide folding. Biochim Biophys Acta. 2002;1559:160–170. doi: 10.1016/s0005-2736(01)00447-3. [DOI] [PubMed] [Google Scholar]
- 15.Ram N, Weiss N, Texier-Nogues I, Aroui S, Andreotti N, Pirollet F, Ronjat M, Sabatier JM, Darbon H, Jacquemond V, De Waard M. Design of a disulfide-less, pharmacologically-inert and chemically-competent analog of maurocalcine for the efficient transport of impermeant compounds into cells. J Biol Chem. 2008 doi: 10.1074/jbc.M804727200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bonadonna G, Zambetti M, Moliterni A, Gianni L, Valagussa P. Clinical relevance of different sequencing of doxorubicin and cyclophosphamide, methotrexate, and Fluorouracil in operable breast cancer. J Clin Oncol. 2004;22:1614–1620. doi: 10.1200/JCO.2004.07.190. [DOI] [PubMed] [Google Scholar]
- 17.Colozza M, de Azambuja E, Cardoso F, Bernard C, Piccart MJ. Breast cancer: achievements in adjuvant systemic therapies in the pre-genomic era. Oncologist. 2006;11:111–125. doi: 10.1634/theoncologist.11-2-111. [DOI] [PubMed] [Google Scholar]
- 18.Wong HL, Bendayan R, Rauth AM, Xue HY, Babakhanian K, Wu XY. A mechanistic study of enhanced doxorubicin uptake and retention in multidrug resistant breast cancer cells using a polymer-lipid hybrid nanoparticle system. J Pharmacol Exp Ther. 2006;317:1372–1381. doi: 10.1124/jpet.106.101154. [DOI] [PubMed] [Google Scholar]
- 19.Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5:219–234. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- 20.Smith L, Watson MB, O’Kane SL, Drew PJ, Lind MJ, Cawkwell L. The analysis of doxorubicin resistance in human breast cancer cells using antibody microarrays. Mol Cancer Ther. 2006;5:2115–2120. doi: 10.1158/1535-7163.MCT-06-0190. [DOI] [PubMed] [Google Scholar]
- 21.Mallory JC, Crudden G, Oliva A, Saunders C, Stromberg A, Craven RJ. A novel group of genes regulates susceptibility to antineoplastic drugs in highly tumorigenic breast cancer cells. Mol Pharmacol. 2005;68:1747–1756. doi: 10.1124/mol.105.016519. [DOI] [PubMed] [Google Scholar]
- 22.de la Torre M, Hao XY, Larsson R, Nygren P, Tsuruo T, Mannervik B, Bergh J. Characterization of four doxorubicin adapted human breast cancer cell lines with respect to chemotherapeutic drug sensitivity, drug resistance associated membrane proteins and glutathione transferases. Anticancer Res. 1993;13:1425–1430. [PubMed] [Google Scholar]
- 23.Kim DS, Park SS, Nam BH, Kim IH, Kim SY. Reversal of drug resistance in breast cancer cells by transglutaminase 2 inhibition and nuclear factor-kappaB inactivation. Cancer Res. 2006;66:10936–10943. doi: 10.1158/0008-5472.CAN-06-1521. [DOI] [PubMed] [Google Scholar]
- 24.Fang Y, Sullivan R, Graham CH. Confluence-dependent resistance to doxorubicin in human MDA-MB-231 breast carcinoma cells requires hypoxia-inducible factor-1 activity. Exp Cell Res. 2007;313:867–877. doi: 10.1016/j.yexcr.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 25.Hruby M, Konak C, Ulbrich K. Polymeric micellar pH-sensitive drug delivery system for doxorubicin. J Control Release. 2005;103:137–148. doi: 10.1016/j.jconrel.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 26.Tewes F, Munnier E, Antoon B, Ngaboni Okassa L, Cohen-Jonathan S, Marchais H, Douziech-Eyrolles L, Souce M, Dubois P, Chourpa I. Comparative study of doxorubicin-loaded poly(lactide-co-glycolide) nanoparticles prepared by single and double emulsion methods. Eur J Pharm Biopharm. 2007;66:488–492. doi: 10.1016/j.ejpb.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 27.Massing U, Fuxius S. Liposomal formulations of anticancer drugs: selectivity and effectiveness. Drug Resist Updat. 2000;3:171–177. doi: 10.1054/drup.2000.0138. [DOI] [PubMed] [Google Scholar]
- 28.Di Stefano G, Lanza M, Kratz F, Merina L, Fiume L. A novel method for coupling doxorubicin to lactosaminated human albumin by an acid sensitive hydrazone bond: synthesis, characterization and preliminary biological properties of the conjugate. Eur J Pharm Sci. 2004;23:393–397. doi: 10.1016/j.ejps.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 29.Di Stefano G, Fiume L, Domenicali M, Busi C, Chieco P, Kratz F, Lanza M, Mattioli A, Pariali M, Bernardi M. Doxorubicin coupled to lactosaminated albumin: Effects on rats with liver fibrosis and cirrhosis. Dig Liver Dis. 2006;38:404–408. doi: 10.1016/j.dld.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 30.Liang JF, Yang VC. Synthesis of doxorubicin-peptide conjugate with multidrug resistant tumor cell killing activity. Bioorganic & medicinal chemistry letters. 2005;15:5071–5075. doi: 10.1016/j.bmcl.2005.07.087. [DOI] [PubMed] [Google Scholar]
