

Abstract
Studies suggest that the inflammatory cytokine, TNF-α plays a role in the prognosis of end-stage renal diseases. We have previously shown that TNF-α inhibition slowed the progression of hypertension and renal damage in angiotensin II salt-sensitive hypertension. Thus, we hypothesize that TNF-α contributes to renal inflammation in a model of mineralocorticoid-induced hypertension. Four groups of rats (n=5-6) were studied for 3 weeks with the following treatments 1) placebo, 2) placebo + TNF-α inhibitor, etanercept (1.25 mg/kg/day, sc), 3) deoxycorticosterone acetate plus 0.9 % NaCl to drink (DOCA-salt), or 4) DOCA-salt + etanercept. Mean arterial blood pressure (MAP) measured by telemetry increased in DOCA-salt rats compared to baseline (177±4 vs. 107±3 mmHg, P<0.05) and TNF-α inhibition had no effect in the elevation of MAP in these rats (177±8 mmHg). Urinary protein excretion significantly increased in DOCA-salt rats compared to placebo (703±76 vs. 198±5 mg/day, respectively); etanercept lowered the proteinuria (514±64 mg/day, P < 0.05 vs. DOCA-salt alone). Urinary albumin excretion followed a similar pattern in each group. Urinary MCP-1 and ET-1 excretion were also increased in DOCA-salt rats compared to placebo (MCP-1: 939±104 vs. 43±7 ng/day, and ET-1: 3.30±0.29 vs. 1.07±0.03 fmol/day, respectively, both P<0.05); TNF-α inhibition significantly decreased both MCP-1 and ET-1 excretion (409±138 ng/day and 2.42±0.22 fmol/day, respectively, both P < 0.05 vs. DOCA-salt alone). Renal cortical NFκB activity also increased in DOCA-salt hypertensive rats and etanercept treatment significantly reduced this effect. These data support the hypothesis that TNF-α contributes to the increase in renal inflammation in DOCA-salt rats.
Keywords: salt, DOCA, renal inflammation, blood pressure, TNF-α, etanercept, NFκB
Introduction
Recently, inflammatory cytokines have been shown to play a role in the pathogenesis of hypertension and its associated end-stage renal disease (7). TNF-α is an inflammatory cytokine that is synthesized primarily by monocytes and macrophages (26). The effects of TNF-α are mediated by specific cell surface receptors; an epithelial cell-type receptor (TNF-R1) and a myeloid cell-type receptor (TNF-R2) that is mainly expressed in immune and endothelial cells (20,26). TNF binding to its receptor activates a number of signaling pathways and increases expression of other cytokines such as MCP-1, IL-6, IL-8, PDGF, and fibronectin, growth factors and cell adhesion molecules leading to vascular inflammation (26). Studies have demonstrated that TNF-α levels are elevated in renal diseases such as glomerulosclerosis, immune complex glomerulonephritis and aminoglycoside nephritis (26). Furthermore, blocking TNF-α activity with neutralizing antibodies or receptor blocker in a variety of models results in reduced renal damage (13,15,18). These data suggest that TNF-α plays an important role in the pathophysiology of renal disease.
The deoxycorticosterone acetate (DOCA)-salt hypertensive rat is a well-established model of mineralcorticoid hypertension with renal dysfunction (1). DOCA-salt hypertension is associated with increased plasma and kidney endothelin-1 (ET-1) levels that are thought to contribute to the increase in blood pressure and oxidative stress via the stimulation of ETA and ETB2 receptors (1,5,8,23). Previous studies also demonstrated that plasma and tissue TNF-α levels are increased in DOCA-salt rats (10) and TNF-α stimulates (ET-1) production (17,21). We have previously shown that TNF-α participates in blood pressure elevation and renal injury in angiotensin II-dependent hypertension (9). Therefore, we hypothesize in the current study that TNF-α contributes to hypertension and its associated renal inflammation in mineralocorticoid-dependent hypertension.
Methods
Experiments were conducted using male Sprague–Dawley rats (200–250 g, Harlan Laboratories, Indianapolis, IN). Animal protocols were in accordance with National Institutes of Health guidelines and approved by the Medical College Institutional Animal Care and Use Committee. Rats were fed a normal rat chow diet (Harlan Teklad Global diets, Wilmington, DE) throughout the experiment and were housed under conditions of constant temperature and humidity with a 12:12-h light–dark cycle. Rats were allowed to adapt to these conditions for several days before starting any experimental procedures.
