

Abstract
Neurexins and neuroligins are emerging as central organizing molecules for excitatory glutamatergic and inhibitory GABAergic synapses in mammalian brain. They function as cell adhesion molecules, bridging the synaptic cleft. Remarkably, each partner can trigger formation of a hemisynapse: neuroligins trigger presynaptic differentiation and neurexins trigger postsynaptic differentiation. Recent protein interaction assays and cell culture studies indicate a selectivity of function conferred by alternative splicing in both partners. An insert at site 4 of β-neurexins selectively promotes GABAergic synaptic function, whereas an insert at site B of neuroligin 1 selectively promotes glutamatergic synaptic function. Initial knockdown and knockout studies indicate that neurexins and neuroligins have an essential role in synaptic transmission, particularly at GABAergic synapses, but further studies are needed to assess the in vivo functions of these complex protein families.
Introduction
Neurexin 1α was first identified in 1992 [1] by affinity chromatography of rat brain extract on a column of α-latrotoxin (a component of black widow spider venom that stimulates massive synaptic vesicle fusion). Over the subsequent 15 years, pioneering studies by Sudhof and colleagues have characterized neurexins and their binding partners the neuroligins. The intense current interest in these proteins was triggered by the finding of Scheiffele et al. [2] that neuroligins presented on the surface of non-neuronal cells induce synaptic vesicle clustering and formation of functional release sites in contacting glutamatergic axons. A complementary study by Graf et al. [3•] then showed that neurexins presented alone to dendrites trigger postsynaptic differentiation, inducing clusters of GABA postsynaptic components even more so than glutamate postsynaptic components. Overexpression and knockdown of neuroligins in culture led to the idea that these molecules control the balance of GABAergic and glutamatergic inputs [4•,5•]. We focus our discussion here on studies from the past two years that address how alternative splicing in both neurexin and neuroligin transcripts regulates their function. We also discuss initial results from studies of knockout mice and disease-linked mutations.
Structure of neurexins and neuroligins
There are three neurexin genes in mammals, each of which has both an upstream promoter that is used to generate the larger α-neurexins and a downstream promoter that is used to generate the β-neurexins (Figure 1) [6]. Alternative splicing at five sites and N- and O-glycosylation contribute additional diversity. By contrast, Drosophila melanogaster and Caenorhabditis elegans have only a single α-neurexin homolog. β-Neurexins contain a single LNS domain (laminin, neurexin, sex-hormone-binding protein domain; also known as a Laminin G domain), whereas α-neurexins contain six LNS domains organized into modules with three EGF-like domains. The crystal structure of the neurexin 1β LNS domain revealed a remarkable conservation with the agrin LNS domain in the position of alternative splice sites [7]. This structural similarity might reflect functional similarity. Alternative splicing of agrin regulates its role as an essential synaptic organizer at the mammalian neuromuscular junction [8]; as discussed later in this review, alternative splicing of neurexin regulates its role at synapses in the CNS. A recent structure–function study has confirmed that binding to neuroligins and synaptogenic activity is mediated by same face of the neurexin 1β LNS domain that contains the splice sites (Figure 1) [9••]. The crystal structure of the second LNS domain of neurexin 1α revealed that this region containing the splice sites forms a highly variable surface surrounding a coordinated Ca2+ ion [10]. Ca2+ binding occurred with low affinity (Kd ~400 μM) and was reduced to below detectable levels by addition of the eight-residue or fifteen-residue splice inserts. Thus, some of the Ca2+-dependent interactions of neurexins might be influenced by reductions in cleft Ca2+ concentrations following synaptic activity (but note that interaction of neurexin 1β with neuroligin 1 was half-maximal at Ca2+ concentrations of 2 μM [11], which is well below the estimated range in the synaptic cleft).
Figure 1.

Structure of neurexins and neuroligins. In humans, there are three neurexin genes and five neuroligin genes. Each neurexin gene uses an upstream promoter to generate the larger α-neurexins and a downstream promoter to generate the smaller β-neurexins. Thus, β-neurexins can be thought of as N-terminally truncated α-neurexins that have a short β-specific leader (βN). In α-neurexins, the LNS (laminin, neurexin, sex-hormone-binding protein) domains are organized with EGF (epidermal growth-factor)-like domains into three homologous modules, I–III. The position of each of five sites of alternative splicing (SS1–SS5) is indicated. Neuroligins contain an extracellular acetylcholinesterase (AChE)-homologous domain that contains one or two sites of alternative splicing (SSA, plus SSB in the case of neuroligin 1). Both neurexins and neuroligins contain a highly glycosylated region (CH) and a transmembrane domain (TM; not present in some splice variants of neurexin 3), and terminate in PDZ-domain-binding sites (PDZ BD). Shown between the neurexins and neuroligins are structures of AChE, a model for the AChE-homologous domain of neuroligins, and the neurexin 1β LNS domain [7]. The position of splice sites SS2–SS4 is shown on a single LNS domain for simplicity, although SS2 and SS3 actually occur in different LNS domains of α-neurexins. Note also that the left face of the neurexin LNS as shown here binds neuroligin [9••] but the precise structure and interacting region of neuroligin has not been reported yet. The structure files E.C.3.1.1.7 (AChE) and d1c4ra (neurexin LNS) were downloaded from the Research Collaboratory for Structural Bioinformatics protein data bank (http://www.rcsb.org/pdb/home/home.do) and visualized using the program Visual Molecular Dynamics [66].
Neurexins have three known extracellular binding partners in the mammalian brain: neuroligins, dystroglycan and neurexophilins. Neuroligins exhibit Ca2+-dependent binding to α-neurexins and β-neurexins, dystroglycan shows Ca2+-dependent binding preferentially to α-neurexins, and neurexophilin binding is Ca2+-independent and specific to α-neurexin. Neuroligins were first identified using an affinity column of neurexin 1β and they are the most intensively studied neurexin binding partners. There are five neuroligin genes in humans: NLGN1, NLGN2, NLGN3, NLGN4 and NLGN4Y [12]. The major extracellular domain of neuroligins is homologous to acetylcholinesterase (AChE) but lacks cholinesterase activity and mediates binding to neurexins. Curiously, overexpression of AChE reduces levels of β-neurexins in vivo and in culture, and impairs genesis of glutamatergic synapses in culture, indicating that there is cross-talk between the two proteins [13,14]. Neuroligins form homo-multimers through the AChE-homologous domain, a structural association that is important for neuroligin function [15,16]. However, hetero-oligomerization versus homo-oligomerization is a potential regulatory step that has not yet been explored. The AChE-homologous region of neuroligins contains alternative splice site A, and there is an additional splice site B within this region specifically in neuroligin 1 (Figure 1). Thus, both neuroligins and neurexins exist in numerous isoforms that derive from multiple genes and alternative splicing. Neuroligins and neurexins both have relatively short intracellular domains that terminate in PDZ-domain-binding sites, which are presumably important for linking them to other synaptic proteins.
