

Protein aggregation, a well-known culprit in human disease,1,2 is also a major problem facing the use of proteins as therapeutic or diagnostic agents.3,4 Insights into the protein aggregation problem have been garnered from the study of natural proteins, where a relationship between solubility and net charge has been noted. For example, it is known that proteins are least soluble at their isoelectric point, where they bear a net charge of zero.5 More recently, small differences in net charge (±3 charge units) have been shown to predict aggregation tendencies among peptide variants.6 In addition, intrinsically disordered proteins,7,8 a class of proteins that are largely unfolded in the cell but that do not lead to pathological aggregation, tend to have large net charges.9,10
We speculated that the relationship between net charge and aggregation resistance might also be applicable to globular proteins, which can aggregate via partial unfolding induced by thermal agitation, chemical treatment, or conformational breathing. Recent evidence that some proteins can tolerate significant changes in net charge (for example, the finding that carbonic anhydrase retains activity after exhaustive acetylation of its surface lysines11) encouraged us to test the hypothesis that the solubility and aggregation resistance of some proteins might be significantly enhanced, without abolishing their folding or function, by extensively mutating their surfaces to dramatically increase their net charge, a process we refer to as “supercharging”.
We began with green fluorescent protein (GFP), an easily assayed protein that undergoes chromophore maturation and becomes fluorescent only when folded correctly. To minimize the possibility that our GFP was unusually delicate and therefore unusually easy to improve, we used a starting GFP (stGFP) based on the state-of-the-art GFP variant called “superfolder”, which has been highly optimized for folding robustness and resistance to denaturants.12 The net charge of the stGFP is −7, similar to that of wild-type GFP (−8). To create a superpositive variant of GFP, we identified 29 positions in the crystal structure that were highly solvent-exposed and mutated these to positively charged amino acids (Lys and Arg), yielding a design with a theoretical net charge of +36 (Figure 1 and Supporting Information). Genes encoding stGFP and GFP(+36) were constructed and expressed in E. coli, and both genes yielded intensely green fluorescent bacteria. Following protein purification, the fluorescence properties of GFP(+36) were measured and found to be very similar to those of stGFP (Supporting Information).
Figure 1.
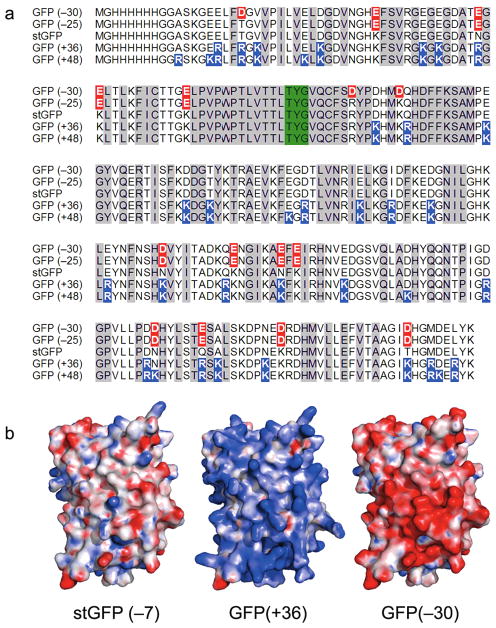
Supercharged green fluorescent proteins (GFPs). (a) Protein sequences of GFP variants, with fluorophore-forming residues highlighted green, negatively charged residues highlighted red, and positively charged residues highlighted blue. (b) Electrostatic surface potentials of the starting GFP (stGFP), GFP(+36), and GFP(−30), colored from −25 kT/e (red) to +25 kT/e (blue).
Encouraged by this finding, we produced and characterized additional supercharged GFPs having net charges of +48, −25, and −30, all of which were also found to have stGFP-like fluorescence (Figures 2a and S1). All supercharged GFP variants exhibited circular dichroism spectra similar to that of stGFP, indicating that the proteins have similar secondary structure content (Figure 2b). The thermodynamic stabilities of the supercharged GFP variants were only modestly lower than that of stGFP (1.0–4.1 kcal/mol, Figure 2c and Table S1) despite the presence of as many as 36 mutations.
Figure 2.
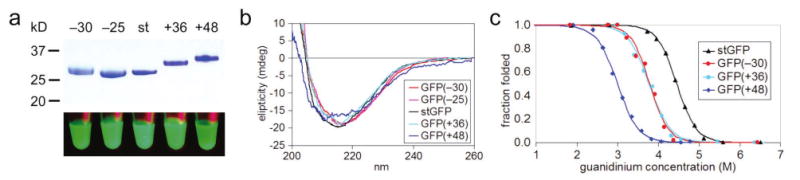
Intramolecular properties of GFP variants. (a) Staining and UV fluorescence of purified GFP variants. Each lane and tube contains 0.2 μg of protein. (b) Circular dichroism spectra of GFP variants. (c) Thermodynamic stability of GFP variants, measured by guanidinium-induced unfolding.