Rats were anesthetized with 100 mg/kg ketamine/20 mg/kg xylazine (i.p.). A right nephrectomy was performed via a retroperitoneal incision. Rats were implanted subcutaneously with either 60-day time-release DOCA or placebo pellet (200 mg; Innovative Research of America, Sarasota, FL). Systolic arterial pressure was recorded using the tail cuff method (n=5–6 per group) as previously described (30). Tail cuff pressure was measured prior to implantation of DOCA pellets and once a week until the end of the experiment 3 weeks later. Following recovery from surgery, rats were classified into 4 groups: 1) placebo, 2) placebo/ plus the TNF-α inhibitor, etanercept, 3) DOCA treatment, and 4), DOCA/ plus etanercept. In addition, DOCA-rats were also given 0.9% NaCl to drink. Etanercept inhibits TNF-α binding to the TNF-α receptor (11). Etanercept was delivered subcutaneously in alzet mini-osmotic pumps at the time of pellets implantation at a dose of 1.25 mg/kg/day (Cupertino, CA). This dose has been shown previously to reduce renal damage effectively in angiotensin II hypertension (9,24). After a 3-week period, rats were placed in metabolic cages for 24 hour urine collection. Rats were then anesthetized with sodium pentobarbital (65 mg/kg ip; Abbott Laboratories), and a terminal blood sample was taken from the abdominal aorta for determination of plasma 8-isoprostane (Cayman Chemical, Ann Arbor, Michigan). Renal cortex was also collected, subdivided into three tubes, and snap frozen in liquid nitrogen for NFκB analysis, real time PCR, and western blotting. Urinary protein concentration was determined using bicinchoninic acid protein assay (Pierce, Rockford, IL) and albumin concentration was measured using a highly sensitive immunoassay SPI-BIO kit (Cayman Chemical, Ann Arbor, Michigan). Urinary MCP-1 concentration was also determined using a commercially available immunoassay kit (BD Biosciences, San Jose, CA) and urinary immunoreactive endothelin () concentration was measured by radioimmunoassay (Amersham Pharmacia Biotech, Arlington Heights, IL).
NFκB transcription factor assay
Whole-cell lysates were obtained from kidney cortex from the above-mentioned groups using the nuclear extract kit (Active Motif, Carlsbad, CA). Protein concentrations were determined using a bicinchoninic acid protein assay (Pierce, Rockford, IL). Twenty µg of whole-cell extract was used for the determination of NFκB activity using the TransAM NFκB p65 transcription factor assay kit (Active Motif, Carlsbad, CA).
Real-time Polymerase Chain Reaction
Total RNA was also isolated from 100 mg of kidney cortex using ultra-pure Trizol reagent according to the procedure described by the manufacturer (GIBCO-BRL, Grand Island, NY) and RNA concentration was determined and equal amounts of total RNA (3 μg) was used in the reverse transcription (RT) using the iScript cDNA synthesis kit (Bio-Rad Laboratories, Hercules, CA). Real-time polymerase chain reaction (PCR) was used to quantify the mRNA expression of NFκB, ICAM-1, VCAM-1, TNF-α, and TGF-β. Amplification was performed using iQ Supermix with the iCycler iQ Real-Time Detection System according to the manufacturers instructions (Bio-Rad Laboratories, Hercules, CA). A standard curve was generated for each primer pair and probe to determine PCR efficiency. Mean threshold cycle (Ct) values for each sample were normalized to GAPDH and calibrated to the control group to obtain the threshold cycle difference (ΔΔCt) with 2-ΔΔCt being the fold change relative to the control group.
Probes and primer sequences were used as follows: NFκB probe 5’-FAM-CGC GAT CAC TAA ATC CAA CAC AGG CAT CAC CCA GAT CGC G-BHQ-3’; NFκB forward 5’-GTA TGG CTT CCC GCA CTA TGG-3’; NFκB reverse 5’-TCG TCA CTC TTG GCA CAA TCT C-3’; ICAM-1 probe 5’-FAM- CCT CCT CCT GAG CCT TCT GTA ACT TGT A-BHQ-3’; ICAM-1 Forward 5’-GTA CTG ATC ATT GCG GGC TT- 3’; ICAM-1 reverse 5’-GGG GCT TGT ACC TTG AGT TT-3’; TGF-B probe 5’-FAM-AGC CCT GTA TTC CGT CTC CTT GGT TC-BHQ-3’; TGF-β forward 5’-AGT CAA CTG TGG AGC AAC AC-3’; TGF-β reverse 5’-ACA AGA GCA GTG AGC ACT GA-3’; VCAM-1 probe 5’-FAM-CCA TCA CTT GAG CAG GTC AGG TTC ACA-BHQ-3’; VCAM-1 forward 5’-CCC AAA CAA AGG CAG AGT AC-3’; VCAM-1 reverse 5’-CCA CAG GAT TTT GGG AGT TG-3’; TNF-α probe 5’-FAM-CGT CGT AGC AAA CCA CCA AGC GGA G-BHQ-3’; TNF-α forward 5’-AGT GAC AAG CCC GTA GCC-3’; TNF-α reverse 5’-GGG TGA GGA GCA CGT AGT C-3’; GAPDH probe 5’-FAM-ACT CCA CGA CAT ACT CAG CAC CAG CA-BHQ-3’; GAPDH forward 5’-CAC GGC AAG TTC AAC GGC-3’; GAPDH reverse 5’-GGT GGT GAA GAC GCC AGT A-3’.
Homogenization of Renal Cortex for Protein Expression
Kidney cortex was homogenized in homogenization buffer containing protease inhibitors and VCAM-1, ICAM-1, TNF-α, and TGF-β protein expression were determined by western blotting as previously described (12). The primary antibodies used were rabbit anti-VCAM-1 (1:500; Santa Cruz, CA), goat anti-rat ICAM-1 (1:500; R&D, Minneapolis, MN), rabbit anti-TNF-α (1:200, Cell Signaling, MA), rabbit anti-TGF-β (1:2000, Santa Cruz, CA), and mouse anti-ß-actin (1:10000, Sigma, MO), respectively. Goat anti-rabbit (Santa Cruz, CA) was used as a secondary antibody for VCAM-1, TNF-α, and TGF-β, donkey anti-goat was used as a secondary antibody for ICAM-1 (Santa Cruz, CA), and goat anti-mouse was used as a secondary antibody for ß-actin (Santa Cruz, CA). Band intensity was measured densitometrically using Scion Image software and the values were normalized to ß-actin.