Synaptic localization and interacting partners
Because neurexin 1α can function as an α-latrotoxin receptor to mediate transmitter release [17], it is assumed that neurexins at least partially localize to the presynaptic terminus; this assumption has been confirmed by antibody labeling and localization of tagged recombinant neurexins [1,3•,16]. Ultrastructural localization using high-quality antibodies is still needed to define the precise localization of neurexins in relation to different synapse types during development. At the cellular level, the six major neurexin forms (1α, 2α, 3α, 1β, 2β and 3β) show fairly broad overlapping expression patterns in brain, such that most neurons express multiple neurexins [18]. Thus, there is not an obvious segregation according to transmitter type.
Extracellularly, the functional significance of neurexins binding to neuroligins is becoming appreciated, as we will go on to discuss extensively, but the significance of the interaction of neurexins with dystroglycan or neurexophilins is not so clear. Dystroglycan binds either to the LNS domain that is common to both α-neurexins and β-neurexins or to the second LNS domain of α-neurexins, and in both cases binds only to LNS domains lacking splice inserts [19]. Mice that have brain-specific deletion of dystroglycan exhibit impaired long-term potentiation and developmental malformations characteristic of muscular dystrophy, but they have relatively normal baseline synaptic transmission [20]. Given the selective concentration of dystroglycan to a subset of mature GABAergic synapses [21,22], it might be informative to study in detail GABA-mediated transmission in mice lacking neuronal dystroglycan, both in the single-mutant mice and in knockouts that also lack neuroligins.
Neurexophilins are secreted peptides that are processed proteolytically and that bind to the second LNS domain of α-neurexin. The two neurexophilins that bind α-neurexins in mice, Nxph1 and Nxph3 — NXPH2 is expressed only in humans — show very restricted expression patterns [23,24]. Each single-knockout mouse shows normal brain morphology, and a Nxph1;Nxph3 double-knockout mouse is viable. However, the increased startle responses and impaired motor coordination of the Nxph3 knockout mice indicate that neurexophilins have a functional role in specific circuits [24].
Intracellularly, the C termini of neurexins bind to synaptotagmin and to the PDZ domains of CASK, syntenin and Mint [25-28]. These interactions constitute a link from neurexins to both synaptic vesicles and the vesicle fusion apparatus.
Neuroligins bind to several postsynaptic components of glutamatergic synapses: PDZ-domain scaffolding proteins such as PSD-95 and related MAGUKs, S-SCAM and related MAGIs, and probably also Shank, PICK1, GOPC and SPAR [29-31]. Thus, it came as a surprise when two groups [3•,32] independently found that neuroligin 2 concentrates not at glutamate postsynaptic sites but at GABA postsynaptic sites. This is in contrast to the glutamatergic postsynaptic localization of neuroligin 1 (Figure 2) [33]. The mechanisms by which neuroligin 2 is inhibited from binding to glutamate-specific PDZ domain proteins such as PSD-95 in neurons, how it localizes to GABAergic synapses and which proteins it binds to in neurons are questions under active investigation. A dendritic-targeting motif identified in the central portion of the neuroligin 1 cytoplasmic domain seems to be conserved among the neuroligins and distinct from the synaptic-targeting regions [34].
Figure 2.

Molecular interactions at glutamatergic and GABAergic synapses that are linked by neurexins and neuroligins. (a) A glutamatergic synapse. (b) A GABAergic synapse. The broken lines between neuroligin 2 and GABA receptors (GABAR) and Gephyrin indicate that there are some links (direct or indirect) but their nature is not yet known. At glutamatergic synapses, α-neurexins can bind neuroligin 1 that lacks an insert at the B splice site (−B) but the majority of neuroligin 1 is in the +B form, which does not bind to α-neurexins. Additional abbreviations: AMPAR, AMPA receptor; NMDAR, NMDA receptors; VGAT, vesicular GABA transporter; VGlut1, vesicular glutamate transporter.
Triggers of presynaptic and postsynaptic differentiation
Neuroligins presented alone on the surface of non-neuronal cells or beads induce localized formation of functional release sites in axons, by aggregating neurexins on the axon surface [2,16]. Neuroligins induce formation of glutamatergic and GABAergic presynaptic specializations, with neuroligin 2 being relatively more active on GABAergic axons [3•,5•,35]. The neuroligin-induced presynaptic specializations exhibit release characteristics remarkably similar to those of bona fide synapses and might be considered as hemisynapses. A striking demonstration of the function of these hemisynapses is the miniature excitatory postsynaptic current (mEPSC)-like events that occur in HEK cells expressing neuroligins and glutamate receptors and co-cultured with neurons [36,37].
Neurexins, like their neuroligin binding partners, also signal trans-synaptically. Neurexins presented alone on the surface of non-neuronal cells or beads induce localized clustering of neurotransmitter receptors and other postsynaptic components, by aggregating neuroligins on the dendrite surface [3•,38]. Neurexins induce clustering of several scaffolding and signaling proteins that are characteristic of glutamatergic and GABAergic synapses, with a selective link from neuroligin 2 to components of GABAergic synapses [3•]. An important exception is AMPA receptors: these were not aggregated by neurexins alone but seem to require additional signals, including stimulation of the aggregated NMDA receptors and perhaps increased levels of PSD-95 [38].
Several studies have assessed the function of neuroligins by overexpression of full-length or truncated versions in neurons; these increase or decrease synaptic protein accumulation, mEPSCs and miniature inhibitory postsynaptic currents (mIPSCs), depending on the construct and expression level (reviewed recently in [39]). A particularly interesting finding of these and related studies is that the level of the interacting protein PSD-95 can influence the level of neuroligins at excitatory versus inhibitory synapses, and the balance of functional excitatory versus inhibitory input [4•,35,40]. For example, overexpression of PSD-95 can redirect neuroligin 2 from excitatory to inhibitory synapses, reducing inhibitory input while strengthening excitatory synapses. The levels of neuroligins 1, 2 and 3 have been reduced using RNA interference (RNAi) in hippocampal cultures [5•]. Knockdown of all three neuroligins, and to a lesser extent knockdown of any one alone, reduced the density of glutamatergic and GABAergic inputs identified by immunofluorescence for the glutamate transporter VGlut1 and the GABA transporter VGAT. Functionally, there was a strong reduction in frequency and amplitude of mIPSC-like events but little reduction in amplitude of mEPSC-like events.