We next examined the effect of supercharging on aggregation resistance. Although stGFP is the product of a long history of GFP optimization,13 like most proteins it can be induced to aggregate by thermal or chemical unfolding. Heating stGFP to 100 °C induced its quantitative precipitation and the irreversible loss of fluorescence (Figures 3a and S2). In contrast, supercharged GFP(+36) and GFP(−30) remained soluble when heated to 100 °C and recovered significant fluorescence (62 and 28% of their initial fluorescence, respectively) upon cooling (Figure 3a). When mixed with 40% TFE, an additive commonly used to examine chemically induced protein aggregation, stGFP lost all fluorescence and began to visibly aggregate (Figures 3b and S3). Within half an hour at 25 °C, aggregation of stGFP was complete as followed by right-angle light scattering. The +36 and −30 supercharged GFP variants also became completely nonfluorescent when exposed to 40% TFE; however, in contrast to stGFP, they suffered no significant aggregation under the same conditions even after several hours (Figure 3b). Taken together, the results of these studies indicate that supercharged GFPs, upon thermal or chemical denaturation, remain entirely soluble.
Figure 3.
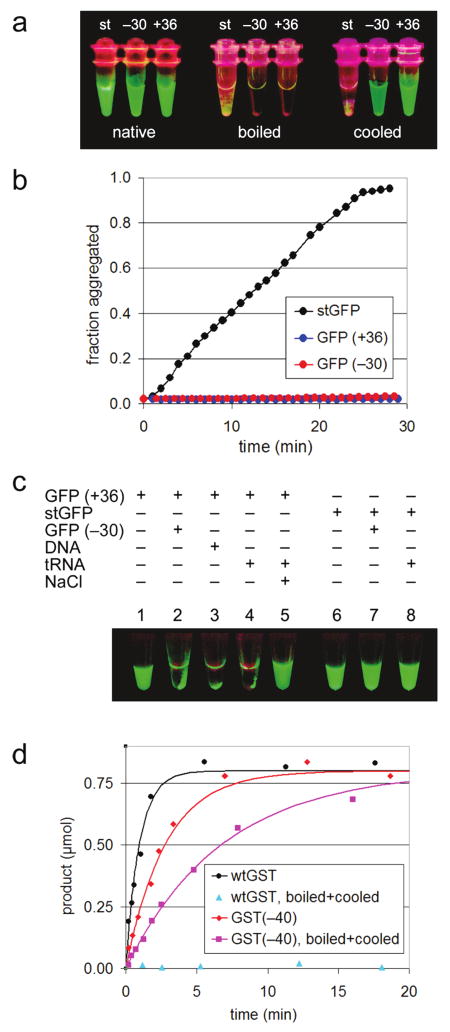
(a–c) Intermolecular properties of supercharged GFPs. (a) UV-illuminated samples of purified GFP variants (“native”), those samples heated 1 min at 100 °C (“boiled”), and those samples subsequently cooled for 2 h at 25 °C (“cooled”). (b) Aggregation of GFP variants was induced with 40% TFE at 25 °C and monitored by right-angle light scattering. (c) Supercharged GFPs adhere reversibly to oppositely charged macromolecules. Sample 1: 6 μg of GFP(+36) in 30 μL of 25 mM Tris pH 7.0 and 100 mM NaCl. Sample 2: 6 μg of GFP(−30) added to sample 1. Sample 3: 30 μg of salmon sperm DNA added to sample 1. Sample 4: 20 μg of E. coli tRNA added to sample 1. Sample 5: addition of NaCl to 1 M of sample 4. Samples 6–8: identical to samples 1, 2, and 4, respectively, except using stGFP instead of GFP(+36). All samples were spun briefly in a microcentrifuge and visualized under UV light. (d) Enzymatic activity of GST variants. Reactions contained 0.1 mg/mL of GST variant, 4 mM chloro-dinitrobenzene, 4 mM glutathione, and 100 mM potassium phosphate pH 6.5. Product formation was monitored at 340 nm, yielding reaction rates (vinit) of 1.0 μmol/min for wild-type GST, 0.36 μmol/min for GST(−40), and 0.15 μmol/min for GST(−40) after being boiled and cooled.
In addition to this aggregation resistance, supercharged GFP variants show a strong, reversible avidity for highly charged macromolecules of the opposite charge (Figures 3c and S4). When mixed together in 1:1 stoichiometry, GFP(+36) and GFP(−30) immediately formed a green fluorescent co-precipitate, indicating the association of folded, functional proteins. GFP(+36) similarly co-precipitated with high concentrations of RNA or DNA. The addition of NaCl was sufficient to dissolve these complexes, consistent with the electrostatic basis of their formation. In contrast, stGFP was unaffected by the addition of GFP(−30), RNA, or DNA (Figure 3c).