Telemetry experiment
To more accurately detect any subtle change in blood pressure, telemetry transmitters (Data Sciences) were implanted in a separate series of rats 2 weeks prior to DOCA-pellets implantation according to manufacturer’s specifications while under sodium pentobarbital anesthesia as previously described (29). In brief, a midline incision was used to expose the abdominal aorta that was occluded to allow insertion of the transmitter catheter. The catheter was secured in place with tissue glue and the transmitter body was sutured to the abdominal wall along the incision line as the incision was closed. The skin was closed with staples that were removed 7 days later after the incision had healed. Rats were allowed to recover from surgery and were returned to individual housing. A baseline arterial pressure was recorded for one week prior to DOCA pellets implantation. A right nephrectomy was performed and rats were implanted subcutaneously with DOCA pellets with or without etanercept treatment for three weeks as previously mentioned (n=6 per group). Mean arterial pressure was continuously recorded in the last two groups for 3 weeks.
Statistical analysis
All data are presented as mean ± SEM. Arterial blood pressure data were analyzed using ANOVA for repeated measures combined with post hoc contrasts. Other data were analyzed using one-way analysis of variance (ANOVA) followed by Tukey’s post-hoc test for multiple group comparisons. Differences were considered statistically significant with P< 0.05 compared to the control. Analyses were performed using GraphPad Prism Version 4.0 software.
Results
Systolic blood pressure increased in DOCA-salt hypertensive rats compared to placebo group and TNF-α inhibition with etanercept treatment did not significantly affect blood pressure in either placebo or DOCA-salt rats (Figure 1A). Consistent with the tail cuff pressure data, DOCA-salt treatment significantly increased MAP when measured by telemetry (180 ± 5 mm Hg on day 21 vs. 107 ± 3 baseline, P<0.05) and etanercept treatment did not affect the increase in MAP in DOCA-salt hypertensive rats (Figure 1B).
Figure 1.
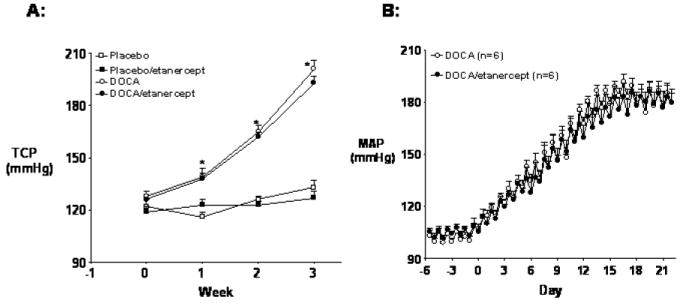
Effect of the TNF-α inhibitor, etanercept, on arterial pressure in DOCA-salt and placebo-treated rats. (A) Systolic blood pressure in placebo and DOCA-salt rats with or without etanercept treatment for 3 weeks. (B) Twelve hour average mean arterial pressure (MAP) in DOCA-salt hypertensive rats with or without etanercept treatment for 3 weeks. Values are means±SEM (n=5-6 per groups). * P< 0.05 vs. baseline values.
Excretion of protein, albumin, and MCP-1 was used as an indirect measure of renal injury. Urinary protein excretion increased 3.5 fold in DOCA-salt hypertensive rats compared to the placebo group and etanercept treatment tended to lower protein excretion in DOCA-salt rats although this change was not significant (Figure 2A). A similar pattern was observed in albumin excretion (Figure 2B). Urinary MCP-1 excretion increased roughly 10 fold in DOCA-salt hypertensive rats and TNF-α inhibition significantly attenuated this increase (Figure 2C). Urinary endothelin excretion also increased 10 fold in DOCA-salt hypertensive rats, and again, etanercept treatment significantly lowered endothelin excretion in DOCA-salt rats (Figure 2D).
Figure 2.
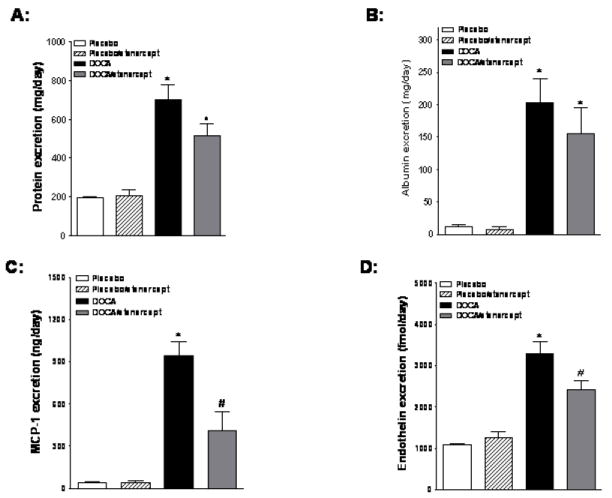
Effect of etanercept treatment on protein, albumin, MCP-1, and endothelin excretion in DOCA-salt treated rats. (A) Protein excretion in placebo and DOCA-salt hypertensive rats with or without etanecrept treatment. (B, C, and D) Albumin, MCP-1, and endothelin excretion, respectively, in the same rat groups (n=5-6 per group). Values are means±SEM. * P< 0.05 vs. placebo group and # P< 0.05 vs. DOCA-salt group.