Regulation by alternative splicing
Neurexins undergo extensive alternate splicing, generating a huge diversity of >2000 potential variants [6,41,42]. The fact that neurexin splice insert sequences and their positions are well conserved among neurexin genes and among species supports the idea that alternative splicing has important functional roles. Three of the five splice sites occur in LNS domains, and a fourth can generate both secreted and transmembrane forms of neurexin 3 [41]. Alternative splicing of neuroligins is much less extensive but also occurs in the key functional domain, the AChE-homologous region. Distinct roles of different neurexin and neuroligin splice variants in cell–cell recognition, synaptic organization and synaptic signaling have been suggested. Testing some of these ideas will require generation and analysis of targeted mutations in vivo. Nonetheless, important insights have come from three recent studies [9••,43••,44••] that focused on how alternative splicing in neurexins and neuroligins alters binding affinity, hemisynapse formation and neuronal function in culture.
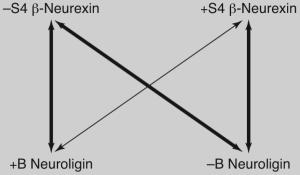
These recent findings indicate that splicing at site 4 in β-neurexins and at site B in neuroligin 1 regulate binding selectivity and synapse function. Presence of the 30-residue insert at site 4 in β-neurexin reduces the affinity of interaction specifically with neuroligin 1 that contains an insert at site B (+B), while maintaining high-affinity interaction with neuroligin 1 that lacks an insert at site B (−B) or with neuroligin 2 (which lacks any B splice site and exists only in the insert-less form) [9••,43••,44••]. The long α-neurexins (with or without an insert at site 4) also bind selectively to −B neuroligins [43••,44••]. Interestingly, it is not the nine amino acid B insert itself but N-linked glycosylation of this insert that regulates the interaction of neuroligin 1 with specific neurexins [15,43••,44••]. Furthermore, +B neuroligin 1, which is more selective than the −B variant, comprises the majority of neuroligin 1 in adult rat hippocampus, cortex and cerebellum [44••]. In chick sympathetic neurons, the ratio of neurexin transcripts containing versus lacking the insert at site 4 (+S4 versus −S4) changes during development and in response to addition of growth factors in culture [45]. Further studies are needed to determine how splicing at these key sites in neuroligin 1 (at site B) and β-neurexins (at site 4) is regulated in specific neuron types, during development and perhaps by activity.
These specific splice alterations of neurexins and neuroligins contribute to differences in function at GABAergic versus glutamatergic synapses (Table 1). In co-culture assays, addition of the site 4 insert to β-neurexin reduces its ability to cluster the glutamate postsynaptic proteins neuroligin 1/3/4 and PSD-95, but not the GABA postsynaptic proteins neuroligin 2 and gephyrin [9••,44••]. Consistent with this finding, and with the low affinity of +S4 β-neurexins for +B neuroligin, addition of the B splice insert to neuroligin 1, or artificially to neuroligin 2, reduces their ability to cluster VGAT but not VGlut1 when overexpressed in neurons [44••]. Also consistent with this idea, neuroligin 2 (which is always −B) promotes VGAT clustering more than +B neuroligin 1 [5•,35]. Thus, +S4 β-neurexins and −B neuroligins together selectively promote differentiation of GABAergic synapses, whereas −S4 β-neurexins and +B neuroligin 1 together selectively promote differentiation of glutamatergic synapses.
Table 1.
Selectivity of −S4 β-neurexins and +B neuroligin for glutamatergic synapses, and +S4 β-neurexins and −B neuroligins for GABAergic synapses
| A. Effect of splicing on relative binding affinitiesa | ||||
 |
||||
| B. Induction of postsynaptic protein clustering by +S4 neurexin 1β in co-culture assaysb | ||||
| Proteins: | Neuroligin 1/3/4 | Neuroligin 2 | PSD-95 | Gephyrin |
| Clustering (%): | 65c | 90c | 50c, 7d | 104c, 110d |
| C. Enhancement of selective presynaptic input onto neurons by overexpression of neuroligin variantse | ||||
| Neuroligin 1 +B | Neuroligin 1 −B | Neuroligin 2 (always −B) | ||
| Enhancement of VGlut1: | 4.7d, 1.5f | 2.5d | 5.3d, 1.5f | |
| Enhancement of VGAT: | 1.5d, 2.2f | 5.0d | 5.6d, 3.0f | |
Line widths reflect affinity of interaction. β-Neurexins that contain an insert at site 4 (+S4) interact preferentially with neuroligins that lack an insert at the B site (−B); they have lower affinity for +B neuroligin. β-Neurexins that lack an insert at site 4 (−S4) interact with −B neuroligins and +B neuroligin with equal affinity [9••,43••,44••].
Induction is reported normalized to 100% induction by neurexin 1β (−S4).
From [9••].
From [44••].
Enhancement is reported as increased density of inputs immunoreactive for VGlut (a marker of glutamatergic presynapses) or VGAT (a marker of GABAergic presynapses) relative to a value of 1.0 for control input density onto neurons expressing enhanced green fluorescent protein (EGFP). The neuroligin forms tested all contained the A-site insert.
From [35].
A few issues raised in these recent studies seem more controversial, including the extent to which splicing regulates localization of neuroligins. Chih et al. [44••] reported that recombinant +B neuroligin 1 is localized preferentially at glutamatergic synapses, whereas −B neuroligin 1 is localized equally at glutamatergic and GABAergic synapses. By contrast, Graf et al. [3•] reported that artificial addition of the B splice insert to neuroligin 2 did not alter exclusive localization of this protein to GABAergic synapses. Furthermore, the role of alternative splicing at the A site of neuroligins is not yet understood. The presence or absence of an insert at this site does not seem to affect binding neuroligins to neurexins [43••]. However, Chih et al. [44••] reported that addition of the A insert in the absence of a B insert promotes localization of neuroligins to GABAergic synapses, and that addition of the A insert to neuroligin 1 but not to neuroligin 2 reduces ability of the neuroligin to cluster VGlut1 but not VGAT. Thus, the A insert might promote neuroligin localization and function at GABAergic synapses by unknown mechanisms.
The binding of α-neurexins (−S4 or +S4) to −B neuroligins raises the question of how the roles of α-neurexins and β-neurexins overlap in synaptic development. In co-culture experiments, −S4 neurexin 1α selectively promoted clustering of GABA postsynaptic proteins, suggesting potential overlap in function with +S4 neurexin 1β [44••]. The phenotype of mice that lack all three α-neurexins but continue to express β-neurexins (discussed further in the following section) indicates that α-neurexins have a unique function that is not provided by β-neurexins. Whether β-neurexins supply a unique function not provided by α-neurexins is not yet known, but is a possibility based on differential binding of the two neurexin forms to +B neuroligin 1. The protein interaction studies of Boucard et al. [43••] strongly suggest that only LNS6 of α-neurexins mediates binding to −B neuroligin 1. However, the presence of alternative splice sites in the equivalent binding surface of LNS2 and LNS4 of α-neurexins leads to speculation that these domains might also mediate regulated binding to other partners. Indeed, only LNS2 and LNS6 of α-neurexins that lack splice inserts bind to dystroglycan [19]. By reducing affinity of α-neurexins for Ca2+, the LNS2 splice inserts [10] might be expected to reduce binding to multiple ligands. Consistent with this idea, neurexophilins bind α-neurexin LNS2 in a Ca2+-independent manner and bind all splice variants [23]. Identification and functional characterization of additional α-neurexin binding partners, and determination of the role of the secreted forms of neurexin 3 that are generated by alternative splicing at site 5, are two avenues that are ripe for exploration.