Because the robust folding of superfolder GFP together with its monomeric nature may have contributed to the ability of stGFP to be supercharged, we next sought to determine whether the supercharging principle could apply to proteins other than GFP, including non-monomeric proteins with binding or catalytic activities. To this end, we applied the supercharging process to two proteins unrelated to GFP.
Streptavidin is a tetramer with a total net charge of −4. Using an entirely automated version of our mutagenesis strategy (Supporting Information), we designed two supercharged streptavidin variants with theoretical net charges of −40 or +52 and expressed and purified the proteins (Figure S5). Both were capable of forming tetramers, as judged by analytical size-exclusion chromatography (Figure S10), and both had significant, albeit greatly reduced, biotin-binding capacity (Figure S11). Like the supercharged GFPs, the supercharged streptavidins were resistant to thermally induced aggregation and underwent no significant aggregation after boiling; in contrast, wild-type streptavidin was 93% aggregated after heating to 100 °C for 1 min (Figure S7 and Table S1).
Glutathione-S-transferase (GST), a dimer with a total net charge of +2, was supercharged to yield a dimer with net charge of −40 (Figure S5). Purified GST(−40) was able to form dimers (Figure S8) and exhibited comparable KM and only 3-fold lower kcat than that of the wild-type enzyme (wtGST: KM = 0.3 ± 0.1 mM, kcat = 46 s−1; GST(−40): KM = 0.25 ± 0.05 mM, kcat = 16 s−1) (Figure S9). Moreover, supercharged GST remained soluble when heated to 100 °C, in contrast to its wild-type counterpart which, like stGFP and wild-type streptavidin, precipitated quantitatively and irreversibly (Figure S7). In addition, GST(−40) recovered 40% of its catalytic activity upon cooling (Figure 3d), unlike wtGST, which was irreversibly aggregated and had no detectable residual activity after cooling.
In summary, we have demonstrated that monomeric and multimeric proteins of varying structures and functions can be “supercharged” by simply replacing their most solvent-exposed residues with like-charged amino acids. Supercharging greatly alters the intermolecular properties of proteins, imparting aggregation resistance and the ability to associate in folded form with oppositely charged macromolecules like “molecular Velcro” (Figure 3c). We note that these unusual intermolecular properties arise from high net charge, rather than from the total number of charged amino acids, which was not significantly changed by the supercharging process (Table S1).
In contrast to the substantial intermolecular effects we observed, the intramolecular properties of the seven supercharged proteins studied here were largely intact: specifically, the supercharged GFPs retained the ability to assume a fluorescent native-like state; the supercharged GST remained dimeric and retained catalytic activity; and the supercharged streptavidins retained some biotin affinity and their propensity to form tetramers. Supercharging therefore may represent a useful approach for reducing the aggregation tendency and improving the solubility of some proteins without abolishing their function.
The high charge density on the surface of the supercharged proteins is likely to perturb the pKa values of some of the charged residues, potentially lowering the actual magnitude of net charge. We found some evidence for this effect: for example, GST(−40), with a predicted pI of 4.8, migrated as a protein with a pI of 5.5 on an isoelectric focusing (IEF) gel (Figure S6); however, GST(+36), with a predicted pI of 10.4, indeed failed to migrate into an IEF gel with a nominal pI range of 3–10. Further, not all supercharged proteins are readily accessible. An early design for a supernegative GFP did not yield fluorescent protein, and a gene encoding a superpositive GST (Supporting Information) failed to express in E. coli.
Protein supercharging illustrates the plasticity of protein surfaces and highlights the opportunities that arise from the mutational tolerance of solvent-exposed residues. For example, it was recently shown that the thermodynamic stability of some proteins can be enhanced by rationally engineering charge–charge interactions,14 and that introducing a positively charged patch of amino acids onto the side of GFP endowed the protein with cell permeability.15 Protein supercharging demonstrates how this plasticity can be exploited in a different way to impart robust resistance to protein aggregation. Our findings are consistent with the results of a complementary study in which removal of all charges from ubiquitin did not prevent folding but significantly impaired its solubility.16
The principles revealed here may be particularly useful in de novo protein design efforts, where unpredictable protein handling properties including aggregation remain a significant challenge.17 In light of the above results of supercharging natural proteins, it is tempting to speculate that the aggregation resistance of designed proteins could also be improved by biasing the design process to increase the frequency of like-charged amino acids at positions predicted to lie on the outside of the folded protein.