TNF-α mRNA and protein expression increased in the kidney cortex of DOCA-salt hypertensive rats compared to placebo and etanercept treatment reduced these changes although it was only significant for mRNA (Figure 3A). Renal cortical TGF-β mRNA expression also increased in DOCA-salt hypertensive rats and etanercept treatment produced a modest decrease in TGF-β expression. However, we did not observe any significant difference in renal cortical TGF-β protein expression among four groups (Figure 3B).
Figure 3.
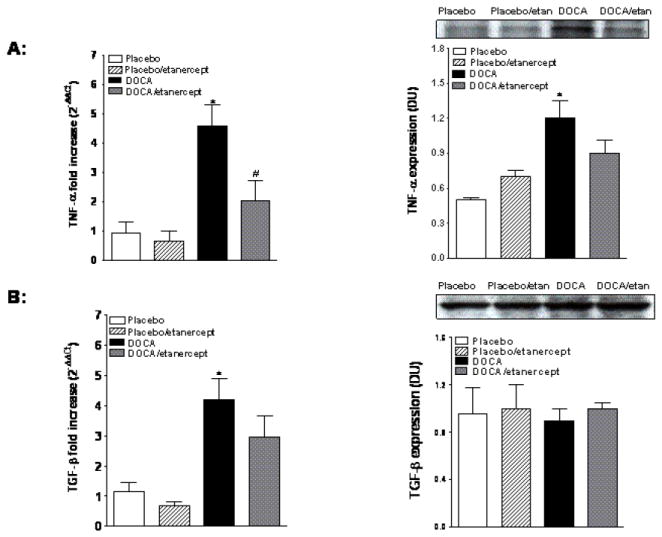
Renal cortical TNF-α and TGF-β mRNA and protein expression in placebo and DOCA-salt treated rats (A and B, respectively). Values are means±SEM. * P< 0.05 vs. placebo group and # P< 0.05 vs. DOCA-salt group.
Plasma 8-isoprostane concentrations, a measure of oxidative stress, were significantly higher in DOCA-salt hypertensive rats when compared with placebo rats (32 ± 5.1 vs. 21 ± 0.1.4 pg/ml, P<0.05). Etanercept treatment had no effect on the DOCA-salt-induced increase in plasma 8-isoprostane (35± 9 pg/ml). Renal cortical NFκB mRNA expression was increased in DOCA-salt hypertensive rats compared to placebo rats (Figure 4A). TNF-α inhibition had no significant effect on NFκB mRNA expression in DOCA-salt rats although there was a trend towards reduced expression. Similar to expression, renal cortical activity of NFκB also increased in DOCA-salt hypertension compared to placebo (1.4 ± 0.4 vs. 0.3 ± 0.08 pg/ng protein, P<0.05). Etanercept treatment significantly lowered the renal activity of NFκB in DOCA-salt hypertension (0.6± 0.08 pg/ng protein) (Figure 4B).
Figure 4.
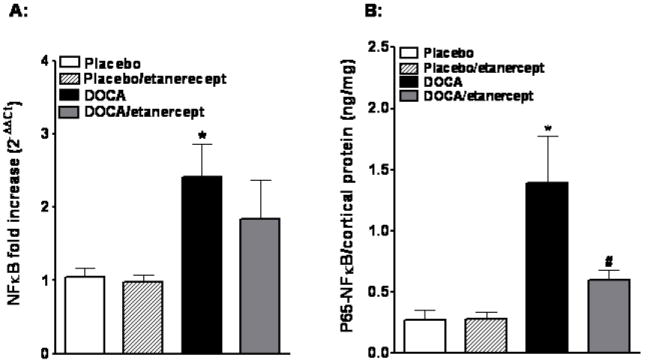
Renal cortical NFκB mRNA expression and activity in DOCA-salt hypertensive rats. (A) Cortical NFκB mRNA expression in placebo, placebo/etanercept, DOCA, DOCA/etanercept rats. (B) P65-NFκB activity in renal cortex of the same groups (n=5-6 per group). Values are means±SEM. * P <0.05 vs. placebo group and # P< 0.05 vs. DOCA-salt group.
We also determined the effect of TNF-α inhibition on expression of cell adhesion molecules in DOCA salt rats. Renal cortical ICAM-1 mRNA and protein expression increased in DOCA-salt hypertension and etanercept treatment reduced the increase in mRNA but not protein expression (Figure 5A). However, we have not seen any changes among these groups in either renal cortical VCAM-1 mRNA or protein expression (Figure 5B).
Figure 5.