In vivo function
The ultimate test for the functional significance of neurexins and neuroligins is to establish whether these proteins are necessary for normal nervous system function in vivo. This is a daunting task in mammals, but it has been addressed heroically by generation and analysis of Nrxn1;Nrxn2;Nrxn3 triple mutant mice that lack all three α-neurexins [46] and of Nlgn1;Nlgn2;Nlgn3 triple neuroligin knockout mice [47••]. Interestingly, knockout of the three α-neurexins, leaving the three β-neurexins intact, leads to perinatal lethality due to loss of presynaptic Ca2+ channel function [46]. Transmitter release that depends on N-type and P/Q-type Ca2+ channels is severely reduced in these mutants [48]. A hypothesis to reconcile these data with those from cell culture studies of neurexins and neuroligins is that all neurexins function as synaptic organizing molecules. α-Neurexins, via their unique extracellular domains, are required for function and perhaps localization of presynaptic Ca2+channels, whereas both α-neurexins and β-neurexins contribute to presynaptic and postsynaptic localization and function of other synaptic components. We look forward to future studies of β-neurexin knockout mice and complete neurexin knockout mice, and eventually animal models altered only in neurexin and neuroligin splice composition.
Although individual neuroligin knockout mice survive and are fertile, the Nlgn1;Nlgn2;Nlgn3 triple knockout mice die shortly after birth owing to respiratory failure [47••]. Synapses seemed to be morphologically normal in these mice, but there were marked functional defects in synaptic transmission. In neurons of the pre-Bötzinger complex, the frequency of spontaneous GABAergic/glycinergic currents was reduced by approximately 90%, and frequency of spontaneous glutamatergic currents reduced by approximately 75%. In Nlgn1;Nlgn2;Nlgn3 knockout mice, the failure rate of evoked transmission was more than tenfold greater than normal at GABAergic/glycinergic synapses, but unchanged at glutamatergic synapses. Reduced postsynaptic clustering of GABAA receptors seemed to be one causative factor, although no changes in clustering of the scaffolding proteins gephyrin or PSD-95 were observed. Reduced levels of several synaptic vesicle proteins and a small reduction in the ratio of VGAT to VGlut clusters indicate that presynaptic defects are a contributing factor. Surprisingly, normal transmission at glutamatergic synapses in cultured neocortical neurons indicates either a region-specific defect or one that is compensated for in culture but not in vivo. These data support the idea that neuroligins are essential for recruitment of key synaptic components or for maintenance of their function. More detailed analyses of the neuroligin knockout mice might provide further important information, especially considering their potential as animal models of autism spectrum disorders [12].
A general question raised by the cell culture and in vivo studies concerns the stage of synapse development at which neurexins and neuroligins function. The co-culture assays indicate that neurexins and neuroligins presented locally at high concentration are sufficient to trigger postsynaptic and presynaptic differentiation. However, given the complex presynaptic and postsynaptic protein networks, it might be that several molecules that bind transmembrane components of such networks can trigger clustering in the co-culture assays. Such molecules might function endogenously as initial triggers of synaptogenesis, but could instead function after the initial membrane adhesion to recruit synaptic components, and/or even later in the process to stabilize synaptic complexes. The interaction of soluble neuroligin 1 with neurexin 1β is of relatively low affinity (Kd ~300 nM [15]) compared with the affinity of other partners such as soluble Eph receptors for ephrins (Kd <3 nM [49]). We suggest that other adhesion molecules such as immunoglobulin-domain and cadherin family proteins mediate the initial contact between appropriate axons and dendrites, and then neurexins and neuroligins reinforce the contact but mainly function to recruit and stabilize presynaptic and postsynaptic proteins. Indeed, the presence of normal numbers of morphological synapses combined with functional defects in the brainstem of newborn Nlgn1;Nlgn2;Nlgn3 knockout mice strongly supports the idea that neuroligins function in the later stages of protein recruitment or stabilization [47••]. Determining precisely when endogenous neurexins and neuroligins cluster relative to other steps of synaptogenesis is a difficult task. By expression of tagged recombinant proteins, it has been shown that synaptic vesicle clusters in axons can precede apposing clusters of PSD-95 [50], and that PSD-95–neuroligin clusters on dendrites can precede apposing synaptic vesicle clusters [51]. A logical prediction is that clustering of neurexins on the presynaptic membrane and neuroligins on the postsynaptic membrane occur simultaneously, because they would be expected to stabilize each other [52]. This might be followed by stabilization of synaptic vesicles by the neurexin clusters and stabilization of postsynaptic scaffolds and receptors by the neuroligin clusters. Neuroligins also function in ongoing synapse maintenance, at least in cell culture studies [3•,4•,5•].
A second general question brought to the forefront recently concerns what roles neurexins and neuroligins have in development of glutamatergic versus GABAergic synapses. The link between α-neurexins and presynaptic Ca2+ channels is essential for function of both glutamatergic and GABAergic synapses in vivo [46]. This functional link of α-neurexins to presynaptic Ca2+ channels is not mimicked by β-neurexins, nor does it seem to involve neuroligins. By contrast, the neurexin–neuroligin link was initially proposed to be specific for glutamatergic synapses on the basis of observed protein interactions [29]. However, results from co-culture of neurons with neurexin-expressing cells [3•], neuroligin knockdown in culture [5•] and initial neuroligin knockout studies in vivo [47••] seem to indicate stronger roles for neurexin–neuroligin interactions in function of GABAergic synapses. Complementary studies on SynCAMs [53], ephrins and Eph receptors [54], synaptic adhesion-like molecules [55,56] and netrin-G ligands [57] indicate that additional cell adhesion molecules are specific for glutamatergic as opposed to GABAergic synapses, and that these molecules can also link to postsynaptic receptors and/or to the presynaptic release mechanism. Perhaps there is a greater redundancy of transmembrane molecular organizing partners at glutamatergic than at GABAergic synapses. A clear area for further investigation is to determine the molecular links among neuroligins and other key postsynaptic elements of GABAergic synapses, including GABAA receptors and gephyrin. The most recent set of studies on alternative splicing suggest that different splice variants of neurexins and neuroligins function selectively at glutamatergic versus GABAergic synapses [9••,43••,44••]. From these culture studies, it can be predicted that elimination of the inserts at site 4 in neurexins in vivo might promote development of excitatory synapses and reduce that of inhibitory ones, whereas elimination of the B insert from neuroligin 1 in vivo might promote development of inhibitory synapses and reduce that of excitatory ones. Whether specific neurexins and neuroligins contribute to the matched alignment of the appropriate postsynaptic receptor type opposite the corresponding transmitter release site is a related open question that might be addressed using knockout or chimera knockin mice.