These observations may also illuminate the modest net-charge distribution of natural proteins.18,19 The net charge of 84% of Protein Data Bank (PDB) polypeptides, for example, falls within ±10. Our results argue against the hypothesis that high net charge creates sufficient electrostatic repulsion to force unfolding. Indeed, GFP(+48) has a higher positive net charge than any polypeptide currently in the PDB, yet retains the ability to fold and fluoresce. Supercharged proteins have a charge distribution reminiscent of micelles, which are stabilized by colloidal forces that are able to overcome charge–charge repulsion.20 Instead, our findings suggest that nonspecific intermolecular adhesions may have disfavored the evolution of too many highly charged natural proteins. Consistent with this hypothesis, almost all natural proteins with very high net charge, such as ribosomal proteins L3 (+36) and L15 (+44), which bind RNA, or calsequestrin (−80), which binds calcium cations, associate with oppositely charged species as part of their essential cellular functions.
Supplementary Material
Acknowledgments
This work was supported by the NIH/ NIGMS (R01 GM65400) and the Howard Hughes Medical Institute.
Footnotes
Supporting Information Available: Details of supercharging design algorithm, experimental methods, and supporting data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Cohen FE, Kelly JW. Nature. 2003;426(6968):905–9. doi: 10.1038/nature02265. [DOI] [PubMed] [Google Scholar]
- 2.Chiti F, Dobson CM. Annu Rev Biochem. 2006;75:333–66. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 3.Fowler SB, Poon S, Muff R, Chiti F, Dobson CM, Zurdo J. Proc Natl Acad Sci USA. 2005;102(29):10105–10. doi: 10.1073/pnas.0501215102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Frokjaer S, Otzen DE. Nat Rev Drug Discovery. 2005;4(4):298–306. doi: 10.1038/nrd1695. [DOI] [PubMed] [Google Scholar]
- 5.Loeb J. J Gen Physiol. 1921;4:547–555. doi: 10.1085/jgp.3.4.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chiti F, Stefani M, Taddei N, Ramponi G, Dobson CM. Nature. 2003;424(6950):805–8. doi: 10.1038/nature01891. [DOI] [PubMed] [Google Scholar]
- 7.Fink AL. Curr Opin Struct Biol. 2005;15(1):35–41. doi: 10.1016/j.sbi.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 8.Sugase K, Dyson H, Wright P. Nature. 2007;447:1021–5. doi: 10.1038/nature05858. [DOI] [PubMed] [Google Scholar]
- 9.Dosztanyi Z, Csizmok V, Tompa P, Simon I. J Mol Biol. 2005;347(4):827–39. doi: 10.1016/j.jmb.2005.01.071. [DOI] [PubMed] [Google Scholar]
- 10.Pawar AP, Dubay KF, Zurdo J, Chiti F, Vendruscolo M, Dobson CM. J Mol Biol. 2005;350(2):379–92. doi: 10.1016/j.jmb.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 11.Gudiksen KL, Gitlin I, Yang J, Urbach AR, Moustakas DT, Whitesides GM. J Am Chem Soc. 2005;127(13):4707–14. doi: 10.1021/ja043804d. [DOI] [PubMed] [Google Scholar]
- 12.Pedelacq JD, Cabantous S, Tran T, Terwilliger TC, Waldo GS. Nat Biotechnol. 2006;24(1):79–88. doi: 10.1038/nbt1172. [DOI] [PubMed] [Google Scholar]
- 13.Giepmans BN, Adams SR, Ellisman MH, Tsien RY. Science. 2006;312(5771):217–24. doi: 10.1126/science.1124618. [DOI] [PubMed] [Google Scholar]
- 14.Strickler SS, Gribenko AV, Keiffer TR, Tomlinson J, Reihle T, Loladze VV, Makhatadze GI. Biochemistry. 2006;45(9):2761–6. doi: 10.1021/bi0600143. [DOI] [PubMed] [Google Scholar]
- 15.Fuchs SM, Raines RT. ACS Chem Biol. 2007;2(3):167–70. doi: 10.1021/cb600429k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loladze VV, Makhatadze GI. Protein Sci. 2002;11(1):174–7. doi: 10.1110/ps.29902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baker D. Personal communication [Google Scholar]
- 18.Knight CG, Kassen R, Hebestreit H, Rainey PB. Proc Natl Acad Sci USA. 2004;101(22):8390–5. doi: 10.1073/pnas.0307270101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gitlin I, Carbeck JD, Whitesides GM. Angew Chem, Int Ed. 2006;45(19):3022–60. doi: 10.1002/anie.200502530. [DOI] [PubMed] [Google Scholar]
- 20.Russel W, Saville D, Schowalter W. Colloidal Dispersions. Cambridge University Press; New York: 1989. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.