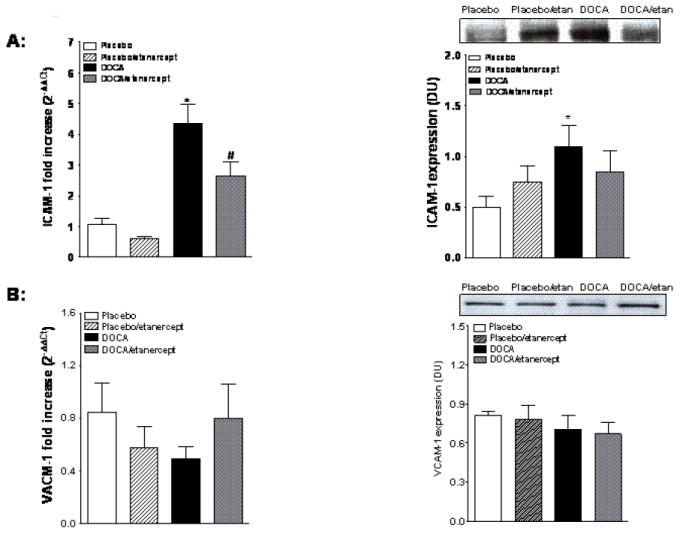
Cell adhesion molecules expression in kidney cortex of DOCA-salt hypertensive rats. ICAM-1 mRNA and protein expression (A) and VCAM-1 mRNA and protein expression (B) in placebo and DOCA hypertensive rats with or without etanercept treatment (n=5-6 per group). Values are means±SEM. * P <0.05 vs. placebo group and # P< 0.05 vs. DOCA-salt group.
Discussion
Compelling evidence supports the hypothesis that inflammatory cytokines play a critical role in the development of hypertension and end-stage renal disease (7). Therefore; we proposed that blocking TNF-α would decrease renal inflammation in the DOCA-salt model of hypertension. TNF-α inhibition with etanercept treatment reduced most indices of inflammation independent of any blood pressure lowering effect as evidenced by the ability of etanercept to reduce MCP-1 excretion in DOCA-salt hypertensive rats. Etanercept also lowered renal cortical NFκB activity in DOCA-salt hypertension. Renal cortical ICAM-1 and TNF-α expression also decreased in DOCA-salt hypertensive rats upon etanercept treatment along with reductions in urinary ET-1 excretion. Together, these data suggest that TNF-α contributes to renal inflammation associated with mineralocorticoid hypertension.
Although our previous finding showed that etanercept was able to slow the progression of blood pressure elevation in angiotensin II hypertension (9), our present study demonstrated that etanercept did not affect systolic blood pressure in placebo and DOCA-salt hypertensive rats. Our initial observation using tail cuff was confirmed using telemetry to determine whether any subtle change in blood pressure upon etanercept treatment could account for changes in other measured variables. Consistent with our tail cuff pressure data, DOCA-salt treatment increased mean arterial pressure and etanercept failed to lower mean arterial pressure in this model. Our data are also consistent with the previous finding of Muller et al. in which etanercept treatment attenuated albuminuria in double transgenic rats harboring both human renin and angiotensinogen (dTGR) without lowering blood pressure (24). Thus, the data in the present study provide evidence that etanercept treatment protects the kidney from damage and this effect was not dependent on the ability of etanercept to lower blood pressure or even slow the progression of hypertension in DOCA-salt treated rats.
Albumin excretion is an important marker for the progression to end-stage renal disease and was found to positively correlate with renal TNF-α levels in a rat model of diabetic nephropathy (25). In our study, DOCA-salt treatment significantly increased urinary albumin and protein excretion. Although not statistically significant, etanercept treatment reduced urinary excretion of albumin and protein in DOCA salt hypertensive rats. These trends are consistent with our previous finding where etanercept reduced albumin and protein excretion in angiotensin salt sensitive hypertension (9). Since these changes are not significant in our study, it is quite possible that much of the albuminuria and proteinuria in the DOCA-salt hypertensive model is driven by the elevations in renal perfusion pressure and mediators aside from TNF-α.
TNF-α is a monocyte and macrophage-derived inflammatory cytokine and can produce vasodilation, increased vascular permeability, and platelet activation (26). For example, infusion of TNF-α induced inflammation of glomerular capillaries in rabbits (3). Etanercept binds to TNF-α and prevents it from interacting with TNF-α receptors (11). Thus, blocking TNF-α may reduce the incidence of cardiac and renal vascular inflammation associated with hypertension. In the present study, renal TNF-α expression increased in DOCA-salt hypertension and inhibiting TNF-α with etanercept reduced renal TNF-α expression and renal damage in the DOCA-salt model. A previous study showed that plasma and tissue TNF-α levels increased in DOCA treated rats and this effect was blocked by the mineralocorticoid antagonist, spiranolactone (10). These data suggest that TNF-α is involved in the inflammatory response and renal damage in mineralocorticoid-dependent hypertension and suggest to us that TNF-α inhibition could be a potential therapy in to reduce hypertension-induced renal inflammation.
There is substantial evidence that TNF-α stimulates ET-1 release, which is a potent vasoconstrictor peptide that contributes to the progression of renal diseases (6,34). For example, Marsden et al have previously demonstrated that TNF-α increased ET-1 release and preproendothelin mRNA content in vitro in bovine renal artery and glomerular capillary endothelial cells (21). Infusion of TNF-α in vivo was found to increase arterial pressure and preproendothelin in kidney, placenta, and aorta of normal pregnant rats and ETA receptor blockade attenuated the increase in blood pressure produced by TNF-α infusion (17). Similarly, ETA receptor blockade also lowered MAP and renal injury in DOCA-salt model (23). In our study, urinary ET-1 excretion was increased in DOCA-salt hypertensive rats. Interestingly, the increase in ET-1 excretion was significantly reduced with etanercept treatment in DOCA-salt hypertension. These data suggest that the TNF-α induced inflammatory response and renal injury could be, at least in part, through the stimulation of renal ET-1 production in DOCA-salt hypertension. However, the relationship between endothelin and TNF-α needs further investigation given the contrasting actions of ETA versus ETB receptors to increase and decrease blood pressure, respectively (35).