Clinical implications
In addition to the obvious link between neurological disorders and the molecules that are fundamental to synapse function, clinical interest in neuroligins was stimulated by a report from Jamain et al. in 2003 [12]. These authors found mutations in the X-linked genes NLGN3 and NLGN4 in siblings who had autism spectrum disorders. The mutation in NLGN4, a frameshift that resulted in a premature termination (396X), occurred de novo in the mother of the affected siblings, thus pointing strongly to a causative role. The mutation in NLGN3 resulted in a single amino acid change (R451C) within the key AChE-homologous domain. Subsequent cell culture studies showed that these two mutations enhance intracellular retention of neuroligins and thus abolish or reduce function in synaptogenesis assays [58-60].
These exciting initial findings were followed by two confirmatory studies. In one study, another mutation in NLGN4 that results in a premature stop (429X) was found in all patients that were afflicted with mental retardation with or without autism in a single family, but not in non-affected family members or control subjects [61]. In the second study, three missense mutations in the AChE-homologous domain (G99S, K378R and V403M) and one in the cytoplasmic domain (R704C) were found in NLGN4 in autistic patients in a study of 148 unrelated autistic individuals [62]. However, a limited prevalence of mutations in NLGN3 or NLGN4 in autism is indicated by additional studies that found no mutations in the coding regions of these genes in a total of 416 additional autistic patients [63-65]. Nonetheless, the association of neuroligin mutations with a subset of autism spectrum disorders and mental retardation opens up a molecular and cellular avenue for further understanding and perhaps treatment of these disorders (see also review by Geschwind and Levitt, in this issue). Specific links between neurexins and neurological disorders have not been reported, although the unusually large size of NRXN1 and NRXN3 (1.1 Mb and 1.7 Mb, respectively [42]) places them as likely candidates.
Conclusions
Accumulating studies are leading to the realization that there might not be any single molecule, or molecular family, that is essential for assembly of CNS synapses. Nonetheless, neurexins and neuroligins are the best candidates for central organizing molecules to stabilize networks of presynaptic and postsynaptic proteins across the synaptic cleft. Recent protein interaction assays and cell culture experiments indicate selective functions of splice variants: +S4 β-neurexins and −B neuroligins at GABAergic synapses versus −S4 β-neurexins and +B neuroligins at glutamatergic synapses. Different affinities of the interactions between different neurexin–neuroligin pairs are combined with a selective linkage to components of glutamatergic versus GABAergic synapses, by mechanisms that are not yet understood. However, the significance of such a shared system is that alterations in stoichiometry of key players can be used to regulate the balance of excitatory and inhibitory inputs onto a neuron [40]. Recent in vivo studies indicate that neurexins and neuroligins have essential roles in synapse development — not in initial adhesion, but in recruitment of molecular components and maturation [47••]. Further studies are needed to explore the functional significance of the rich diversity of neurexin and neuroligin variants; particularly useful will be targeted in vivo mutagenesis and analysis of the structure, molecular composition, function and long-term stability of glutamatergic and GABAergic synapses in defined circuits.
Acknowledgements
We thank Ethan Graf and members of the Craig laboratory for helpful discussions. Work in our laboratory is supported by the National Institutes of Health (NIH) grant MH070860, Canadian Institutes for Health Research (CIHR) grant MOP69096, Michael Smith Foundation for Health Research, and Canada Research Chair Program.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Ushkaryov YA, Petrenko AG, Geppert M, Sudhof TC. Neurexins: synaptic cell surface proteins related to the alpha-latrotoxin receptor and laminin. Science. 1992;257:50–56. doi: 10.1126/science.1621094. [DOI] [PubMed] [Google Scholar]
- 2.Scheiffele P, Fan J, Choih J, Fetter R, Serafini T. Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell. 2000;101:657–669. doi: 10.1016/s0092-8674(00)80877-6. [DOI] [PubMed] [Google Scholar]
- 3•.Graf ER, Zhang X, Jin SX, Linhoff MW, Craig AM. Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell. 2004;119:1013–1026. doi: 10.1016/j.cell.2004.11.035.Using co-cultures of neurons with COS cells expressing neurexins, this study showed that signaling occurs from neurexins to the postsynaptic site to mediate clustering of receptors and scaffolding proteins through neuroligins. In contrast to prior work focusing on glutamate synapses, these authors showed that neurexin signaling occurs at GABA synapses selectively through neuroligin 2.
- 4•.Prange O, Wong TP, Gerrow K, Wang YT, El-Husseini A. A balance between excitatory and inhibitory synapses is controlled by PSD-95 and neuroligin. Proc Natl Acad Sci USA. 2004;101:13915–13920. doi: 10.1073/pnas.0405939101.These authors altered the levels of neuroligin 1 and PSD-95 in cultured neurons and showed that the ratio of these proteins affects not only glutamate synapses but also GABA synapses. For example, increased expression of PSD-95 not only enhanced glutamate-mediated transmission but also reduced GABA-mediated input. Thus, the level of neuroligins and key interacting partners can control the balance of glutamate versus GABA inputs on a neuron.
- 5•.Chih B, Engelman H, Scheiffele P. Control of excitatory and inhibitory synapse formation by neuroligins. Science. 2005;307:1324–1328. doi: 10.1126/science.1107470.This study followed on key earlier work [2] showing that presentation of neuroligins to glutamatergic axons is sufficient to induce presynaptic differentiation. Here, overexpression and knockdown of neuroligins in cultured neurons was reported to alter glutamatergic and GABAergic inputs. The major functional effect of knockdown of neuroligins 1, 2 and 3 together was a reduction in mIPSC-like events.