- 31.Hande KR. Clinical applications of anticancer drugs targeted to topoisomerase II. Biochim Biophys Acta. 1998;1400:173–184. doi: 10.1016/s0167-4781(98)00134-1. [DOI] [PubMed] [Google Scholar]
- 32.Shen F, Chu S, Bence AK, Bailey B, Xue X, Erickson PA, Montrose MH, Beck WT, Erickson LC. Quantitation of doxorubicin uptake, efflux, and modulation of multidrug resistance (MDR) in MDR human cancer cells. J Pharmacol Exp Ther. 2008;324:95–102. doi: 10.1124/jpet.107.127704. [DOI] [PubMed] [Google Scholar]
- 33.Modok S, Mellor HR, Callaghan R. Modulation of multidrug resistance efflux pump activity to overcome chemoresistance in cancer. Curr Opin Pharmacol. 2006;6:350–354. doi: 10.1016/j.coph.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 34.Sharom FJ. ABC multidrug transporters: structure, function and role in chemoresistance. Pharmacogenomics. 2008;9:105–127. doi: 10.2217/14622416.9.1.105. [DOI] [PubMed] [Google Scholar]
- 35.Nielsen D, Maare C, Skovsgaard T. Cellular resistance to anthracyclines. Gen Pharmacol. 1996;27:251–255. doi: 10.1016/0306-3623(95)02013-6. [DOI] [PubMed] [Google Scholar]
- 36.Schuurhuis GJ, van Heijningen TH, Cervantes A, Pinedo HM, de Lange JH, Keizer HG, Broxterman HJ, Baak JP, Lankelma J. Changes in subcellular doxorubicin distribution and cellular accumulation alone can largely account for doxorubicin resistance in SW-1573 lung cancer and MCF-7 breast cancer multidrug resistant tumour cells. Br J Cancer. 1993;68:898–908. doi: 10.1038/bjc.1993.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McLellan LI, Wolf CR. Glutathione and glutathione-dependent enzymes in cancer drug resistance. Drug Resist Updat. 1999;2:153–164. doi: 10.1054/drup.1999.0083. [DOI] [PubMed] [Google Scholar]
- 38.Ziegler A, Seelig J. High affinity of the cell-penetrating peptide HIV-1 Tat-PTD for DNA. Biochemistry. 2007;46:8138–8145. doi: 10.1021/bi700416h. [DOI] [PubMed] [Google Scholar]
- 39.van Hensbergen Y, Broxterman HJ, Elderkamp YW, Lankelma J, Beers JC, Heijn M, Boven E, Hoekman K, Pinedo HM. A doxorubicin-CNGRC-peptide conjugate with prodrug properties. Biochem Pharmacol. 2002;63:897–908. doi: 10.1016/s0006-2952(01)00928-5. [DOI] [PubMed] [Google Scholar]
- 40.Boisseau S, Mabrouk K, Ram N, Garmy N, Collin V, Tadmouri A, Mikati M, Sabatier JM, Ronjat M, Fantini J, De Waard M. Cell penetration properties of maurocalcine, a natural venom peptide active on the intracellular ryanodine receptor. Biochim Biophys Acta. 2006;1758:308–319. doi: 10.1016/j.bbamem.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 41.Gusarova GA, Wang IC, Major ML, Kalinichenko VV, Ackerson T, Petrovic V, Costa RH. A cell-penetrating ARF peptide inhibitor of FoxM1 in mouse hepatocellular carcinoma treatment. J Clin Invest. 2007;117:99–111. doi: 10.1172/JCI27527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim Y, Lillo AM, Steiniger SC, Liu Y, Ballatore C, Anichini A, Mortarini R, Kaufmann GF, Zhou B, Felding-Habermann B, Janda KD. Targeting heat shock proteins on cancer cells: selection, characterization, and cell-penetrating properties of a peptidic GRP78 ligand. Biochemistry. 2006;45:9434–9444. doi: 10.1021/bi060264j. [DOI] [PubMed] [Google Scholar]
- 43.Perea SE, Reyes O, Puchades Y, Mendoza O, Vispo NS, Torrens I, Santos A, Silva R, Acevedo B, Lopez E, Falcon V, Alonso DF. Antitumor effect of a novel proapoptotic peptide that impairs the phosphorylation by the protein kinase 2 (casein kinase 2) Cancer Res. 2004;64:7127–7129. doi: 10.1158/0008-5472.CAN-04-2086. [DOI] [PubMed] [Google Scholar]
- 44.Hirose M, Takatori M, Kuroda Y, Abe M, Murata E, Isada T, Ueda K, Shigemi K, Shibazaki M, Shimizu F, Hirata M, Fukazawa K, Sakaguchi M, Kageyama K, Tanaka Y. Effect of synthetic cell-penetrating peptides on TrkA activity in PC12 cells. J Pharmacol Sci. 2008;106:107–113. doi: 10.1254/jphs.fpz070263. [DOI] [PubMed] [Google Scholar]