TNF-α has been shown to activate the nuclear factor NFκB, which modulates gene expression of many inflammatory genes, such as cytokines, chemokines, and growth factors (12,14,36). Normally, NFκB is composed of two subunits that are present in the cytoplasm as inactive heterodimers (2). Once stimulated, NFκB translocates to the nucleus and regulates the transcription of cell adhesion molecules and chemoattractant factors including monocyte chemoattractant protein (MCP-1) leading to vascular inflammation and organ damage (2,37,38). To explore whether TNF-α-mediated renal damage may operate through a NFκB-dependent mechanism in DOCA-salt hypertension, we measured renal cortical NFκB expression and activity in each of the treatment groups. Both NFκB expression and activity were increased in DOCA-salt rats and etanercept treatment tended to reduce the increase in expression, but more importantly, renal cortical NFκB activity was significantly reduced by etanercept in the kidneys of DOCA-salt hypertensive rats. Previous findings demonstrated that renal NFκB was increased in DOCA salt hypertensive rats (4,31). Muller et al has also shown that NFκB activity increased in dTGR rats and this effect was reduced with etanerecept treatment (24). Clinically, NFκB activity was reduced in epidermis of psoriasis patients upon etanercept treatment (19). These data suggest that down-regulation of NFκB is a potential mechanism of action of TNF-α inhibitors.
Because TNF-α-induced NFκB activation can also increase production of down-stream inflammatory markers such as adhesion molecules and MCP-1 (22,31), we also determined renal expression of TGF-β, ICAM-1, and VCAM-1 as well as urinary MCP-1 excretion. Our results suggest that TNF-α contributes to the increase in renal TGF-β and ICAM-1 expression increased in DOCA-salt hypertension and was reduced with etanercept treatment; however, we did not see any changes in VCAM-1 or TGF-β expression among treated the various groups. Urinary MCP-1 excretion also was increased in DOCA-salt hypertension and this effect was reduced with etanercept treatment. Previous studies have shown that TGF-β, adhesion molecules, and MCP-1 are increased in kidney and heart of DOCA hypertensive rats (16.27,28,31). Rodriguez et al also showed that the increased in NFκB activity was accompanied by increased ICAM-1 and MCP-1 mRNA expression in kidney of SHR rats and these effects were reduced with NFκB inhibition (32,33). These data suggest that down-regulation of NFκB with etanercept treatment protects the kidney from at least some of the factors stimulated by NFκB such as ICAM-1 and MCP-1.
In conclusion, our data indicate that TNF-α contributes to the progression of renal damage in DOCA-salt hypertension and this effect is independent of blood pressure lowering. Overall, these results suggest that reduction of the TNF-α mediated inflammatory response may be a potential therapeutic strategy for the prevention of renal inflammation and the development of end organ damage.
Acknowledgments
These studies were supported by grants from the National Heart Lung and Blood Institute (HL59699, HL64776, and HL74167), American Heart Association Established Investigator Awards to J.D. Imig, J.S. Pollock and D.M. Pollock and Post-doctoral Fellowships from the Southeast Affiliate of the American Heart Association awarded to A. Elmarakby.
References
- 1.Allcock GH, Venema RC, Pollock DM. ETA receptor blockade attenuates the hypertension but not renal dysfunction in DOCA-salt rats. Am J Physiol. 1998;275:R245–R252. doi: 10.1152/ajpregu.1998.275.1.R245. [DOI] [PubMed] [Google Scholar]
- 2.Baldwin AS., Jr The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 3.Bertani T, Abbate M, Zoja C, Corna D, Perico N, Ghezzi P, Remuzzi G. Tumor necrosis factor induces glomerular damage in the rabbit. Am J Pathol. 1989;134:419–430. [PMC free article] [PubMed] [Google Scholar]
- 4.Beswick RA, Zhang H, Marable D, Catravas JD, Hill WD, Webb RC. Long-term antioxidant administration attenuates mineralocorticoid hypertension and renal inflammatory response. Hypertension. 2001;37:781–786. doi: 10.1161/01.hyp.37.2.781. [DOI] [PubMed] [Google Scholar]
- 5.Callera GE, Tostes RC, Yogi A, Montezano AC, Touyz RM. Endothelin-1-induced oxidative stress in DOCA-salt hypertension involves NADPH-oxidase-independent mechanisms. Clin Sci (Lond) 2006;110:243–253. doi: 10.1042/CS20050307. [DOI] [PubMed] [Google Scholar]