- 6.Tabuchi K, Sudhof TC. Structure and evolution of neurexin genes: insight into the mechanism of alternative splicing. Genomics. 2002;79:849–859. doi: 10.1006/geno.2002.6780. [DOI] [PubMed] [Google Scholar]
- 7.Rudenko G, Nguyen T, Chelliah Y, Sudhof TC, Deisenhofer J. The structure of the ligand-binding domain of neurexin Ibeta: regulation of LNS domain function by alternative splicing. Cell. 1999;99:93–101. doi: 10.1016/s0092-8674(00)80065-3. [DOI] [PubMed] [Google Scholar]
- 8.Ferns MJ, Campanelli JT, Hoch W, Scheller RH, Hall Z. The ability of agrin to cluster AChRs depends on alternative splicing and on cell surface proteoglycans. Neuron. 1993;11:491–502. doi: 10.1016/0896-6273(93)90153-i. [DOI] [PubMed] [Google Scholar]
- 9••.Graf ER, Kang Y, Hauner AM, Craig AM. Structure function and splice site analysis of the synaptogenic activity of the neurexin-1 beta LNS domain. J Neurosci. 2006;26:4256–4265. doi: 10.1523/JNEUROSCI.1253-05.2006.Along with [43••,44••], this is one of a series of recent studies that explored the functional effects of alternative splicing of neurexins. This study reported that addition of the site 4 insert to neurexin 1β selectively reduces binding to neuroligin 1 and reduces clustering of neuroligins 1/3/4 and PSD-95 in the co-culture assay, but maintains high-affinity binding to neuroligin 2 and clustering of neuroligin 2 and gephyrin. Thus, β-neurexin +S4 variants might be selective for GABAergic synapses, and −S4 variants selective for glutamatergic synapses.
- 10.Sheckler LR, Henry L, Sugita S, Sudhof TC, Rudenko G. Crystal structure of the second LNS/LG domain from neurexin 1alpha: Ca2+ binding and the effects of alternative splicing. J Biol Chem. 2006;281:22896–22905. doi: 10.1074/jbc.M603464200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nguyen T, Sudhof TC. Binding properties of neuroligin 1 and neurexin 1beta reveal function as heterophilic cell adhesion molecules. J Biol Chem. 1997;272:26032–26039. doi: 10.1074/jbc.272.41.26032. [DOI] [PubMed] [Google Scholar]
- 12.Jamain S, Quach H, Betancur C, Rastam M, Colineaux C, Gillberg IC, Soderstrom H, Giros B, Leboyer M, Gillberg C, et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet. 2003;34:27–29. doi: 10.1038/ng1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Andres C, Beeri R, Friedman A, Lev-Lehman E, Henis S, Timberg R, Shani M, Soreq H. Acetylcholinesterase-transgenic mice display embryonic modulations in spinal cord choline acetyltransferase and neurexin Ibeta gene expression followed by late-onset neuromotor deterioration. Proc Natl Acad Sci USA. 1997;94:8173–8178. doi: 10.1073/pnas.94.15.8173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dong H, Xiang YY, Farchi N, Ju W, Wu Y, Chen L, Wang Y, Hochner B, Yang B, Soreq H, et al. Excessive expression of acetylcholinesterase impairs glutamatergic synaptogenesis in hippocampal neurons. J Neurosci. 2004;24:8950–8960. doi: 10.1523/JNEUROSCI.2106-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Comoletti D, Flynn R, Jennings LL, Chubykin A, Matsumura T, Hasegawa H, Sudhof TC, Taylor P. Characterization of the interaction of a recombinant soluble neuroligin-1 with neurexin-1beta. J Biol Chem. 2003;278:50497–50505. doi: 10.1074/jbc.M306803200. [DOI] [PubMed] [Google Scholar]
- 16.Dean C, Scholl FG, Choih J, DeMaria S, Berger J, Isacoff E, Scheiffele P. Neurexin mediates the assembly of presynaptic terminals. Nat Neurosci. 2003;6:708–716. doi: 10.1038/nn1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Geppert M, Khvotchev M, Krasnoperov V, Goda Y, Missler M, Hammer RE, Ichtchenko K, Petrenko AG, Sudhof TC. Neurexin I alpha is a major alpha-latrotoxin receptor that cooperates in alpha-latrotoxin action. J Biol Chem. 1998;273:1705–1710. doi: 10.1074/jbc.273.3.1705. [DOI] [PubMed] [Google Scholar]
- 18.Ullrich B, Ushkaryov YA, Sudhof TC. Cartography of neurexins: more than 1000 isoforms generated by alternative splicing and expressed in distinct subsets of neurons. Neuron. 1995;14:497–507. doi: 10.1016/0896-6273(95)90306-2. [DOI] [PubMed] [Google Scholar]
- 19.Sugita S, Saito F, Tang J, Satz J, Campbell K, Sudhof TC. A stoichiometric complex of neurexins and dystroglycan in brain. J Cell Biol. 2001;154:435–445. doi: 10.1083/jcb.200105003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moore SA, Saito F, Chen J, Michele DE, Henry MD, Messing A, Cohn RD, Ross-Barta SE, Westra S, Williamson RA, et al. Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature. 2002;418:422–425. doi: 10.1038/nature00838. [DOI] [PubMed] [Google Scholar]
- 21.Knuesel I, Mastrocola M, Zuellig RA, Bornhauser B, Schaub MC, Fritschy JM. Short communication: altered synaptic clustering of GABAA receptors in mice lacking dystrophin (mdx mice) Eur J Neurosci. 1999;11:4457–4462. doi: 10.1046/j.1460-9568.1999.00887.x. [DOI] [PubMed] [Google Scholar]
- 22.Levi S, Grady RM, Henry MD, Campbell KP, Sanes JR, Craig AM. Dystroglycan is selectively associated with inhibitory GABAergic synapses but is dispensable for their differentiation. J Neurosci. 2002;22:4274–4285. doi: 10.1523/JNEUROSCI.22-11-04274.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Missler M, Hammer RE, Sudhof TC. Neurexophilin binding to alpha-neurexins. A single LNS domain functions as an independently folding ligand-binding unit. J Biol Chem. 1998;273:34716–34723. doi: 10.1074/jbc.273.52.34716. [DOI] [PubMed] [Google Scholar]
- 24.Beglopoulos V, Montag-Sallaz M, Rohlmann A, Piechotta K, Ahmad M, Montag D, Missler M. Neurexophilin 3 is highly localized in cortical and cerebellar regions and is functionally important for sensorimotor gating and motor coordination. Mol Cell Biol. 2005;25:7278–7288. doi: 10.1128/MCB.25.16.7278-7288.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hata Y, Davletov B, Petrenko AG, Jahn R, Sudhof TC. Interaction of synaptotagmin with the cytoplasmic domains of neurexins. Neuron. 1993;10:307–315. doi: 10.1016/0896-6273(93)90320-q. [DOI] [PubMed] [Google Scholar]
- 26.Hata Y, Butz S, Sudhof TC. CASK: a novel dlg/PSD95 homolog with an N-terminal calmodulin-dependent protein kinase domain identified by interaction with neurexins. J Neurosci. 1996;16:2488–2494. doi: 10.1523/JNEUROSCI.16-08-02488.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Biederer T, Sudhof TC. Mints as adaptors. Direct binding to neurexins and recruitment of munc18. J Biol Chem. 2000;275:39803–39806. doi: 10.1074/jbc.C000656200. [DOI] [PubMed] [Google Scholar]
- 28.Grootjans JJ, Reekmans G, Ceulemans H, David G. Syntenin–syndecan binding requires syndecan-synteny and the co-operation of both PDZ domains of syntenin. J Biol Chem. 2000;275:19933–19941. doi: 10.1074/jbc.M002459200. [DOI] [PubMed] [Google Scholar]
- 29.Irie M, Hata Y, Takeuchi M, Ichtchenko K, Toyoda A, Hirao K, Takai Y, Rosahl TW, Sudhof TC. Binding of neuroligins to PSD-95. Science. 1997;277:1511–1515. doi: 10.1126/science.277.5331.1511. [DOI] [PubMed] [Google Scholar]
- 30.Iida J, Hirabayashi S, Sato Y, Hata Y. Synaptic scaffolding molecule is involved in the synaptic clustering of neuroligin. Mol Cell Neurosci. 2004;27:497–508. doi: 10.1016/j.mcn.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 31.Meyer G, Varoqueaux F, Neeb A, Oschlies M, Brose N. The complexity of PDZ domain-mediated interactions at glutamatergic synapses: a case study on neuroligin. Neuropharmacology. 2004;47:724–733. doi: 10.1016/j.neuropharm.2004.06.023. [DOI] [PubMed] [Google Scholar]
- 32.Varoqueaux F, Jamain S, Brose N. Neuroligin 2 is exclusively localized to inhibitory synapses. Eur J Cell Biol. 2004;83:449–456. doi: 10.1078/0171-9335-00410. [DOI] [PubMed] [Google Scholar]
- 33.Song JY, Ichtchenko K, Sudhof TC, Brose N. Neuroligin 1 is a postsynaptic cell-adhesion molecule of excitatory synapses. Proc Natl Acad Sci USA. 1999;96:1100–1105. doi: 10.1073/pnas.96.3.1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rosales CR, Osborne KD, Zuccarino GV, Scheiffele P, Silverman MA. A cytoplasmic motif targets neuroligin-1 exclusively to dendrites of cultured hippocampal neurons. Eur J Neurosci. 2005;22:2381–2386. doi: 10.1111/j.1460-9568.2005.04400.x. [DOI] [PubMed] [Google Scholar]
- 35.Levinson JN, Chery N, Huang K, Wong TP, Gerrow K, Kang R, Prange O, Wang YT, El-Husseini A. Neuroligins mediate excitatory and inhibitory synapse formation: involvement of PSD-95 and neurexin-1beta in neuroligin-induced synaptic specificity. J Biol Chem. 2005;280:17312–17319. doi: 10.1074/jbc.M413812200. [DOI] [PubMed] [Google Scholar]
- 36.Fu Z, Washbourne P, Ortinski P, Vicini S. Functional excitatory synapses in HEK293 cells expressing neuroligin and glutamate receptors. J Neurophysiol. 2003;90:3950–3957. doi: 10.1152/jn.00647.2003. [DOI] [PubMed] [Google Scholar]
- 37.Sara Y, Biederer T, Atasoy D, Chubykin A, Mozhayeva MG, Sudhof TC, Kavalali ET. Selective capability of SynCAM and neuroligin for functional synapse assembly. J Neurosci. 2005;25:260–270. doi: 10.1523/JNEUROSCI.3165-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nam CI, Chen L. Postsynaptic assembly induced by neurexin-neuroligin interaction and neurotransmitter. Proc Natl Acad Sci USA. 2005;102:6137–6142. doi: 10.1073/pnas.0502038102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dean C, Dresbach T. Neuroligins and neurexins: linking cell adhesion, synapse formation and cognitive function. Trends Neurosci. 2006;29:21–29. doi: 10.1016/j.tins.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 40.Levinson JN, El-Husseini A. Building excitatory and inhibitory synapses: balancing neuroligin partnerships. Neuron. 2005;48:171–174. doi: 10.1016/j.neuron.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 41.Missler M, Sudhof TC. Neurexins: three genes and 1001 products. Trends Genet. 1998;14:20–26. doi: 10.1016/S0168-9525(97)01324-3. [DOI] [PubMed] [Google Scholar]
- 42.Rowen L, Young J, Birditt B, Kaur A, Madan A, Philipps DL, Qin S, Minx P, Wilson RK, Hood L, et al. Analysis of the human neurexin genes: alternative splicing and the generation of protein diversity. Genomics. 2002;79:587–597. doi: 10.1006/geno.2002.6734. [DOI] [PubMed] [Google Scholar]
- 43••.Boucard AA, Chubykin AA, Comoletti D, Taylor P, Sudhof TC. A splice code for trans-synaptic cell adhesion mediated by binding of neuroligin 1 to alpha- and beta-neurexins. Neuron. 2005;48:229–236. doi: 10.1016/j.neuron.2005.08.026.Along with [9•• ,44••], this study explored the functional effects of alternative splicing of neurexins and neuroligins. The authors reported that addition of the site 4 insert to β-neurexins selectively inhibits binding to +B neuroligin 1 but maintains high-affinity binding to −B neuroligins. α-Neurexins also bind specifically to −B neuroligins. Splicing at the neuroligin A site did not regulate binding to neurexins.
- 44••.Chih B, Gollan L, Scheiffele P. Alternative splicing controls selective trans-synaptic interactions of the neuroligin–neurexin complex. Neuron. 2006;51:171–178. doi: 10.1016/j.neuron.2006.06.005.Along with [9•• ,43••], this study explored the functional effects of alternative splicing of neurexins and neuroligins. These authors showed that addition of the B insert to neuroligin 1 reduced its ability to recruit GABAergic terminals in a neuron overexpression assay and increased its targeting to glutamatergic synapses. The effect of the B insert occurs by reduced binding to +S4 β-neurexin. Addition of the A insert to neuroligins was reported to increase targeting to GABAergic synapses and promote function of these synapses by unknown mechanisms.
- 45.Patzke H, Ernsberger U. Expression of neurexin Ialpha splice variants in sympathetic neurons: selective changes during differentiation and in response to neurotrophins. Mol Cell Neurosci. 2000;15:561–572. doi: 10.1006/mcne.2000.0853. [DOI] [PubMed] [Google Scholar]
- 46.Missler M, Zhang W, Rohlmann A, Kattenstroth G, Hammer RE, Gottmann K, Sudhof TC. Alpha-neurexins couple Ca2+ channels to synaptic vesicle exocytosis. Nature. 2003;424:939–948. doi: 10.1038/nature01755. [DOI] [PubMed] [Google Scholar]
- 47••.Varoqueaux F, Aramuni G, Rawson RL, Mohrmann R, Missler M, Gottmann K, Zhang W, Sudhof TC, Brose N. Neuroligins determine synapse maturation and function. Neuron. 2006;51:741–754. doi: 10.1016/j.neuron.2006.09.003.This is a landmark paper showing that neuroligins are essential for synapse function in vivo. Although individual neuroligin knockout mice are viable, Nlgn1;Nlgn2;Nlgn3 triple knockout mice die at birth owing to respiratory failure. Their synapses seem to be morphologically normal, but transmission was dramatically reduced at GABAergic/glycinergic synapses, and also reduced at glutamatergic synapses, in the brainstem. This was accompanied by reduced clustering of GABAA receptors, reduced levels of synaptic vesicle proteins and a slight reduction in the ratio of VGAT puncta to VGlut puncta, but clustering of the scaffolding proteins PSD-95 and gephyrin was normal.