- 6.Chang MY, Parker E, El Nahas M, Haylor JL, Ong AC. Endothelin B receptor blockade accelerates disease progression in a murine model of autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2007;18:560–569. doi: 10.1681/ASN.2006090994. [DOI] [PubMed] [Google Scholar]
- 7.Chatziantoniou C, Boffa JJ, Tharaux PL, Flamant M, Ronco P, Dussaule JC. Progression and regression in renal vascular and glomerular fibrosis. Int J Exp Pathol. 2004;85:1–11. doi: 10.1111/j.0959-9673.2004.00376.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elmarakby AA, Morsing P, Pollock JS, Pollock DM. Omapatrilat increases renal endothelin in deoxycorticosterone acetate-salt hypertensive rats. Vascul Pharmacol. 2003;40:253–259. doi: 10.1016/j.vph.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 9.Elmarakby AA, Quigley JE, Pollock DM, Imig JD. Tumor necrosis factor alpha blockade increases renal Cyp2c23 expression and slows the progression of renal damage in salt-sensitive hypertension. Hypertension. 2006;47:557–562. doi: 10.1161/01.HYP.0000198545.01860.90. [DOI] [PubMed] [Google Scholar]
- 10.Francis J, Beltz T, Johnson AK, Felder RB. Mineralocorticoids act centrally to regulate blood-borne tumor necrosis factor-alpha in normal rats. Am J Physiol Regul Integr Comp Physiol. 2003;285:R1402–R1409. doi: 10.1152/ajpregu.00027.2003. [DOI] [PubMed] [Google Scholar]
- 11.Goffe B, Cather JC. Etanercept: An overview. J Am Acad Dermatol. 2003;49:S105–111. doi: 10.1016/mjd.2003.554. [DOI] [PubMed] [Google Scholar]
- 12.Greenberg AS, McDaniel ML. Identifying the links between obesity, insulin resistance and beta-cell function: potential role of adipocyte-derived cytokines in the pathogenesis of type 2 diabetes. Eur J Clin Invest. 2002;32:24–34. doi: 10.1046/j.1365-2362.32.s3.4.x. [DOI] [PubMed] [Google Scholar]
- 13.Hasegawa G, Nakano K, Sawada M, Uno K, Shibayama Y, Ienaga K, Kondo M. Possible role of tumor necrosis factor and interleukin-1 in the development of diabetic nephropathy. Kidney Int. 1991;40:1007–1012. doi: 10.1038/ki.1991.308. [DOI] [PubMed] [Google Scholar]
- 14.Higuchi Y, Otsu K, Nishida K, Hirotani S, Nakayama H, Yamaguchi O, Matsumura Y, Ueno H, Tada M, Hori M. Involvement of reactive oxygen species-mediated NF-kappa B activation in TNF-alpha-induced cardiomyocyte hypertrophy. J Mol Cell Cardiol. 2002;34:233–40. doi: 10.1006/jmcc.2001.1505. [DOI] [PubMed] [Google Scholar]
- 15.Hruby ZW, Shirota K, Jothy S, Lowry RP. Antiserum against tumor necrosis factor-alpha and a protease inhibitor reduce immune glomerular injury. Kidney Int. 1991;40:43–45. doi: 10.1038/ki.1991.177. [DOI] [PubMed] [Google Scholar]
- 16.Kagitani S, Ueno H, Hirade S, Takahashi T, Takata M, Inoue H. Tranilast attenuates myocardial fibrosis in association with suppression of monocyte/macrophage infiltration in DOCA/salt hypertensive rats. J Hypertens. 2004;22:1007–1115. doi: 10.1097/00004872-200405000-00024. [DOI] [PubMed] [Google Scholar]
- 17.LaMarca BB, Cockrell K, Sullivan E, Bennett W, Granger JP. Role of endothelin in mediating tumor necrosis factor-induced hypertension in pregnant rats. Hypertension. 2005;46:82–86. doi: 10.1161/01.HYP.0000169152.59854.36. [DOI] [PubMed] [Google Scholar]
- 18.Little MA, Bhangal G, Smyth CL, Nakada MT, Cook HT, Nourshargh S, Pusey CD. Therapeutic effect of anti-TNF-alpha antibodies in an experimental model of anti-neutrophil cytoplasm antibody-associated systemic vasculitis. J Am Soc Nephrol. 2006;17:160–169. doi: 10.1681/ASN.2005060616. [DOI] [PubMed] [Google Scholar]
- 19.Lizzul PF, Aphale A, Malaviya R, Sun Y, Masud S, Dombrovskiy V, Gottlieb AB. Differential expression of phosphorylated NF-kappaB/RelA in normal and psoriatic epidermis and downregulation of NF-kappaB in response to treatment with etanercept. J Invest Dermatol. 2005;124:1275–1283. doi: 10.1111/j.0022-202X.2005.23735.x. [DOI] [PubMed] [Google Scholar]
- 20.Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001;104:487–501. doi: 10.1016/s0092-8674(01)00237-9. [DOI] [PubMed] [Google Scholar]
- 21.Marsden PA, Brenner BM. Transcriptional regulation of the endothelin-1 gene by TNF-alpha. Am J Physiol. 1992;262:C854–C861. doi: 10.1152/ajpcell.1992.262.4.C854. [DOI] [PubMed] [Google Scholar]
- 22.Martin T, Cardarelli PM, Parry GC, Felts KA, Cobb RR. Cytokine induction of monocyte chemoattractant protein-1 gene expression in human endothelial cells depends on the cooperative action of NF-kappa B and AP-1. Eur J Immunol. 1997;27:1091–1097. doi: 10.1002/eji.1830270508. [DOI] [PubMed] [Google Scholar]
- 23.Matsumura Y, Hashimoto N, Taira S, Kuro T, Kitano R, Ohkita M, Opgenorth TJ, Takaoka M. Different contributions of endothelin-A and endothelin-B receptors in the pathogenesis of deoxycorticosterone acetate-salt-induced hypertension in rats. Hypertension. 1999;33(2):759–765. doi: 10.1161/01.hyp.33.2.759. [DOI] [PubMed] [Google Scholar]
- 24.Muller DN, Shagdarsuren E, Park JK, Dechend R, Mervaala E, Hampich F, Fiebeler A, Ju X, Finckenberg P, Theuer J, Viedt C, Kreuzer J, Heidecke H, Haller H, Zenke M, Luft FC. Immunosuppressive treatment protects against angiotensin II-induced renal damage. Am J Pathol. 2002;161:1679–1693. doi: 10.1016/S0002-9440(10)64445-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Navarro JF, Milena FJ, Mora C, Leon C, Claverie F, Flores C, Garcia J. Tumor necrosis factor-alpha gene expression in diabetic nephropathy: relationship with urinary albumin excretion and effect of angiotensin-converting enzyme inhibition. Kidney Int. 2005;99:S98–102. doi: 10.1111/j.1523-1755.2005.09918.x. [DOI] [PubMed] [Google Scholar]
- 26.Navarro JF, Mora-Fernandez C. The role of TNF-alpha in diabetic nephropathy: pathogenic and therapeutic implications. Cytokine Growth Factor Rev. 2006;17:441–450. doi: 10.1016/j.cytogfr.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 27.Ogata T, Miyauchi T, Sakai S, Takanashi M, Irukayama-Tomobe Y, Yamaguchi I. Myocardial fibrosis and diastolic dysfunction in deoxycorticosterone acetate-salt hypertensive rats is ameliorated by the peroxisome proliferator-activated receptor-alpha activator fenofibrate, partly by suppressing inflammatory responses associated with the nuclear factor-kappa-B pathway. J Am Coll Cardiol. 2004;43:1481–1488. doi: 10.1016/j.jacc.2003.11.043. [DOI] [PubMed] [Google Scholar]
- 28.Oishi T, Ogura T, Yamauchi T, Harada K, Ota Z. Effect of renin-angiotensin inhibition on glomerular injuries in DOCA-salt hypertensive rats. Regul Pept. 1996;62:89–95. doi: 10.1016/0167-0115(95)00166-2. [DOI] [PubMed] [Google Scholar]
- 29.Pollock DM, Pollock JS. Evidence for endothelin involvement in the response to high salt. Am J Physiol Renal Physiol. 2001;281:F144–F150. doi: 10.1152/ajprenal.2001.281.1.F144. [DOI] [PubMed] [Google Scholar]
- 30.Pollock DM, Rekito A. Hypertensive response to chronic NO synthase inhibition is different in Sprague-Dawley rats from two suppliers. Am J Physiol Regul Integr Comp Physiol. 1998;275:R1719–R1723. doi: 10.1152/ajpregu.1998.275.5.R1719. [DOI] [PubMed] [Google Scholar]
- 31.Pu Q, Amiri F, Gannon P, Schiffrin EL. Dual angiotensin-converting enzyme/neutral endopeptidase inhibition on cardiac and renal fibrosis and inflammation in DOCA-salt hypertensive rats. J Hypertens. 2005;23:401–409. doi: 10.1097/00004872-200502000-00023. [DOI] [PubMed] [Google Scholar]
- 32.Rodriguez-Iturbe B, Ferrebuz A, Vanegas V, Quiroz Y, Mezzano S, Vaziri ND. Early and sustained inhibition of nuclear factor-kappaB prevents hypertension in spontaneously hypertensive rats. J Pharmacol Exp Ther. 2005;315:51–57. doi: 10.1124/jpet.105.088062. [DOI] [PubMed] [Google Scholar]
- 33.Rodriguez-Iturbe B, Quiroz Y, Ferrebuz A, Parra G, Vaziri ND. Evolution of renal interstitial inflammation and NF-kappaB activation in spontaneously hypertensive rats. Am J Nephrol. 2004;24:587–594. doi: 10.1159/000082313. [DOI] [PubMed] [Google Scholar]
- 34.Schiffrin EL. Vascular endothelin in hypertension. Vascul Pharmacol. 2005;43:19–29. doi: 10.1016/j.vph.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 35.Schneider MP, Boesen EI, Pollock DM. Contrasting actions of endothelin ETA and ETB receptors in cardiovascular disease. Ann Rev Pharmacol. 2007;47:731–759. doi: 10.1146/annurev.pharmtox.47.120505.105134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang L, Reinach P, Lu L. TNF-alpha promotes cell survival through stimulation of K+ channel and NFkappaB activity in corneal epithelial cells. Exp Cell Res. 2005;311:39–48. doi: 10.1016/j.yexcr.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yamaoka S, Courtois G, Bessia C, Whiteside ST, Weil R, Agou F, Kirk HE, Kay RJ, Israel A. Complementation cloning of NEMO, a component of the IkappaB kinase complex essential for NF-kappaB activation. Cell. 1998;7:1231–1240. doi: 10.1016/s0092-8674(00)81466-x. [DOI] [PubMed] [Google Scholar]
- 38.Zandi E, Chen Y, Karin M. Direct phosphorylation of IkappaB by IKKalpha and IKKbeta: discrimination between free and NF-kappaB-bound substrate. Science. 1998;281:1360–1363. doi: 10.1126/science.281.5381.1360. [DOI] [PubMed] [Google Scholar]