- 48.Zhang W, Rohlmann A, Sargsyan V, Aramuni G, Hammer RE, Sudhof TC, Missler M. Extracellular domains of alpha-neurexins participate in regulating synaptic transmission by selectively affecting N- and P/Q-type Ca2+ channels. J Neurosci. 2005;25:4330–4342. doi: 10.1523/JNEUROSCI.0497-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gale NW, Holland SJ, Valenzuela DM, Flenniken A, Pan L, Ryan TE, Henkemeyer M, Strebhardt K, Hirai H, Wilkinson DG, et al. Eph receptors and ligands comprise two major specificity subclasses and are reciprocally compartmentalized during embryogenesis. Neuron. 1996;17:9–19. doi: 10.1016/s0896-6273(00)80276-7. [DOI] [PubMed] [Google Scholar]
- 50.Okabe S, Miwa A, Okado H. Spine formation and correlated assembly of presynaptic and postsynaptic molecules. J Neurosci. 2001;21:6105–6114. doi: 10.1523/JNEUROSCI.21-16-06105.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gerrow K, Romorini S, Nabi SM, Colicos MA, Sala C, El-Husseini A. A preformed complex of postsynaptic proteins is involved in excitatory synapse development. Neuron. 2006;49:547–562. doi: 10.1016/j.neuron.2006.01.015. [DOI] [PubMed] [Google Scholar]
- 52.Craig AM, Graf ER, Linhoff MW. How to build a central synapse: clues from cell culture. Trends Neurosci. 2006;29:8–20. doi: 10.1016/j.tins.2005.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Biederer T, Sara Y, Mozhayeva M, Atasoy D, Liu X, Kavalali ET, Sudhof TC. SynCAM, a synaptic adhesion molecule that drives synapse assembly. Science. 2002;297:1525–1531. doi: 10.1126/science.1072356. [DOI] [PubMed] [Google Scholar]
- 54.Dalva MB, Takasu MA, Lin MZ, Shamah SM, Hu L, Gale NW, Greenberg ME. EphB receptors interact with NMDA receptors and regulate excitatory synapse formation. Cell. 2000;103:945–956. doi: 10.1016/s0092-8674(00)00197-5. [DOI] [PubMed] [Google Scholar]
- 55.Wang CY, Chang K, Petralia RS, Wang YX, Seabold GK, Wenthold RJ. A novel family of adhesion-like molecules that interacts with the NMDA receptor. J Neurosci. 2006;26:2174–2183. doi: 10.1523/JNEUROSCI.3799-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ko J, Kim S, Chung HS, Kim K, Han K, Kim H, Jun H, Kaang BK, Kim E. SALM synaptic cell adhesion-like molecules regulate the differentiation of excitatory synapses. Neuron. 2006;50:233–245. doi: 10.1016/j.neuron.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 57.Kim S, Burette A, Chung HS, Kwon SK, Woo J, Lee HW, Kim K, Kim H, Weinberg RJ, Kim E. NGL family PSD-95-interacting adhesion molecules regulate excitatory synapse formation. Nat Neurosci. 2006;9:1294–1301. doi: 10.1038/nn1763. [DOI] [PubMed] [Google Scholar]
- 58.Chih B, Afridi SK, Clark L, Scheiffele P. Disorder-associated mutations lead to functional inactivation of neuroligins. Hum Mol Genet. 2004;13:1471–1477. doi: 10.1093/hmg/ddh158. [DOI] [PubMed] [Google Scholar]
- 59.Comoletti D, De Jaco A, Jennings LL, Flynn RE, Gaietta G, Tsigelny I, Ellisman MH, Taylor P. The Arg451Cys-neuroligin-3 mutation associated with autism reveals a defect in protein processing. J Neurosci. 2004;24:4889–4893. doi: 10.1523/JNEUROSCI.0468-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chubykin AA, Liu X, Comoletti D, Tsigelny I, Taylor P, Sudhof TC. Dissection of synapse induction by neuroligins: effect of a neuroligin mutation associated with autism. J Biol Chem. 2005;280:22365–22374. doi: 10.1074/jbc.M410723200. [DOI] [PubMed] [Google Scholar]
- 61.Laumonnier F, Bonnet-Brilhault F, Gomot M, Blanc R, David A, Moizard MP, Raynaud M, Ronce N, Lemonnier E, Calvas P, et al. X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am J Hum Genet. 2004;74:552–557. doi: 10.1086/382137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yan J, Oliveira G, Coutinho A, Yang C, Feng J, Katz C, Sram J, Bockholt A, Jones IR, Craddock N, et al. Analysis of the neuroligin 3 and 4 genes in autism and other neuropsychiatric patients. Mol Psychiatry. 2005;10:329–332. doi: 10.1038/sj.mp.4001629. [DOI] [PubMed] [Google Scholar]
- 63.Vincent JB, Kolozsvari D, Roberts WS, Bolton PF, Gurling HM, Scherer SW. Mutation screening of X-chromosomal neuroligin genes: no mutations in 196 autism probands. Am J Med Genet B Neuropsychiatr Genet. 2004;129:82–84. doi: 10.1002/ajmg.b.30069. [DOI] [PubMed] [Google Scholar]
- 64.Gauthier J, Bonnel A, St-Onge J, Karemera L, Laurent S, Mottron L, Fombonne E, Joober R, Rouleau GA. NLGN3/NLGN4 gene mutations are not responsible for autism in the Quebec population. Am J Med Genet B Neuropsychiatr Genet. 2005;132:74–75. doi: 10.1002/ajmg.b.30066. [DOI] [PubMed] [Google Scholar]
- 65.Blasi F, Bacchelli E, Pesaresi G, Carone S, Bailey AJ, Maestrini E. Absence of coding mutations in the X-linked genes neuroligin 3 and neuroligin 4 in individuals with autism from the IMGSAC collection. Am J Med Genet B Neuropsychiatr Genet. 2006;141:220–221. doi: 10.1002/ajmg.b.30287. [DOI] [PubMed] [Google Scholar]
- 66.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14(33–38):27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]